
A Novel Lid-Covering Peptide Inhibitor of Nicotinic Acetylcholine Receptors Derived from αD-Conotoxin GeXXA

 , ,
, ,  and
and
Abstract
:1. Introduction
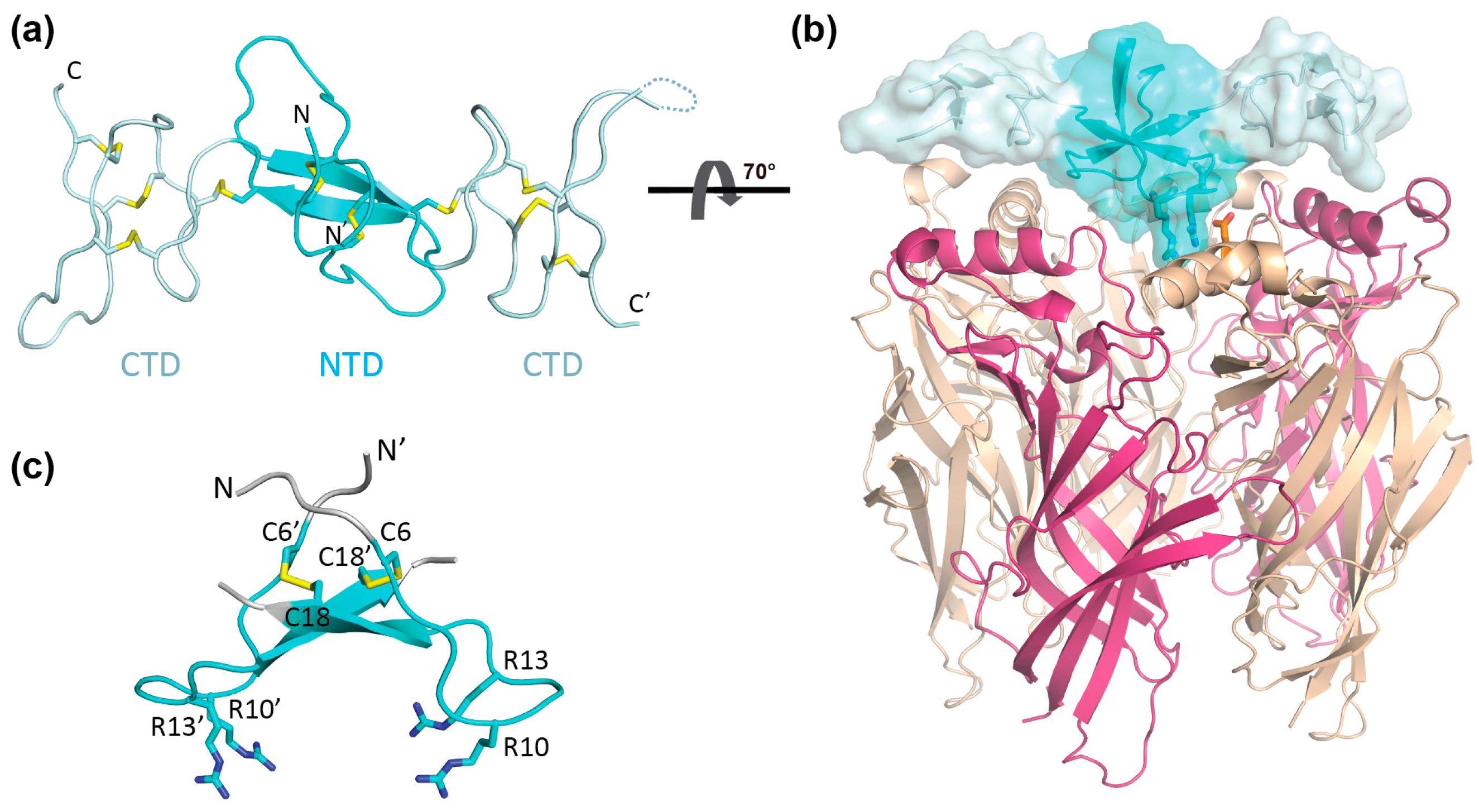
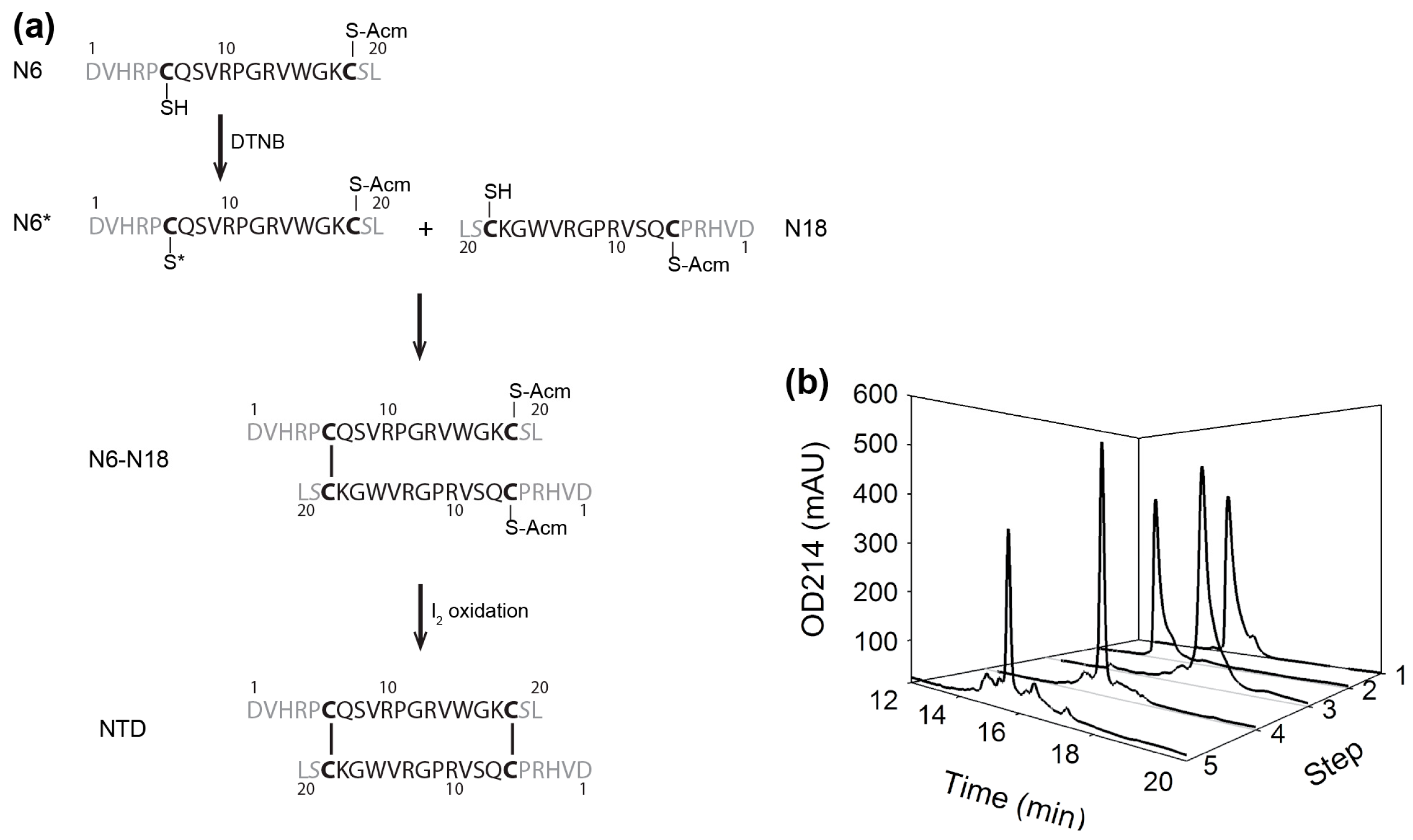
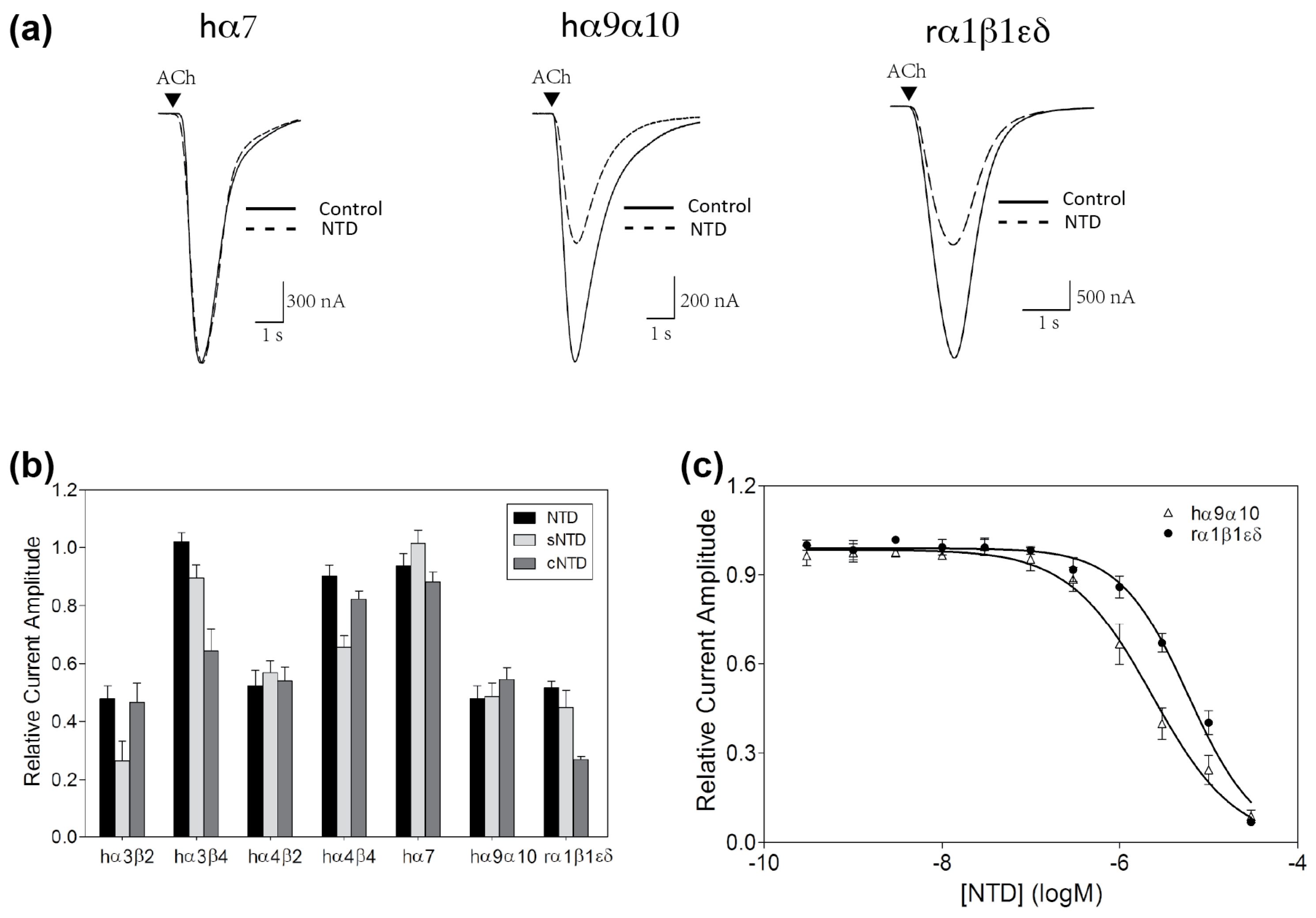
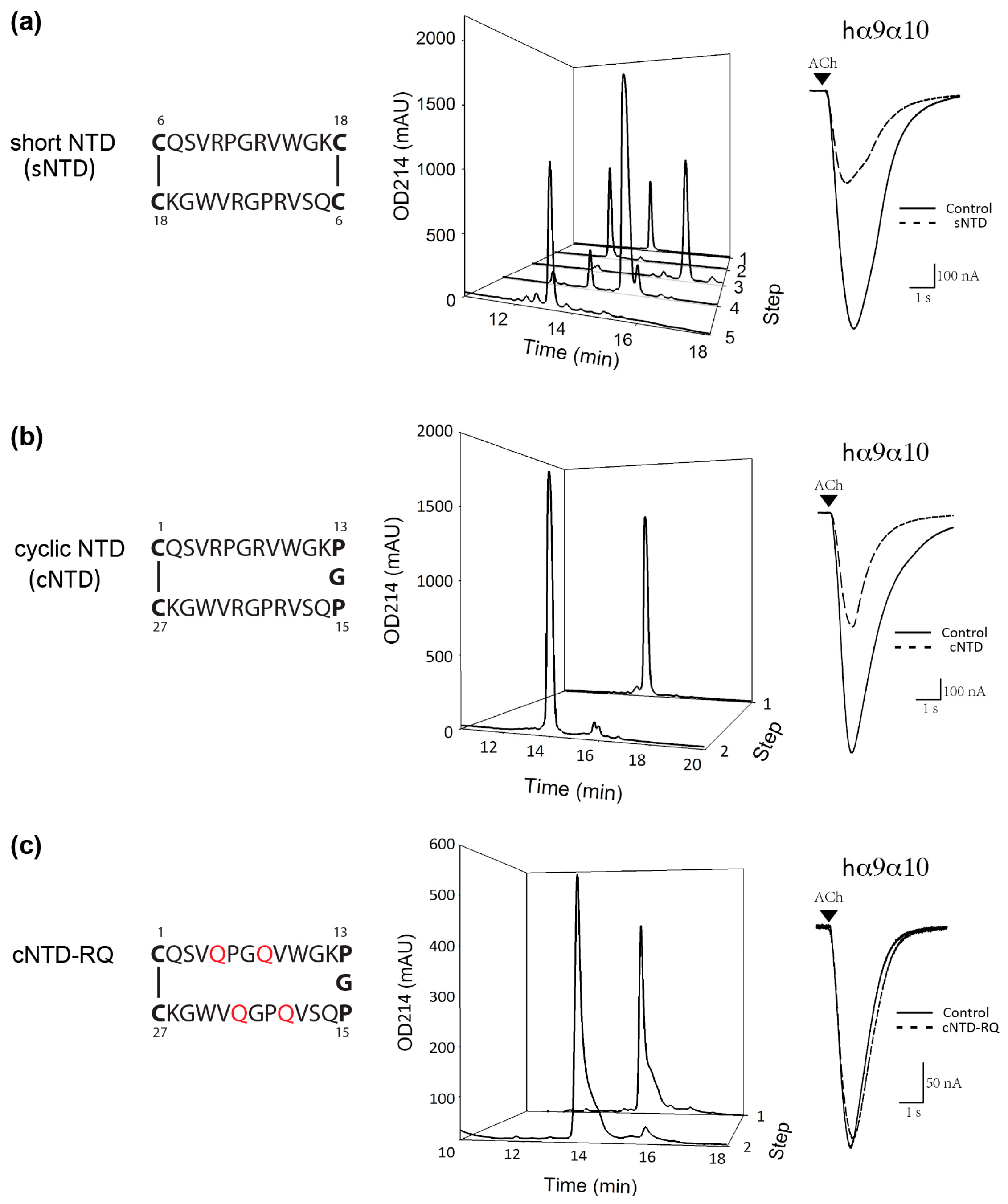
2. Results
2.1. Preparation of αD-GeXXA NTD
2.2. nAChR-Inhibitory Activity of NTD
2.3. Preparation and Activity of Truncated NTD
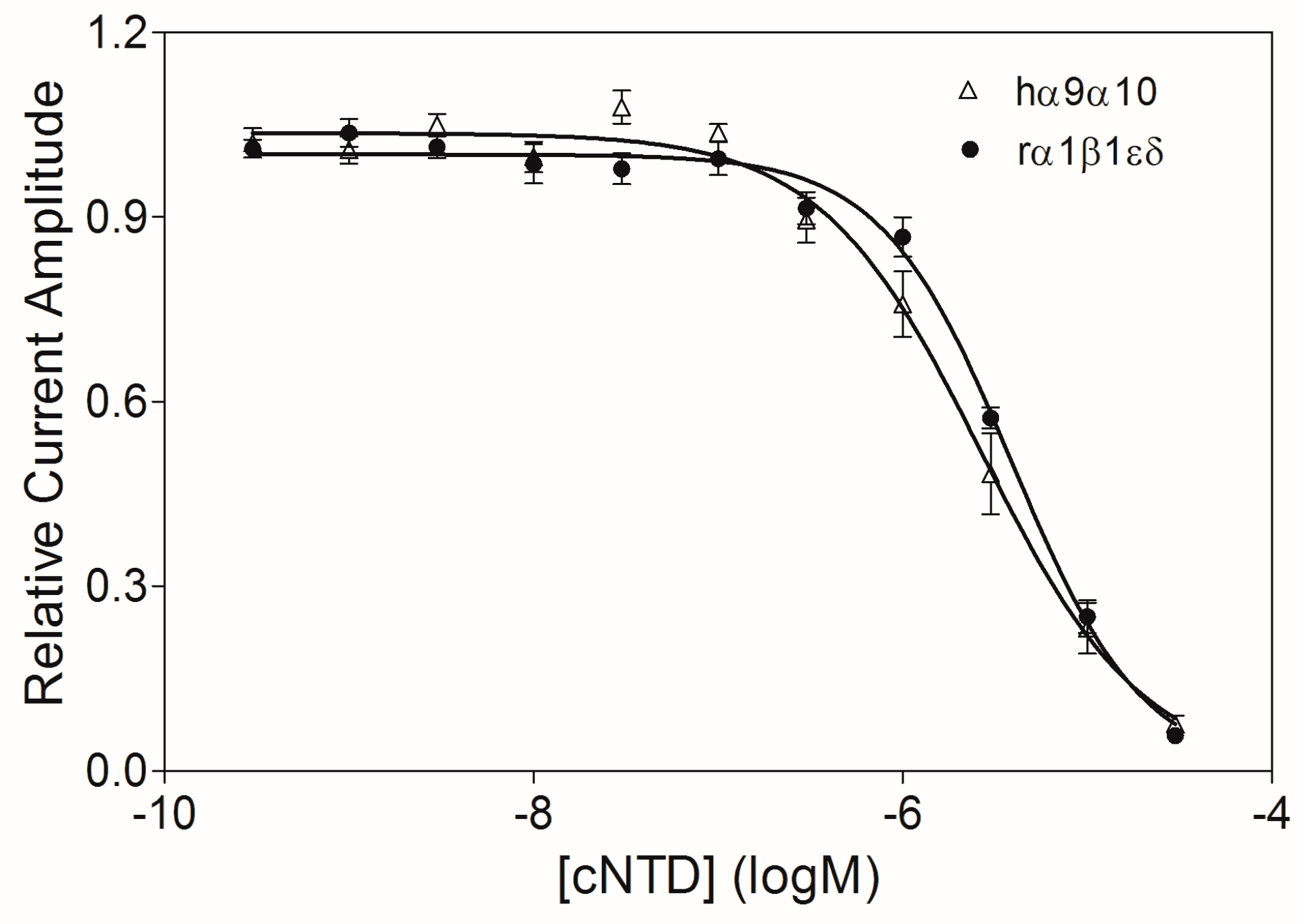
2.4. Preparation and Activity of Cyclic NTD
3. Discussion
4. Materials and Methods
4.1. Preparation of NTD and sNTD
4.2. Preparation of cNTD and cNTD-RQ
4.3. Electrophysiological Recordings from nAChRs Exogenously Expressed in Xenopus Oocytes
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Kalamida, D.; Poulas, K.; Avramopoulou, V.; Fostieri, E.; Lagoumintzis, G.; Lazaridis, K.; Sideri, A.; Zouridakis, M.; Tzartos, S.J. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J. 2007, 274, 3799–3845. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.S.; Jayakar, S.S.; Hamouda, A.K. Orthosteric and allosteric ligands of nicotinic acetylcholine receptors for smoking cessation. Front. Mol. Neurosci. 