Biogenetic Relationships of Bioactive Sponge Merotriterpenoids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
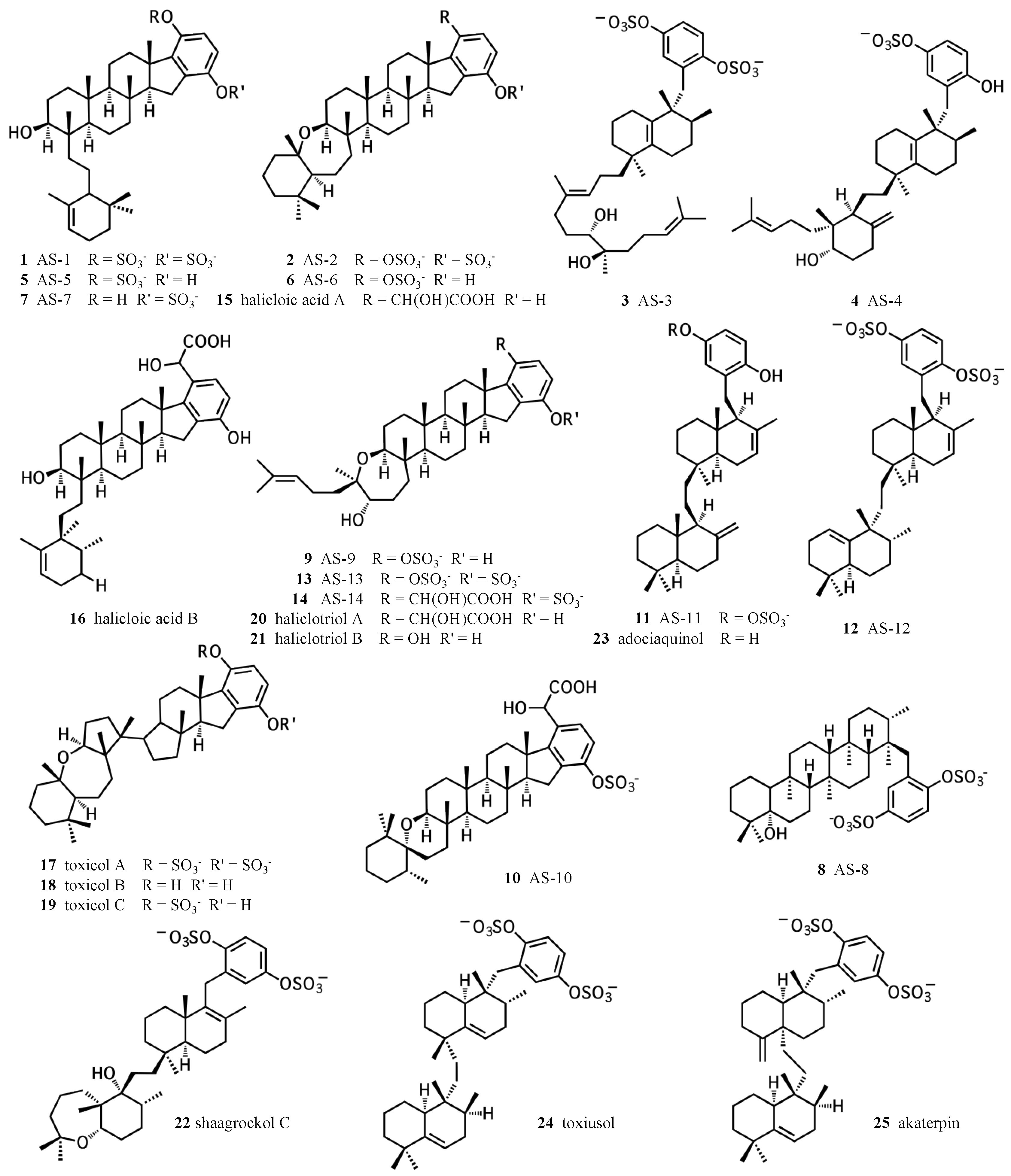
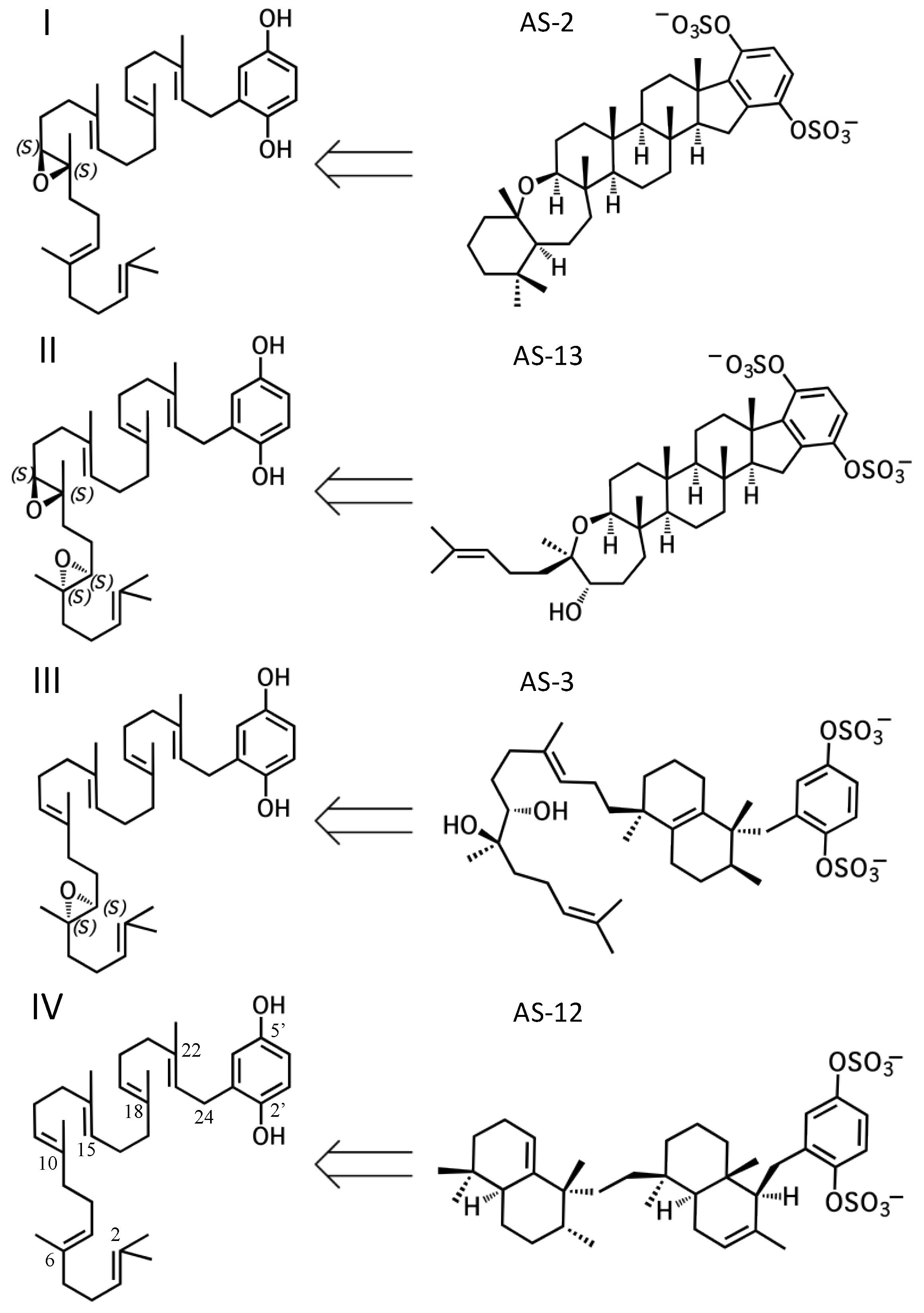
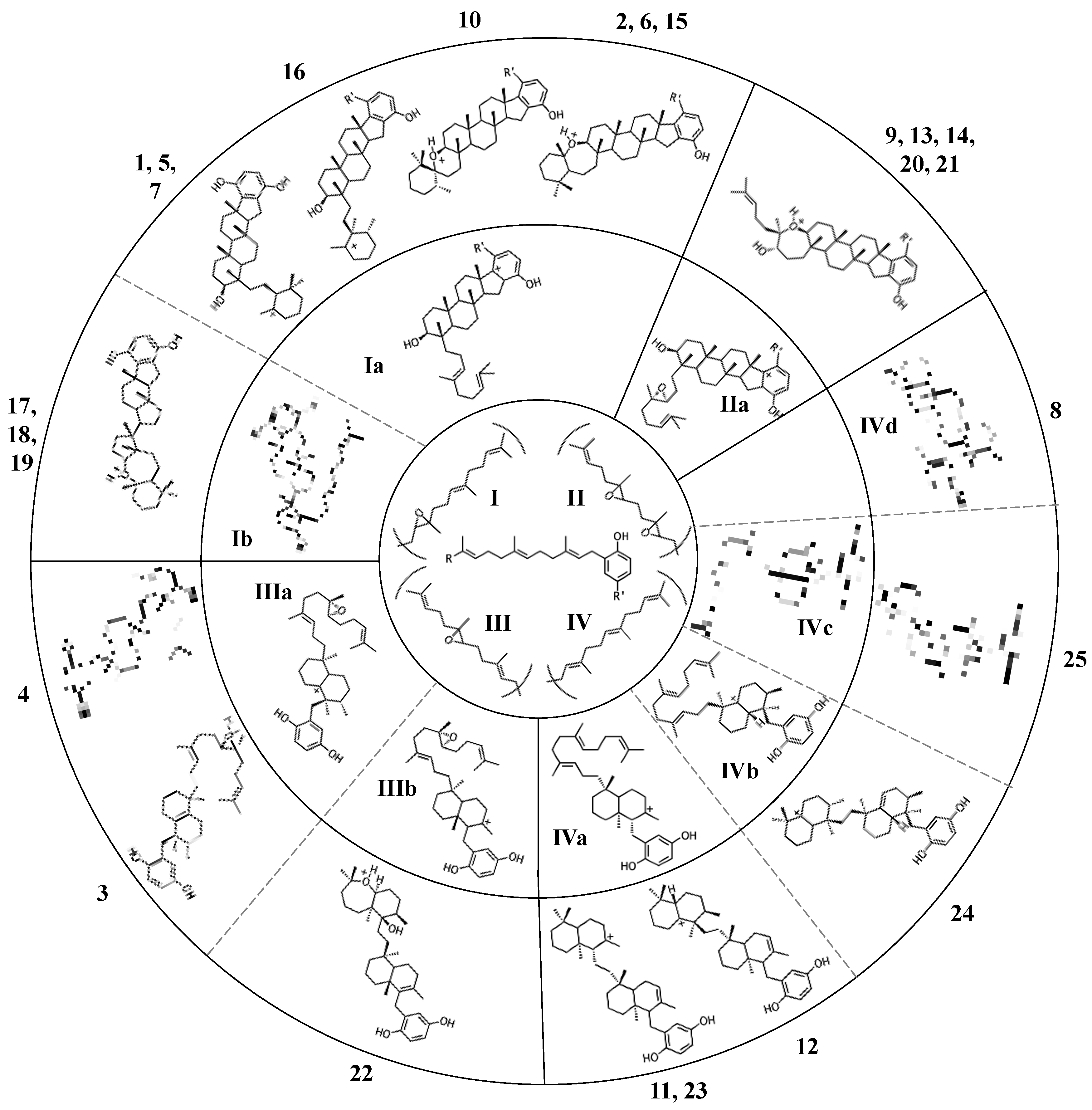
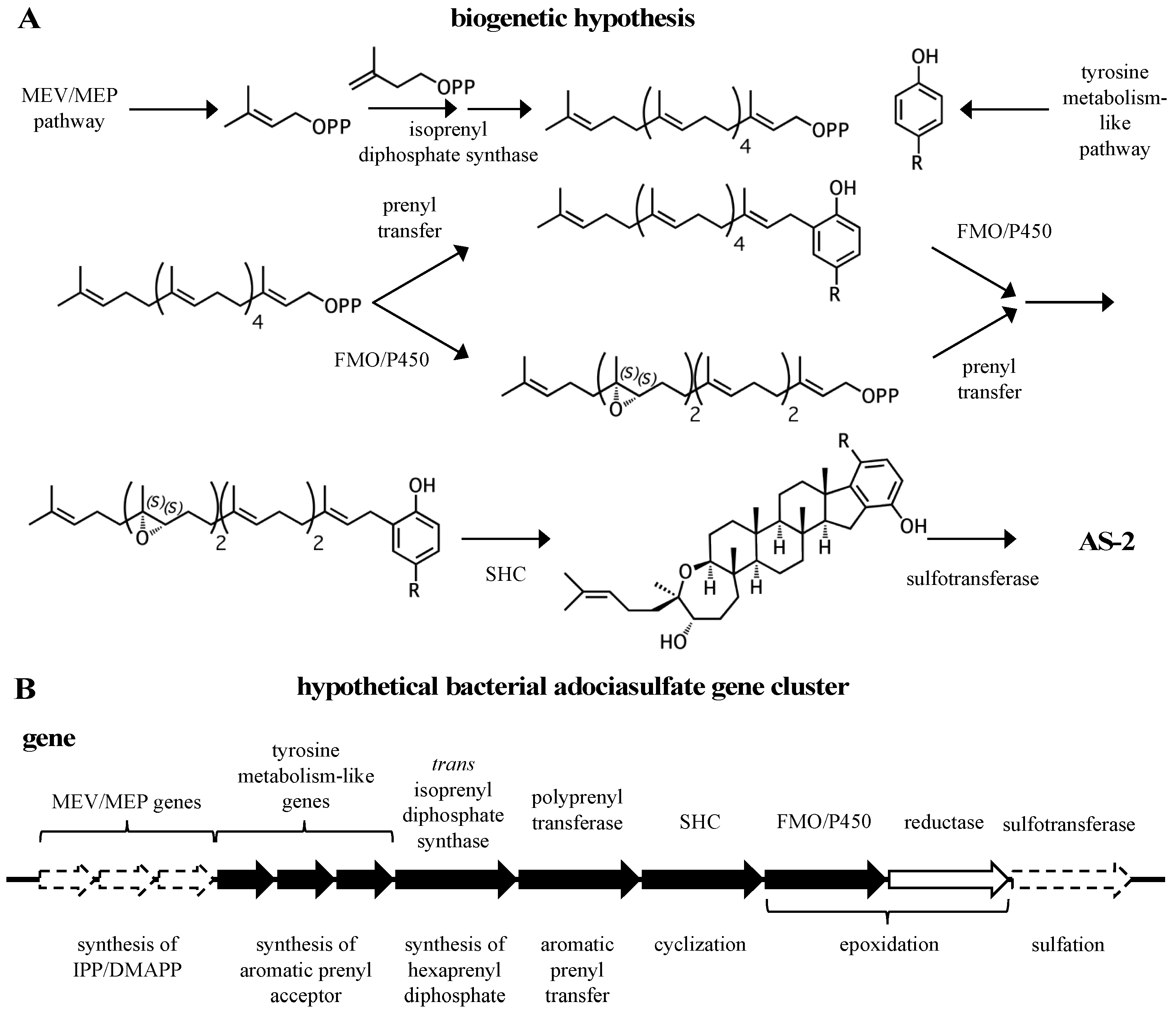
2. A proposed biosynthetic route for sponge hydroquinone merotriterpenoids
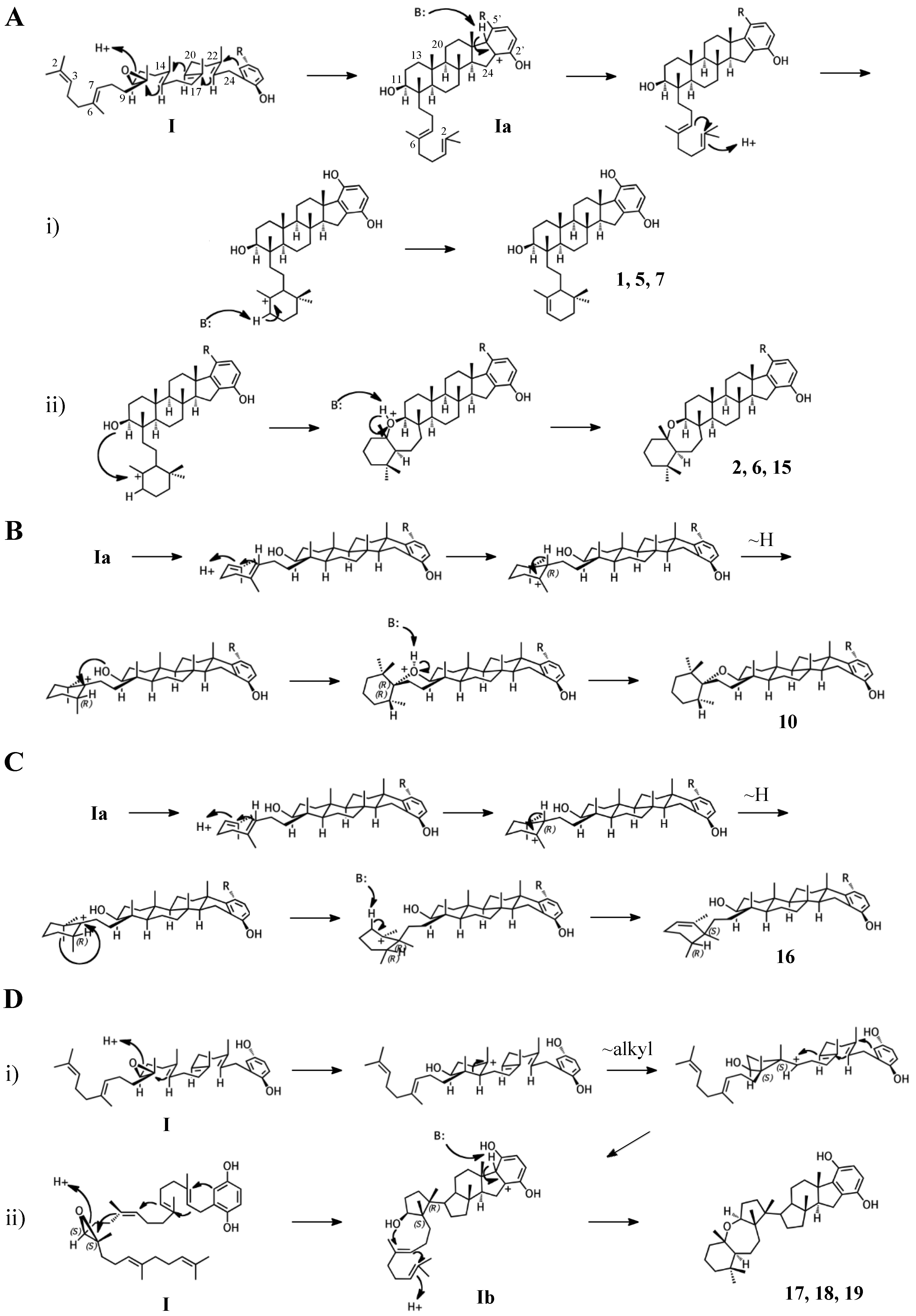
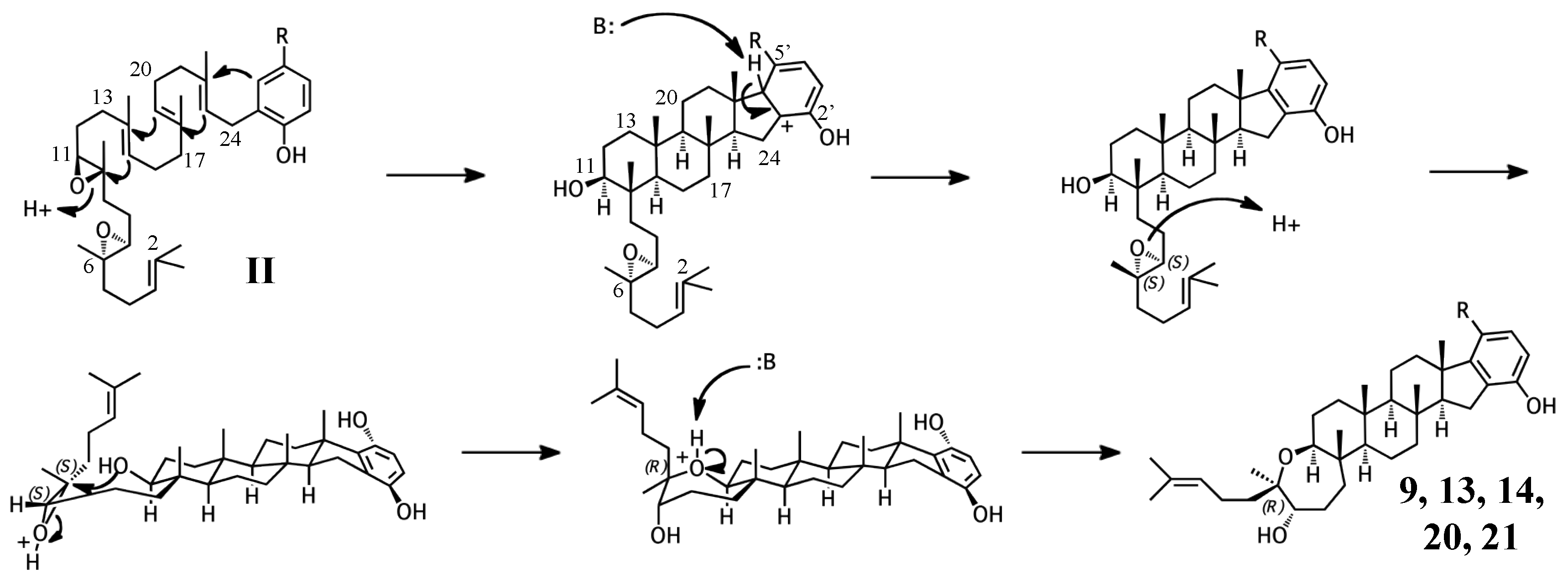
2.1. Group I compounds
2.2. Group II compounds
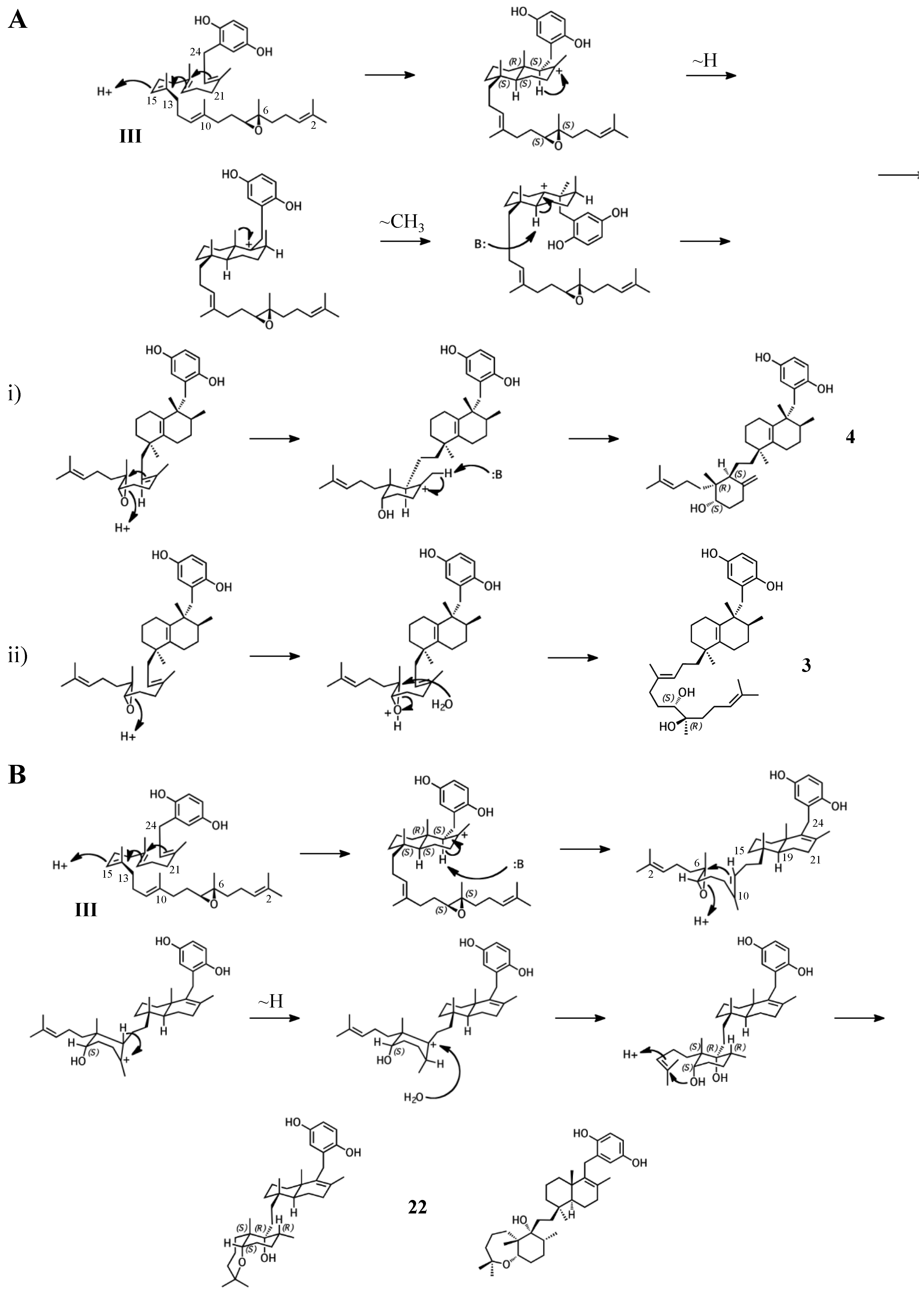
2.3. Group III compounds
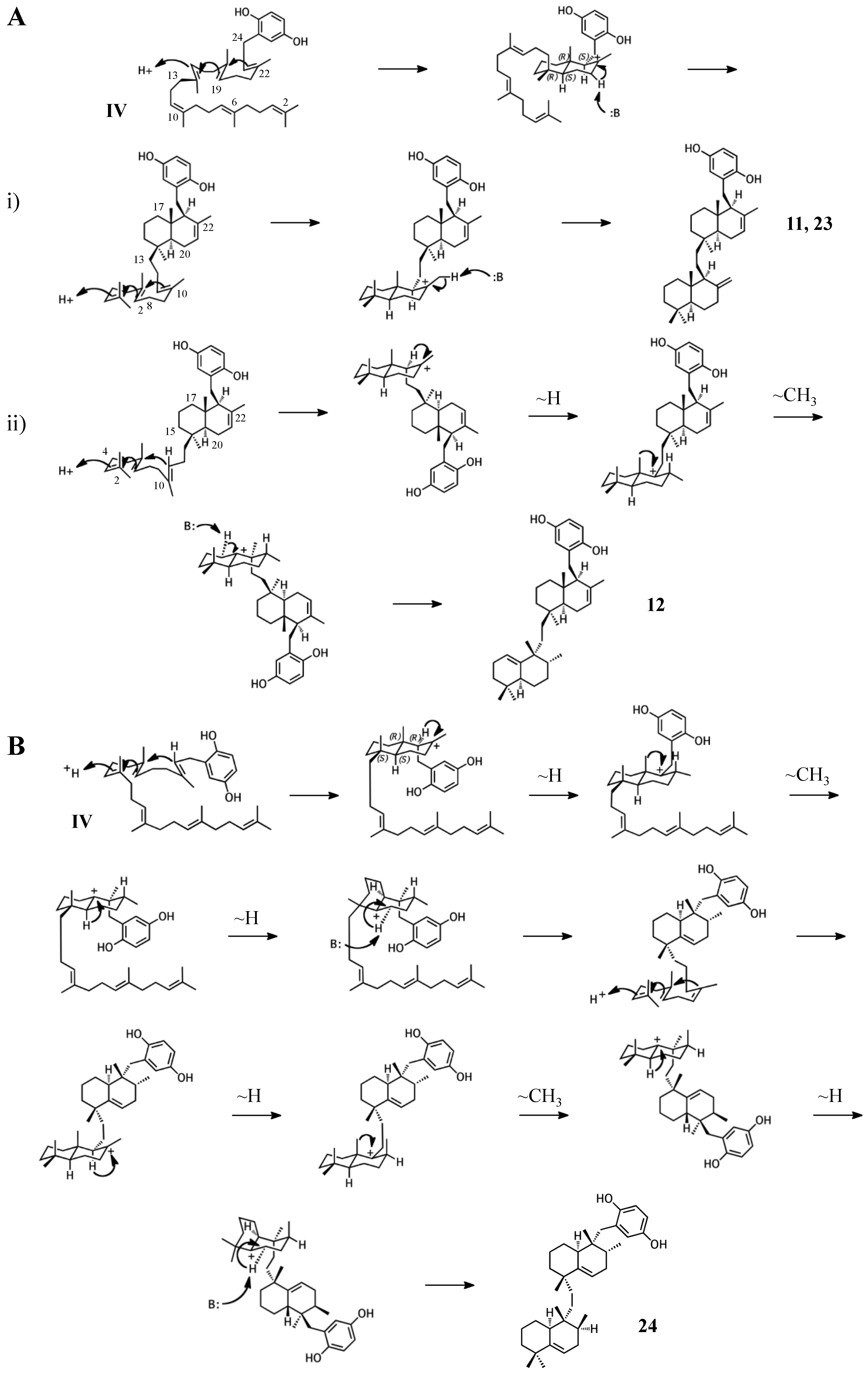
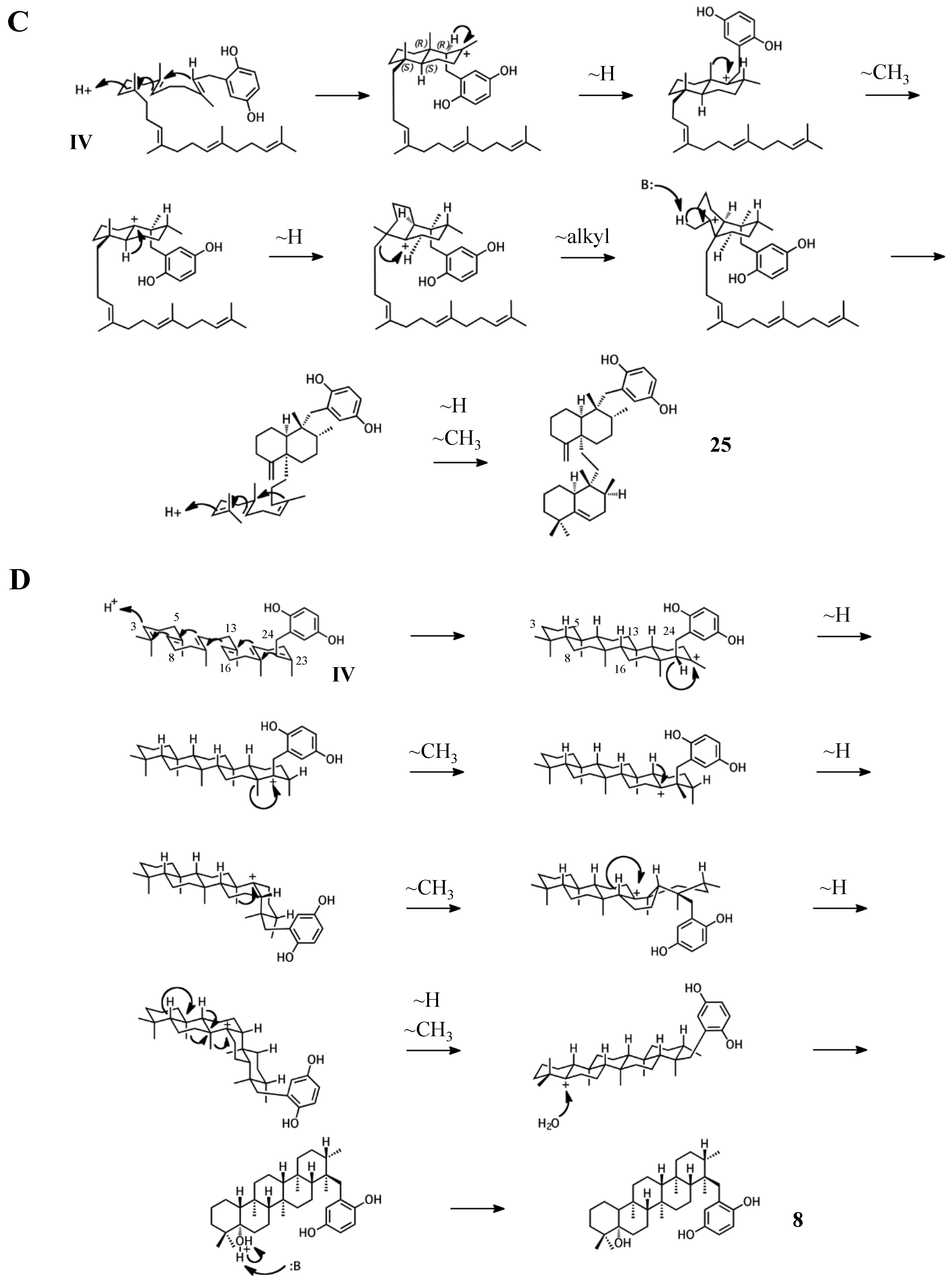
2.4. Group IV compounds
3. Considerations of the enzymatic origin of sponge merotriterpenoids
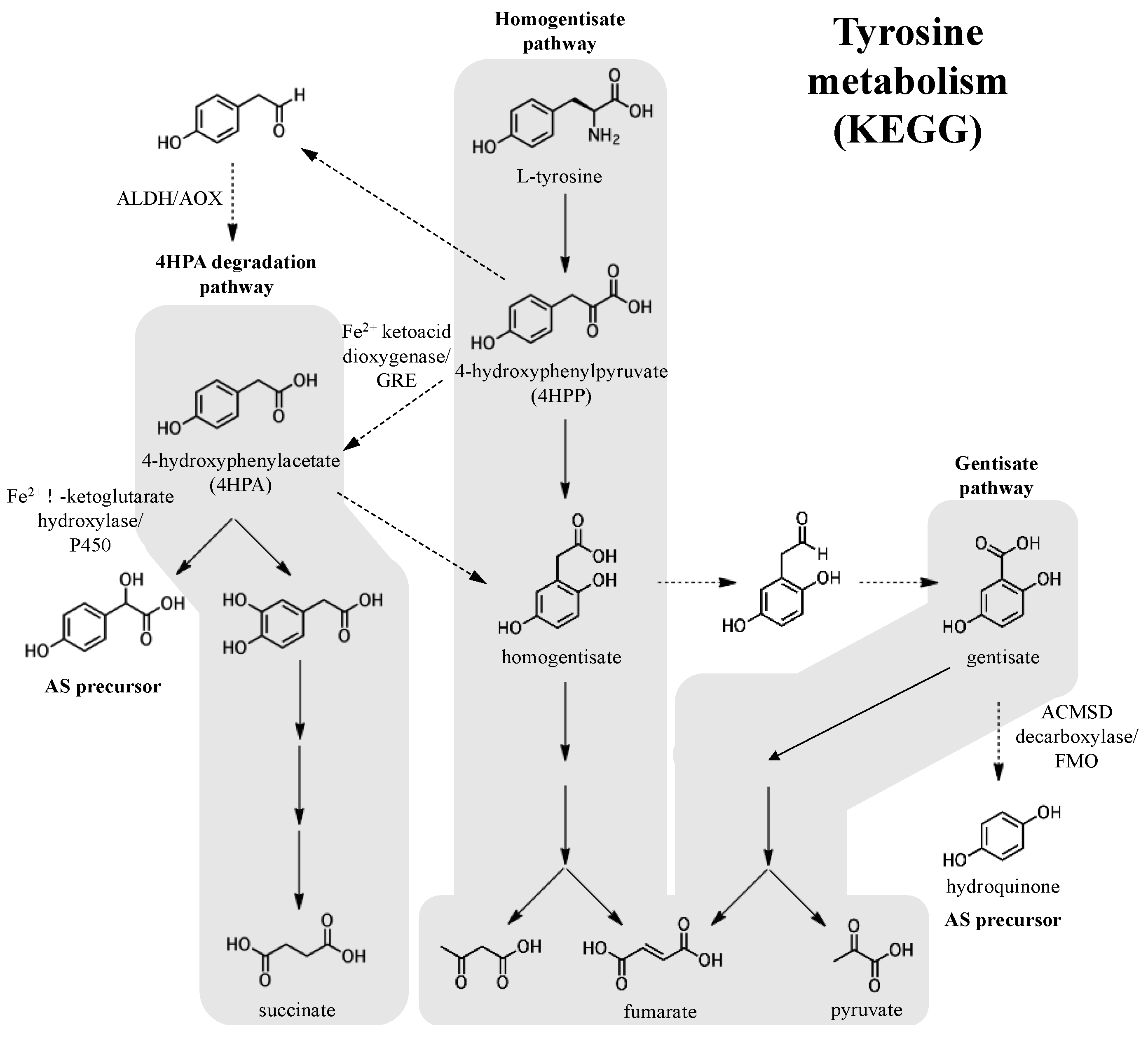
3.1. Origin of precursors
3.2. Prenylation
3.3. Cyclization
3.4. Epoxidation
3.5. Sulfation
4. Concluding remarks
Acknowledgments
Conflicts of Interest
References
