HPLC-HRMS Quantification of the Ichthyotoxin Karmitoxin from Karlodinium armiger

 , , , and
, , , and
Abstract
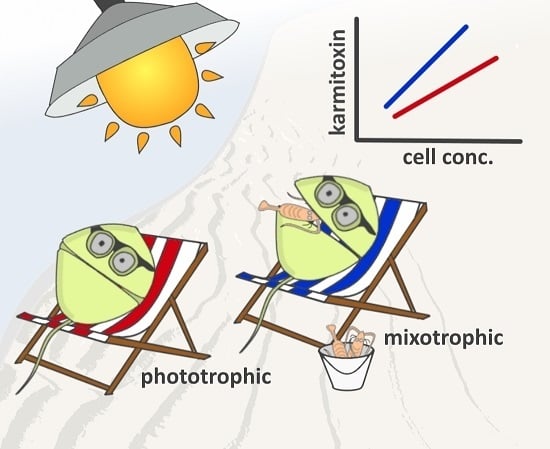
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
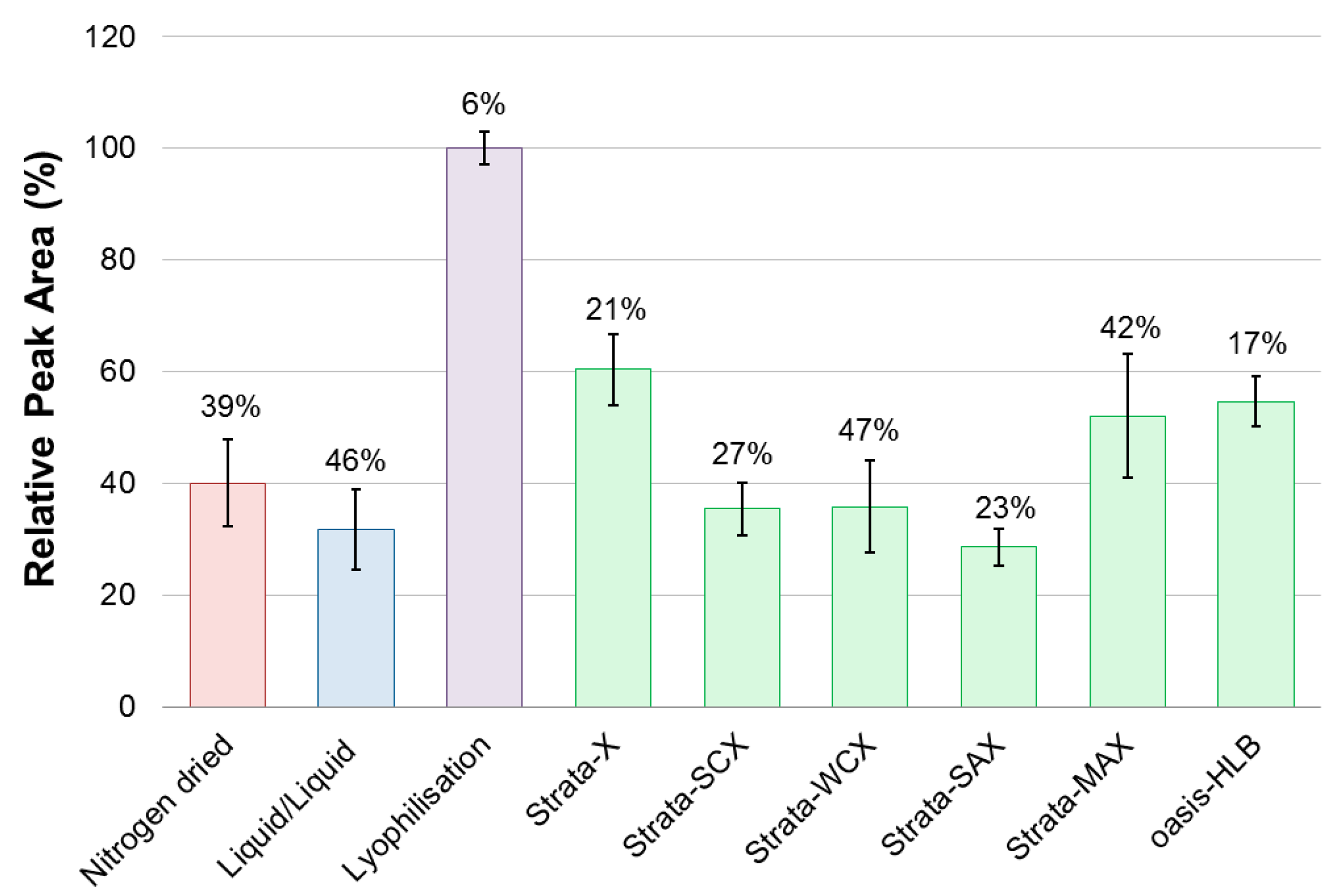