2015, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, D.; Shelukhina, I.; Vulfius, C.; Makarieva, T.; Stonik, V.; Zhmak, M.; Ivanov, I.; Kasheverov, I.; Utkin, Y.; Tsetlin, V. Natural compounds interacting with nicotinic acetylcholine receptors: From low-molecular weight ones to peptides and proteins. Toxins 2015, 7, 1683–1701. [Google Scholar] [CrossRef] [PubMed]
- Tsetlin, V.; Utkin, Y.; Kasheverov, I. Polypeptide and peptide toxins, magnifying lenses for binding sites in nicotinic acetylcholine receptors. Biochem. Pharmacol. 2009, 78, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.; Peigneur, S.; Wijesekara, I.; Tytgat, J. Conotoxins targeting nicotinic acetylcholine receptors: An overview. Mar. Drugs 2014, 12, 2970–3004. [Google Scholar] [CrossRef] [PubMed]
- Hamouda, A.K.; Jayakar, S.S.; Chiara, D.C.; Cohen, J.B. Photoaffinity labeling of nicotinic receptors: Diversity of drug binding sites! J. Mol. Neurosci. 2014, 53, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Spurny, R.; Debaveye, S.; Farinha, A.; Veys, K.; Vos, A.M.; Gossas, T.; Atack, J.; Bertrand, S.; Bertrand, D.; Danielson, U.H.; et al. Molecular blueprint of allosteric binding sites in a homologue of the agonist-binding domain of the α7 nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2015, 112, E2543–E2552. [Google Scholar] [CrossRef] [PubMed]
- Chatzidaki, A.; Millar, N.S. Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 97, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Wermeling, D.P. Ziconotide, an intrathecally administered N-type calcium channel antagonist for the treatment of chronic pain. Pharmacotherapy 2005, 25, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Essack, M.; Bajic, V.B.; Archer, J.A. Conotoxins that confer therapeutic possibilities. Mar. Drugs 2012, 10, 1244–1265. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.; Karim, S.; Kamal, M.A.; Wilson, C.M.; Mirza, Z. Conotoxins: Structure, therapeutic potential and pharmacological applications. Curr. Pharm. Des. 2016, 22, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.A.; Christie, M.J. Conotoxin interactions with α9α10-nAChRs: Is the α9α10-nicotinic acetylcholine receptor an important therapeutic target for pain management? Toxins 2015, 7, 3916–3932. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Brust, A.; Jin, A.H.; Alewood, P.F.; Dutertre, S.; Lewis, R.J. Cone snail venomics: From novel biology to novel therapeutics. Future Med. Chem. 2014, 6, 1659–1675. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, T.; Kompella, S.N.; Yan, M.; Lu, A.; Wang, Y.; Shao, X.; Chi, C.; Adams, D.J.; Ding, J.; et al. Conotoxin αD-GeXXA utilizes a novel strategy to antagonize nicotinic acetylcholine receptors. Sci. Rep. 2015, 5, 14261. [Google Scholar] [CrossRef] [PubMed]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human α4β2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.C.; Olivera, B.M.; Teichert, R.W. αC-Conotoxin PrXA: A new family of nicotinic acetylcholine receptor antagonists. Biochemistry 2007, 46, 8717–8724. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.B.; Bandyopadhyay, P.K.; Olivera, B.M.; McIntosh, J.M. αS-conotoxin GVIIIB potently and selectively blocks α9α10 nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 96, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Christensen, S.; Zhangsun, D.; Wu, Y.; Hu, Y.; Zhu, X.; Chhabra, S.; Norton, R.S.; McIntosh, J.M. A novel inhibitor of α9α10 nicotinic acetylcholine receptors from Conus vexillum delineates a new conotoxin superfamily. PLoS ONE 2013, 8, e54648. [Google Scholar] [CrossRef] [PubMed]
- Shon, K.J.; Grilley, M.; Jacobsen, R.; Cartier, G.E.; Hopkins, C.; Gray, W.R.; Watkins, M.; Hillyard, D.R.; Rivier, J.; Torres, J.; et al. A noncompetitive peptide inhibitor of the nicotinic acetylcholine receptor from Conus purpurascens venom. Biochemistry 1997, 36, 9581–9587. [Google Scholar] [CrossRef] [PubMed]
- Loughnan, M.; Nicke, A.; Jones, A.; Schroeder, C.I.; Nevin, S.T.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Identification of a novel class of nicotinic receptor antagonists: Dimeric conotoxins VxXIIA, VxXIIB, and VxXIIC from Conus vexillum. J. Biol. Chem. 2006, 281, 24745–24755. [Google Scholar] [CrossRef] [PubMed]
- Loughnan, M.L.; Nicke, A.; Lawrence, N.; Lewis, R.J. Novel αD-conopeptides and their precursors identified by cDNA cloning define the D-conotoxin superfamily. Biochemistry 2009, 48, 3717–3729. [Google Scholar] [CrossRef] [PubMed]