- Menna, M.; Imperatore, C.; D’Aniello, F.; Aiello, A. Meroterpenes from marine invertebrates: Structures, occurrence, and ecological implications. Mar. Drugs 2013, 11, 1602–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, Y.; Abe, I. Biosynthesis of fungal meroterpenoids. Nat. Prod. Rep. 2015, 33, 26–53. [Google Scholar] [CrossRef] [PubMed]
- Loya, S.; Tal, R.; Hizi, A. Hexaprenoid hydroquinones, novel inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. J. Nat. Prod. 1993, 56, 2120–2125. [Google Scholar] [CrossRef] [PubMed]
- Fukami, A.; Ikeda, Y.; Kondo, S.; Naganawa, H.; Takeuchi, T.; Furuya, S.; Hirabayashi, Y.; Shimoike, K.; Hosaka, S.; Watanabe, Y.; et al. Akaterpin, a Novel Bioactive Triterpene from the Marine Sponge Callyspongia sp. Tetrahedron Lett. 1997, 38, 1201–1202. [Google Scholar] [CrossRef]
- Williams, D.E.; Steino, A.; de Voogd, N.J.; Mauk, A.G.; Andersen, R.J. Halicloic acids A and B isolated from the marine sponge Haliclona sp. collected in the Philippines inhibit indoleamine 2,3-dioxygenase. J. Nat. Prod. 2012, 75, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Crews, P.; Harrison, B. New triterpene-ketides (merotriterpenes), haliclotriol A and B, from an Indo-Pacific Haliclona sponge. Tetrahedron 2000, 56, 9039–9046. [Google Scholar] [CrossRef]
- Isaacs, S.; Hizi, A.; Kashman, Y. Toxicols A-C and Toxiusol—New Bioactive Hexaprenoid Hydroquinones from Toxiclona toxius. Tetrahedron 1993, 49, 4275–4282. [Google Scholar] [CrossRef]
- Blackburn, C.L.; Faulkner, D.J. Adociasulfate 10, a new merohexaprenoid sulfate from the sponge Haliclona (aka Adocia) sp. Tetrahedron 2000, 56, 8429–8432. [Google Scholar] [CrossRef]
- Blackburn, C.L.; Hopmann, C.; Sakowicz, R.; Berdelis, M.S.; Goldstein, L.S.B.; Faulkner, D.J. Adociasulfates 1–6, inhibitors of kinesin motor proteins from the sponge Haliclona (aka Adocia) sp. J. Org. Chem. 1999, 64, 5565–5570. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzis, J.A.; Leone, P.; Harris, L.; Butler, M.S.; Ngo, A.; Hooper, J.N.A.; Quinn, R.J. Adociasulfates 1, 7, and 8: New bioactive hexaprenoid hydroquinones from the marine sponge Adocia sp. J. Org. Chem. 1999, 64, 5571–5574. [Google Scholar] [CrossRef] [PubMed]
- Sakowicz, R.; Berdelis, M.S.; Ray, K.; Blackburn, C.L.; Hopmann, C.; Faulkner, D.J.; Goldstein, L.S.B. A marine natural product inhibitor of kinesin motors. Science 1998, 280, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.E.; Hong, W.; Zachariah, M.M.; Harper, M.K.; Matainaho, T.K.; Van Wagoner, R.M.; Ireland, C.M.; Vershinin, M. Single-molecule inhibition of human kinesin by adociasulfate-13 and -14 from the sponge Cladocroce aculeata. Proc. Natl. Acad. Sci. USA 2013, 110, 18880–18885. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.C.; Vale, R.D.; Kuntz, I.D. Inhibitors of kinesin activity from structure-based computer screening. Biochemistry 2000, 39, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Learman, S.S.; Kim, C.D.; Stevens, N.S.; Kim, S.; Wojcik, E.J.; Walker, R.A. NSC 622124 Inhibits Human Eg5 and Other Kinesins via Interaction with the Conserved Microtubule-Binding Site. Biochemistry 2009, 48, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, K.; Pillay, S.; Bin Ahmad, N.R.; Bikadi, Z.; Hazai, E.; Yan, L.; Kolatkar, P.R.; Pervushin, K.; Jauch, R. Identification of a polyoxometalate inhibitor of the DNA binding activity of Sox2. ACS Chem. Biol. 2011, 6, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Moucadel, V.; Laudet, B.; Barette, C.; Lafanechere, L.; Hasenknopf, B.; Li, J.; Bareyt, S.; Lacote, E.; Thorimbert, S.; et al. Identification of polyoxometalates as nanomolar noncompetitive inhibitors of protein kinase CK2. Chem. Biol. 2008, 15, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.V.; Flanagan, L.A.; Janmey, P.A.; Leterrier, J.