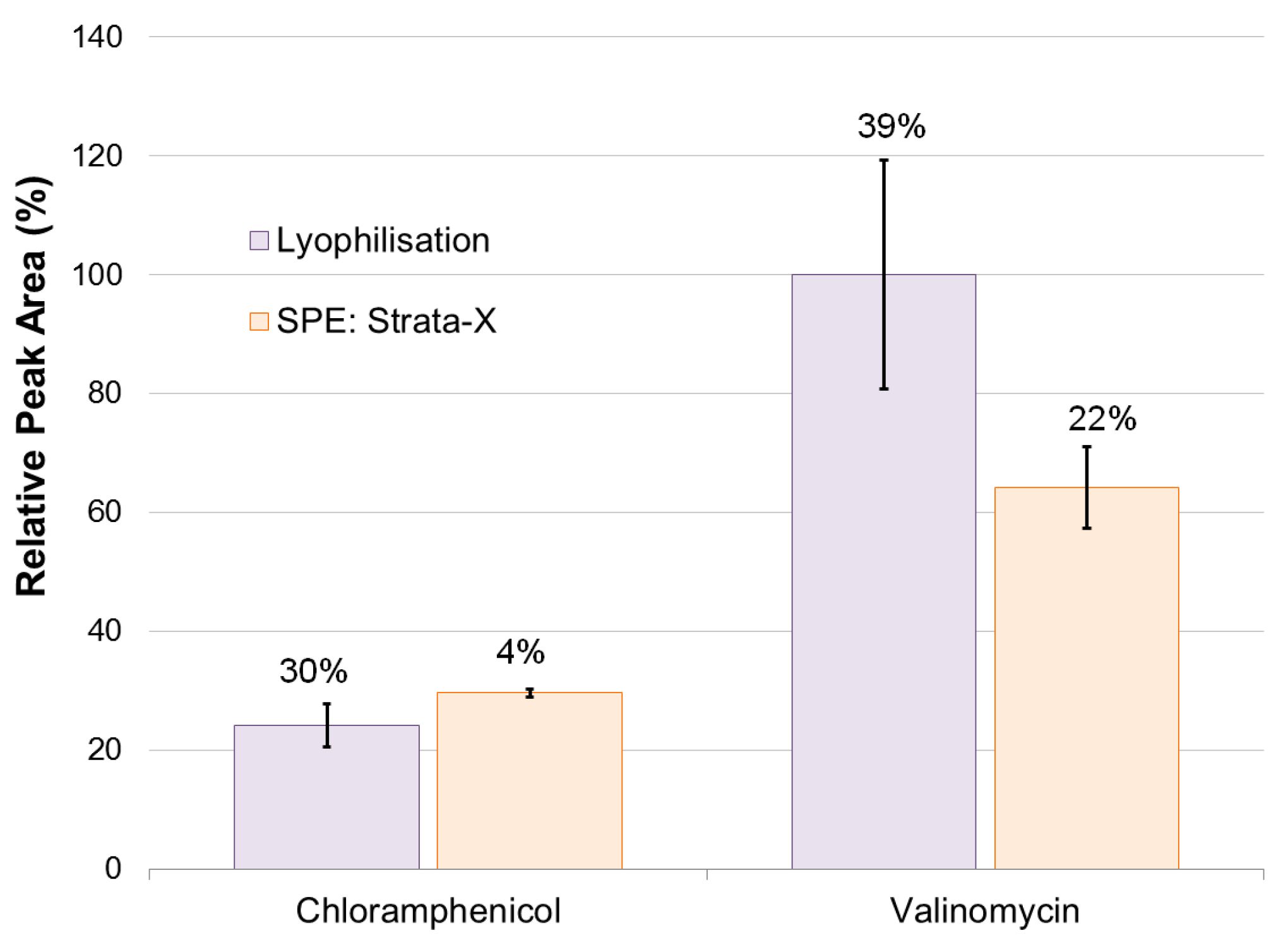
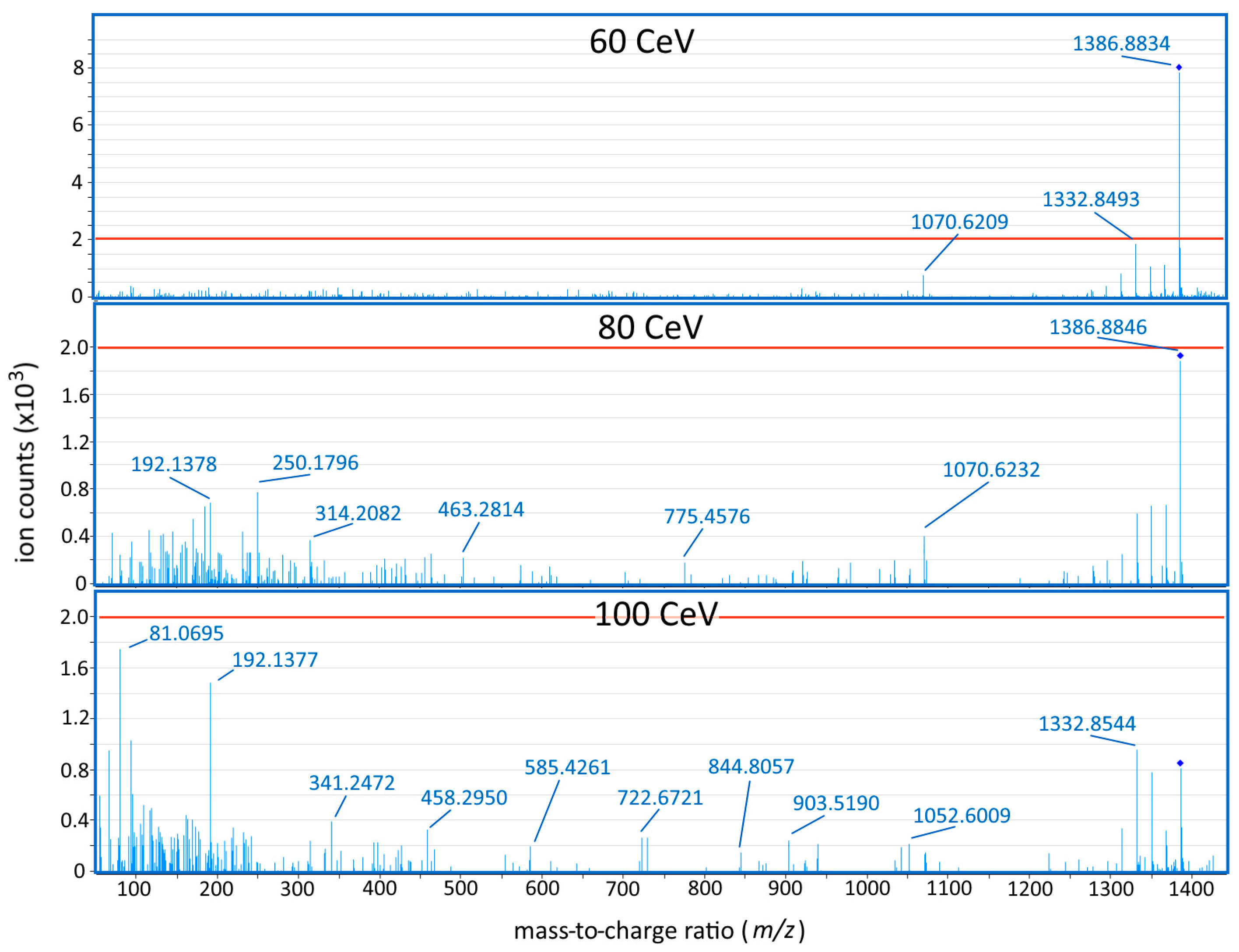
2.1. Method Development
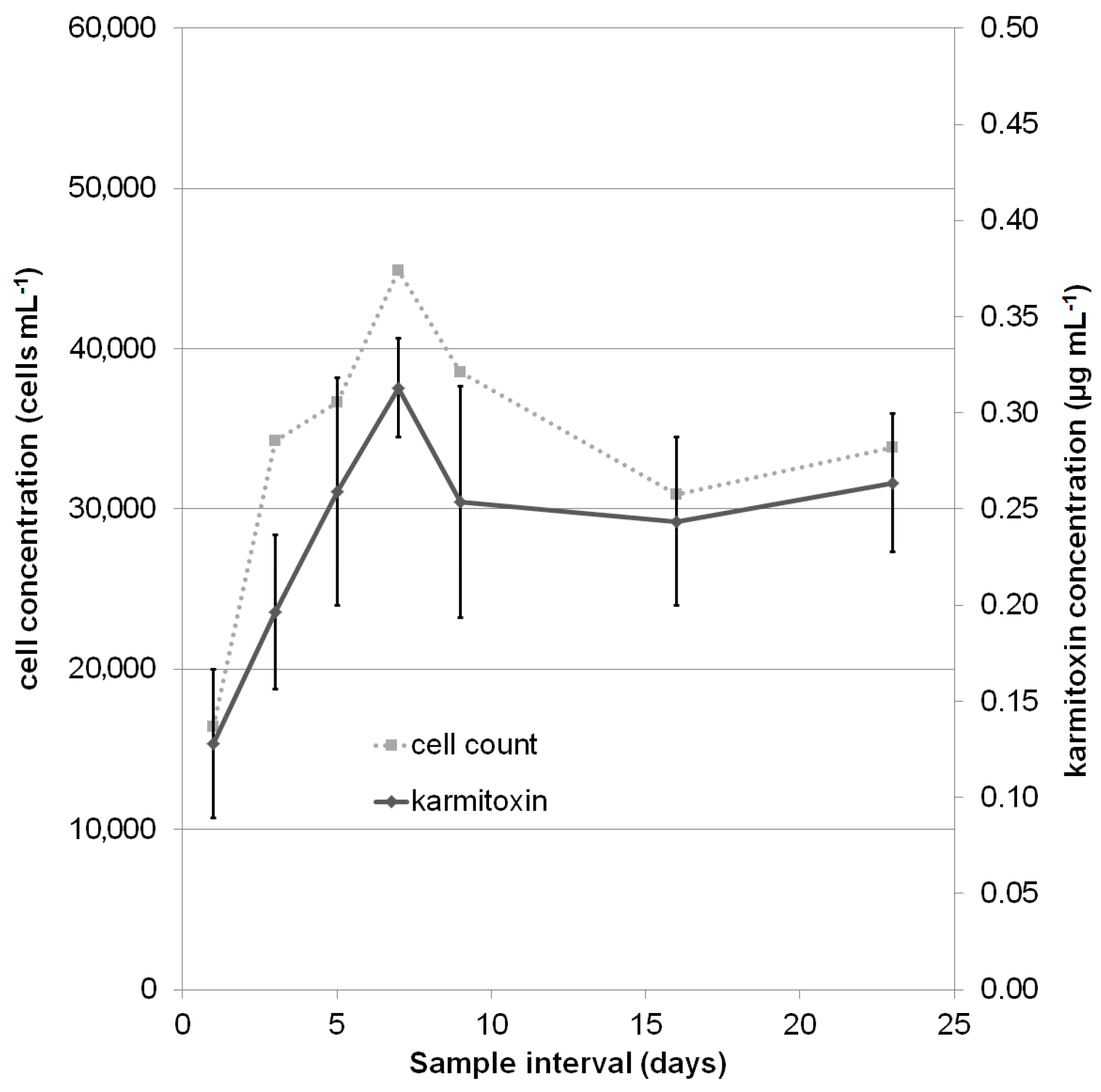
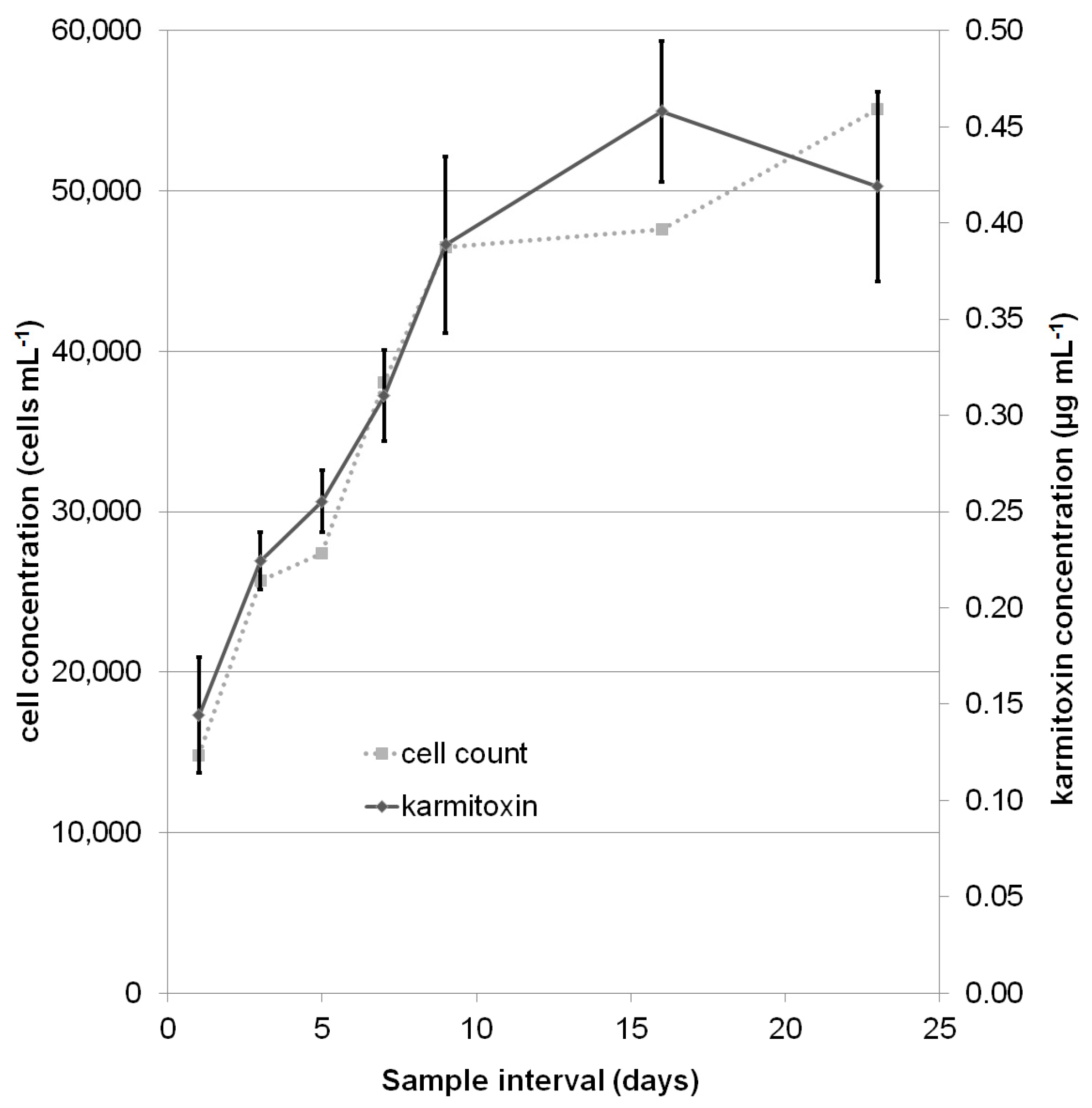
2.2. Quantification
3. Materials and Methods
3.1. Chemicals, Standards and Materials
3.2. Algal Cultures
3.3. Karmitoxin Standard Quantification
3.4. Culture Sampling
- (1)
- The culture subsample (3 mL) was directly dried under a N2 stream (25 °C). The dried sample was dissolved in MeOH (1 mL) and placed in an ultrasonic bath (5 min). A subsample (800 μL) was transferred to an Eppendorf tube (1.5 mL) and centrifuged (10 min, 9000 RCF). The supernatant (500 μL) was transferred to a glass HPLC vial for analysis by LC-MS. The method was performed in five replicates.