- Zouridakis, M.; Giastas, P.; Zarkadas, E.; Chroni-Tzartou, D.; Bregestovski, P.; Tzartos, S.J. Crystal structures of free and antagonist-bound states of human α9 nicotinic receptor extracellular domain. Nat. Struct. Mol. Biol. 2014, 21, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Ulens, C.; Hogg, R.C.; Celie, P.H.; Bertrand, D.; Tsetlin, V.; Smit, A.B.; Sixma, T.K. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc. Natl. Acad. Sci. USA 2006, 103, 3615–3620. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P.; Edelstein, S.J. Allosteric receptors after 30 years. Neuron 1998, 21, 959–980. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | hα9α10 | rα1β1εδ | ||
|---|---|---|---|---|
| IC50 (95% CI) | Hill Slope (nH) | IC50 (95% CI) | Hill Slope (nH) | |
| αD-GeXXA 1 | 28 nM (22–35) | −1.3 | 743 nM (606–911) | −1.6 |
| CTD 1 | 2.02 μM (1.82–2.25) | −1.7 | - 2 | - 2 |
| NTD | 2.33 μM (1.92–2.83) | −0.9 | 5.88 μM (4.71–7.34) | −1.1 |
| cNTD | 2.66 μM (2.15–3.29) | −1.0 | 3.91 μM (3.35–4.56) | −1.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Tae, H.-S.; Fan, Z.; Shao, X.; Xu, S.; Zhao, S.; Adams, D.J.; Wang, C. A Novel Lid-Covering Peptide Inhibitor of Nicotinic Acetylcholine Receptors Derived from αD-Conotoxin GeXXA. Mar. Drugs 2017, 15, 164. https://doi.org/10.3390/md15060164
Yang L, Tae H-S, Fan Z, Shao X, Xu S, Zhao S, Adams DJ, Wang C. A Novel Lid-Covering Peptide Inhibitor of Nicotinic Acetylcholine Receptors Derived from αD-Conotoxin GeXXA. Marine Drugs. 2017; 15(6):164. https://doi.org/10.3390/md15060164
Chicago/Turabian StyleYang, Longjin, Han-Shen Tae, Zhou Fan, Xiaoxia Shao, Shaoqiong Xu, Suwen Zhao, David J. Adams, and Chunguang Wang. 2017. "A Novel Lid-Covering Peptide Inhibitor of Nicotinic Acetylcholine Receptors Derived from αD-Conotoxin GeXXA" Marine Drugs 15, no. 6: 164. https://doi.org/10.3390/md15060164
APA StyleYang, L., Tae, H. -S., Fan, Z., Shao, X., Xu, S., Zhao, S., Adams, D. J., & Wang, C. (2017). A Novel Lid-Covering Peptide Inhibitor of Nicotinic Acetylcholine Receptors Derived from αD-Conotoxin GeXXA. Marine Drugs, 15(6), 164. https://doi.org/10.3390/md15060164