-F. Bidirectional Translocation of Neurofilaments along Microtubules Mediated in Part by Dynein/Dynactin. Mol. Biol. Cell 2000, 11, 3495–3508. [Google Scholar] [CrossRef] [PubMed]
- Dictenberg, J.B.; Swanger, S.A.; Antar, L.N.; Singer, R.H.; Bassell, G.J. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 2008, 14, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Ewald, A.; Zünkler, C.; Lourim, D.; Dabauvalle, M.-C. Microtubule-dependent assembly of the nuclear envelope in Xenopus laevis egg extract. Eur. J. Cell Biol. 2001, 80, 678–691. [Google Scholar] [CrossRef]
- Qiu, D.; Cheng, S.-M.; Wozniak, L.; McSweeney, M.; Perrone, E.; Levin, M. Localization and loss-of-function implicates ciliary proteins in early, cytoplasmic roles in left-right asymmetry. Dev. Dynam. 2005, 234, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Brier, S.; Carletti, E.; DeBonis, S.; Hewat, E.; Lemaire, D.; Kozielski, F. The marine natural product adociasulfate-2 as a tool to identify the MT-binding region of kinesins. Biochemistry 2006, 45, 15644–15653. [Google Scholar] [CrossRef] [PubMed]
- Bogenstätter, M.; Limberg, A.; Overman, L.E.; Tomasi, A.L. Enantioselective Total Synthesis of the Kinesin Motor Protein Inhibitor Adociasulfate 1. J. Am. Chem. Soc. 1999, 121, 12206–12207. [Google Scholar] [CrossRef]
- Darne, C.P. Kinesin Motor Protein Inhibitors: Toward the Synthesis of Adociasulfate Analogs. Master’s Thesis, The University of Georgia, Athens, GA, USA, May 2005. [Google Scholar]
- Erben, F.; Specowius, V.; Wölfling, J.; Schneider, G.; Langer, P. Benzo-Annulated Steroids: Synthesis of Octahydro-indeno-phenanthrenes by Formal [3+3] Cyclocondensation Reaction with 1,3-Bis[(trimethylsilyl)oxy]buta-1,3-dienes. Helv. Chim. Acta 2013, 96, 924–930. [Google Scholar] [CrossRef]
- Hosoi, H.; Kawai, N.; Hagiwara, H.; Suzuki, T.; Nakazaki, A.; Takao, K.