- (2)
- Ethlyacetate (EtOAc) (3 mL) was added to the culture subsample (3 mL) and placed in an ultrasonic bath (5 min). The EtOAc layer (2.8 mL) was dried under a N2 stream (25 °C). The dried sample was dissolved in MeOH (1 mL) and placed in an ultrasonic bath (5 min). A subsample (800 μL) was transferred to an Eppendorf tube (1.5 mL) and centrifuged (10 min, 9000 RCF). The supernatant (500 μL) was transferred to a glass HPLC vial for analysis by LC-MS. The method was performed in five replicates.
- (3)
- The culture subsample (3 mL) was frozen to −80 °C and then lyophilised. The lyophilised sample was dissolved in MeOH (2 mL) and placed in an ultrasonic bath (5 min). A subsample (800 μL) was transferred to an Eppendorf tube (1.5 mL) and centrifuged (10 min, 9000 RCF). The supernatant (500 μL) was transferred to a glass HPLC vial for analysis by LC-MS. The method was performed in five replicates.
- (4)
- The culture subsample (3 mL) was directly loaded onto a conditioned (MeOH, 2 mL) and equilibrated (Milli-Q H2O, 2 mL) SPE cartridge (30 mg). This was followed by a salt removal washing step (H2O, 3 mL) and flushing with air. Sample collection then followed with elution (MeOH, 1 mL) and a final flushing with air. A subsample (500 μL) was transferred to a glass HPLC vial for analysis. The method was performed in triplicates on six different solid phases: Strata-X, Strata-SCX, Strata-WCX, Strata-SAX, Strata-MAX, and Oasis-HLB.
3.5. Culture Quantification
3.6. Liquid Chromatography—High Resolution Mass Spectrometry
3.7. Liquid Chromatography—Tandem Mass Spectrometry
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sellner, K.G.; Doucette, G.J.; Kirkpatrick, G.J. Harmful algal blooms: Causes, impacts and detection. J. Ind. Microbiol. Biotechnol. 2003, 30, 383–406. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Glibert, P.M.; Burkholder, J.M. Harmful algal blooms and eutrophication: Nutrient sources, composition, and consequences. Estuaries 2002, 25, 704–726. [Google Scholar] [CrossRef]
- Heisler, J.; Glibert, P.M.; Burkholder, J.M.; Anderson, D.M.; Cochlan, W.; Dennison, W.C.; Dortch, Q.; Gobler, C.J.; Heil, C.A.; Humphries, E.; et al. Eutrophication and harmful algal blooms: A scientific consensus. Harmful Algae 2008, 8, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, P.; Anderson, D.M.; Kaoru, Y.; White, A.W. The economic effects of harmful algal blooms in the United States: Estimates, assessment issues, and information needs. Estuaries 2002, 25, 819–837. [Google Scholar] [CrossRef]
- Garcı́a, C.; del Carmen Bravo, M.; Lagos, M.; Lagos, N. Paralytic shellfish poisoning: Post-mortem analysis of tissue and body fluid samples from human victims in the Patagonia fjords. Toxicon 2004, 43, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic Alkaloids: Saxitoxin and Its Analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed]
- Scholin, C.A.; Gulland, F.; Doucette, G.J.; Benson, S.; Busman, M.; Chavez, F.P.; Cordaro, J.; DeLong, R.; De Vogelaere, A.; Harvey, J.; et al. Mortality of sea lions along the central California coast linked to a toxic diatom bloom. Nature 2000, 403, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Starr, M.; Lair, S.; Michaud, S.; Scarratt, M.; Quilliam, M.; Lefaivre, D.; Robert, M.; Wotherspoon, A.; Michaud, R.; Ménard, N.; et al. Multispecies mass mortality of marine fauna linked to a toxic dinoflagellate bloom. PLoS ONE 2017, 12, e0176299. [Google Scholar] [CrossRef] [PubMed]
- Geraci, J.R.; Anderson, D.M.; Timperi, R.J.; St. Aubin, D.J.; Early, G.A.; Prescott, J.H.; Mayo, C.A. Humpback Whales (Megaptera novaeangliae) Fatally Poisoned by Dinoflagellate Toxin. Can. J. Fish. Aquat. Sci. 1989, 46, 1895–1898. [Google Scholar] [CrossRef]
- Flewelling, L.J.; Naar, J.P.; Abbott, J.P.; Baden, D.G.; Barros, N.B.; Bossart, G.D.; Bottein, M.-Y.D.; Hammond, D.G.; Haubold, E.M.; Heil, C.A.; et al. Brevetoxicosis: Red tides and marine mammal mortalities. Nature 2005, 435, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Bordner, J.; Thiessen, W.E.; Bates, H.A.; Rapoport, H. Structure of a crystalline derivative of saxitoxin. Structure of saxitoxin. J. Am. Chem. Soc. 1975, 97, 6008–6012. [Google Scholar] [CrossRef] [PubMed]
- Schantz, E.J.; Ghazarossian, V.E.; Schnoes, H.K.; Strong, F.M.; Springer, J.P.; Pezzanite, J.O.; Clardy, J. Structure of saxitoxin. J. Am. Chem. Soc. 1975, 97, 1238–1239. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.L.; Oesterlin, R.; Rapoport, H. The structure of saxitoxin. J. Am. Chem. Soc. 1971, 93, 7344–7345. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Scheuer, P.J.; Tsukitani, Y.; Kikuchi, H.; Van Engen, D.; Clardy, J.; Gopichand, Y.; Schmitz, F.J. Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria. J. Am. Chem. Soc. 1981, 103, 2469–2471. [Google Scholar] [CrossRef]
- Murata, M.; Shimatani, M.; Sugitani, H.; Oshima, Y.; Yasumoto, T. Isolation and structural elucidation of the causative toxin of the diarrhetic shellfish poisoning. Nippon Suisan Gakkaishi 1982, 48, 549–552. [Google Scholar] [CrossRef]
- Rao, D.V.S.; Quilliam, M.A.; Pocklington, R. Domoic acid: A neurotoxic amino acid produced by the marine diatom Nitzschia pungens in culture. Can. J. Fish. Aquat. Sci. 1988, 45, 2076–2079. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Andersen, A.J.C.; Andersen, N.G.; Nielsen, K.F.; Hansen, P.J.; Larsen, T.O. Chemical Diversity, Origin, and Analysis of Phycotoxins. J. Nat. Prod. 2016, 79, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.L.; Oh, J.; Place, A.R.; Hamann, M.T. Stereochemical Studies of the Karlotoxin Class Using NMR Spectroscopy and DP4 Chemical-Shift Analysis: Insights into their Mechanism of Action. Angew. Chem. Int. Ed. 2015, 54, 15705–15710. [Google Scholar] [CrossRef] [PubMed]
- Bachvaroff, T.R.; Adolf, J.E.; Squier, A.H.; Harvey, H.R.; Place, A.R. Characterization and quantification of karlotoxins by liquid chromatography–mass spectrometry. Harmful Algae 2008, 7, 473–484. [Google Scholar] [CrossRef]
- La Claire, J.W., II; Manning, S.R.; Talarski, A.E. Semi-quantitative assay for polyketide prymnesins isolated from Prymnesium parvum (Haptophyta) cultures. Toxicon 2015, 102, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Twiner, M.J.; Bottein Dechraoui, M.-Y.; Wang, Z.; Mikulski, C.M.; Henry, M.S.; Pierce, R.H.; Doucette, G.J. Extraction and analysis of lipophilic brevetoxins from the red tide dinoflagellate Karenia brevis. Anal. Biochem. 2007, 369, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Fire, S.E.; Fauquier, D.; Flewelling, L.J.; Henry, M.; Naar, J.; Pierce, R.; Wells, R.S. Brevetoxin exposure in bottlenose dolphins (Tursiops truncatus) associated with Karenia brevis blooms in Sarasota Bay, Florida. Mar. Biol. 2007, 152, 827–834. [Google Scholar] [CrossRef]
- Yogi, K.; Oshiro, N.; Inafuku, Y.; Hirama, M.; Yasumoto, T. Detailed LC-MS/MS analysis of ciguatoxins revealing distinct regional and species characteristics in fish and causative alga from the pacific. Anal. Chem. 2011, 83, 8886–8891. [Google Scholar] [CrossRef] [PubMed]
- Campora, C.E.; Dierking, J.; Tamaru, C.S.; Hokama, Y.; Vincent, D. Detection of ciguatoxin in fish tissue using sandwich ELISA and neuroblastoma cell bioassay. J. Clin. Lab. Anal. 2008, 22, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.K.K.; Johansen, S.S.; Dalsgaard, P.W.; Linnet, K. Simultaneous screening and quantification of 52 common pharmaceuticals and drugs of abuse in hair using UPLC-TOF-MS. Forensic Sci. Int. 2010, 196, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Ouni, Y.; Taamalli, A.; Gómez-Caravaca, A.M.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Zarrouk, M. Characterisation and quantification of phenolic compounds of extra-virgin olive oils according to their geographical origin by a rapid and resolutive LC-ESI-TOF MS method. Food Chem. 2011, 127, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Masiá, A.; Blasco, C.; Picó, Y. Last trends in pesticide residue determination by liquid chromatography–mass spectrometry. Trends Environ. Anal. Chem. 2014, 2, 11–24. [Google Scholar] [CrossRef]
- Guibal, R.; Lissalde, S.; Charriau, A.; Poulier, G.; Mazzella, N.; Guibaud, G. Coupling passive sampling and time of flight mass spectrometry for a better estimation of polar pesticide freshwater contamination: Simultaneous target quantification and screening analysis. J. Chromatogr. A 2015, 1387, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Fountain, S.T.; Bi, H.; Rossi, D.T. Quantification and rapid metabolite identification in drug discovery using API time-of-flight LC/MS. Anal. Chem. 2000, 72, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.A.; Binzer, S.B.; Hoeck, C.; Meier, S.; de Medeiros, L.S.; Andersen, N.G.; Place, A.; Nielsen, K.F.; Hansen, P.J.; Larsen, T.O. Karmitoxin: An Amine-Containing Polyhydroxy-Polyene Toxin from the Marine Dinoflagellate Karlodinium armiger. J. Nat. Prod. 2017, 80, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Cai, P.; He, S.; Zhou, C.; Place, A.R.; Haq, S.; Ding, L.; Chen, H.; Jiang, Y.; Guo, C.; Xu, Y.; et al. Two new karlotoxins found in Karlodinium veneficum (strain GM2) from the East China Sea. Harmful Algae 2016, 58, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Ishibashi, M.; Nakamichi, H.; Kosaka, T.; Ishikawa, T.; Kobayashi, J. Luteophanol A, a New Polyhydroxyl Compound from Symbiotic Marine Dinoflagellate Amphidinium sp. J. Org. Chem. 1997, 62, 3820–3823. [Google Scholar] [CrossRef]
- Echigoya, R.; Rhodes, L.; Oshima, Y.; Satake, M. The structures of five new antifungal and hemolytic amphidinol analogs from Amphidinium carterae collected in New Zealand. Harmful Algae 2005, 4, 383–389. [Google Scholar] [CrossRef]
- Hanif, N.; Ohno, O.; Kitamura, M.; Yamada, K.; Uemura, D. Symbiopolyol, a VCAM-1 inhibitor from a symbiotic dinoflagellate of the jellyfish Mastigias papua. J. Nat. Prod. 2010, 73, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Houdai, T.; Matsuoka, S.; Murata, M.; Satake, M.; Ota, S.; Oshima, Y.; Rhodes, L.L. Acetate labeling patterns of dinoflagellate polyketides, amphidinols 2, 3 and 4. Tetrahedron 2001, 57, 5551–5555. [Google Scholar] [CrossRef]
- Huang, S.-J.; Kuo, C.-M.; Lin, Y.-C.; Chen, Y.-M.; Lu, C.-K. Carteraol E, a potent polyhydroxyl ichthyotoxin from the dinoflagellate Amphidinium carterae. Tetrahedron Lett. 2009, 50, 2512–2515. [Google Scholar] [CrossRef]
- Huang, X.C.; Zhao, D.; Guo, Y.W.; Wu, H.M.; Lin, L.P.; Wang, Z.H.; Ding, J.; Lin, Y.S. Lingshuiol, a novel polyhydroxyl compound with strongly cytotoxic activity from the marine dinoflagellate Amphidinium sp. Bioorg. Med. Chem. Lett. 2004, 14, 3117–3120. [Google Scholar] [PubMed]
- Huang, X.C.; Zhao, D.; Guo, Y.W.; Wu, H.M.; Trivellone, E.; Cimino, G. Lingshuiols A and B, two new polyhydroxy compounds from the Chinese marine dinoflagellate Amphidinium sp. Tetrahedron Lett. 2004, 45, 5501–5504. [Google Scholar] [CrossRef]
- Hwang, B.S.; Yoon, E.Y.; Kim, H.S.; Yih, W.; Park, J.Y.; Jeong, H.J.; Rho, J.-R. Ostreol A: A new cytotoxic compound isolated from the epiphytic dinoflagellate Ostreopsis cf. ovata from the coastal waters of Jeju Island, Korea. Bioorg. Med. Chem. Lett. 2013, 23, 3023–3027. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, T.; Yamada, K.; Uemura, D. Amdigenols E and G, long carbon-chain polyol compounds, isolated from the marine dinoflagellate Amphidinium sp. Tetrahedron Lett. 2014, 55, 6319–6323. [Google Scholar] [CrossRef]
- Inuzuka, T.; Yamamoto, Y.; Yamada, K.; Uemura, D. Amdigenol A, a long carbon-backbone polyol compound, produced by the marine dinoflagellate Amphidinium sp. Tetrahedron Lett. 2012, 53, 239–242. [Google Scholar] [CrossRef]
- Kubota, T.; Tsuda, M.; Doi, Y.; Takahashi, A.; Nakamichi, H.; Ishibashi, M.; Fukushi, E.; Kawabata, J.; Kobayashi, J. Luteophanols B and C, new polyhydroxyl compounds from marine dinoflagellate Amphidinium sp. Tetrahedron 1998, 54, 14455–14464. [Google Scholar] [CrossRef]
- Kubota, T.; Takahashi, A.; Tsuda, M.; Kobayashi, J. Luteophanol D, New Polyhydroxyl Metabolite from Marine Dinoflagellate Amphidinium sp. Mar. Drugs 2005, 3, 113–118. [Google Scholar] [CrossRef]
- Meng, Y.; Van Wagoner, R.M.; Misner, I.; Tomas, C.; Wright, J.L.C. Structure and biosynthesis of amphidinol 17, a hemolytic compound from Amphidinium carterae. J. Nat. Prod. 2010, 73, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Morsy, N.; Matsuoka, S.; Houdai, T.; Matsumori, N.; Adachi, S.; Murata, M.; Iwashita, T.; Fujita, T. Isolation and structure elucidation of a new amphidinol with a truncated polyhydroxyl chain from Amphidinium klebsii. Tetrahedron 2005, 61, 8606–8610. [Google Scholar] [CrossRef]
- Morsy, N.; Houdai, T.; Matsuoka, S.; Matsumori, N.; Adachi, S.; Oishi, T.; Murata, M.; Iwashita, T.; Fujita, T. Structures of new amphidinols with truncated polyhydroxyl chain and their membrane-permeabilizing activities. Bioorg. Med. Chem. 2006, 14, 6548–6554. [Google Scholar] [CrossRef] [PubMed]
- Swasono, R.T.; Kanemoto, M.; Matsumori, N. Structural Reevaluations of Amphidinol 3, a Potent Antifungal Compound from Dinoflagellate. Heterocycles 2010, 82, 1359. [Google Scholar]
- Nuzzo, G.; Cutignano, A.; Sardo, A.; Fontana, A. Antifungal Amphidinol 18 and Its 7-Sulfate Derivative from the Marine Dinoflagellate Amphidinium carterae. J. Nat. Prod. 2014, 77, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Paul, G.K.; Matsumori, N.; Konoki, K.; Murata, M.; Tachibana, K. Chemical structures of amphidinols 5 and 6 isolated from marine dinof1agellate Amphidinium klebsii and their cholesterol-dependent membrane disruption. J. Mar. Biotechnol. 1997, 5, 124–128. [Google Scholar]
- Paul, G.K.; Matsumori, N.; Murata, M.; Tachibana, K. Isolation and chemical structure of amphidinol 2, a potent hemolytic compound from marine dinoflagellate Amphidinium klebsii. Tetrahedron Lett. 1995, 36, 6279–6282. [Google Scholar] [CrossRef]
- Peng, J.; Place, A.R.; Yoshida, W.; Anklin, C.; Hamann, M.T. Structure and Absolute Configuration of Karlotoxin-2, an Ichthyotoxin from the Marine Dinoflagellate Karlodinium veneficum. J. Am. Chem. Soc. 2010, 132, 3277–3279. [Google Scholar] [CrossRef] [PubMed]
- Satake, M.; Murata, M.; Yasumoto, T.; Fujita, T.; Naoki, H. Amphidinol, a polyhydroxypolyene antifungal agent with an unprecedented structure, from a marine dinoflagellate, Amphidinium klebsii. J. Am. Chem. Soc. 1991, 113, 9859–9861. [Google Scholar] [CrossRef]
- Van Wagoner, R.M.; Deeds, J.R.; Satake, M.; Ribeiro, A.A.; Place, A.R.; Wright, J.L.C. Isolation and characterization of karlotoxin 1, a new amphipathic toxin from Karlodinium veneficum. Tetrahedron Lett. 2008, 49, 6457–6461. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, R.M.; Deeds, J.R.; Tatters, A.O.; Place, A.R.; Tomas, C.R.; Wright, J.L.C. Structure and relative potency of several karlotoxins from Karlodinium veneficum. J. Nat. Prod. 2010, 73, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Washida, K.; Koyama, T.; Yamada, K.; Kita, M.; Uemura, D. Karatungiols A and B, two novel antimicrobial polyol compounds, from the symbiotic marine dinoflagellate Amphidinium sp. Tetrahedron Lett. 2006, 47, 2521–2525. [Google Scholar] [CrossRef]
- Berge, T.; Hansen, P.J.; Moestrup, Ø. Feeding mechanism, prey specificity and growth in light and dark of the plastidic dinoflagellate Karlodinium armiger. Aquat. Microb. Ecol. 2008, 50, 279–288. [Google Scholar] [CrossRef]
- Robledo, J.; Deeds, J.; Place, A.; Vasta, G.; Saito, K. A Decade of Research on Pfiesteria Spp. and Their Toxins. In Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection; CRC Press: Boca Raton, FL, USA, 2008; pp. 717–751. [Google Scholar]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersen, A.J.C.; De Medeiros, L.S.; Binzer, S.B.; Rasmussen, S.A.; Hansen, P.J.; Nielsen, K.F.; Jørgensen, K.; Larsen, T.O. HPLC-HRMS Quantification of the Ichthyotoxin Karmitoxin from Karlodinium armiger. Mar. Drugs 2017, 15, 278. https://doi.org/10.3390/md15090278
Andersen AJC, De Medeiros LS, Binzer SB, Rasmussen SA, Hansen PJ, Nielsen KF, Jørgensen K, Larsen TO. HPLC-HRMS Quantification of the Ichthyotoxin Karmitoxin from Karlodinium armiger. Marine Drugs. 2017; 15(9):278. https://doi.org/10.3390/md15090278
Chicago/Turabian StyleAndersen, Aaron John Christian, Lívia Soman De Medeiros, Sofie Bjørnholt Binzer, Silas Anselm Rasmussen, Per Juel Hansen, Kristian Fog Nielsen, Kevin Jørgensen, and Thomas Ostenfeld Larsen. 2017. "HPLC-HRMS Quantification of the Ichthyotoxin Karmitoxin from Karlodinium armiger" Marine Drugs 15, no. 9: 278. https://doi.org/10.3390/md15090278
APA StyleAndersen, A. J. C., De Medeiros, L. S., Binzer, S. B., Rasmussen, S. A., Hansen, P. J., Nielsen, K. F., Jørgensen, K., & Larsen, T. O. (2017). HPLC-HRMS Quantification of the Ichthyotoxin Karmitoxin from Karlodinium armiger. Marine Drugs, 15(9), 278. https://doi.org/10.3390/md15090278