-I.; Umezawa, K.; Kobashi, S. Determination of the Absolute Structure of (+)-Akaterpin. Chem. Pharm. Bull. 2012, 60, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzis, J.A.; Quinn, R.J. Adociasulfate-9, a new hexaprenoid hydroquinone from the Great Barrier Reef sponge Adocia aculeata. J. Nat. Prod. 1999, 62, 1682–1684. [Google Scholar] [CrossRef]
- Isaacs, S.; Kashman, Y. Shaagrockol B and C; Two Hexaprenylhydroquinone disulfates from the Red Sea Sponge Toxiclona toxius. Tetrahedron Lett. 1992, 33, 2227–2230. [Google Scholar] [CrossRef]
- West, L.M.; Faulkner, D.J. Hexaprenoid Hydroquinones from thr Sponge Haliclona (aka Adocia) sp. J. Nat. Prod. 2006, 69, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Bonitz, T.; Zubeil, F.; Grond, S.; Heide, L. Unusual N-prenylation in diazepinomicin biosynthesis: The farnesylation of a benzodiazepine substrate is catalyzed by a new member of the ABBA prenyltransferase superfamily. PLoS ONE 2013, 8, e85707. [Google Scholar] [CrossRef] [PubMed]
- Haagen, Y.; Gluck, K.; Fay, K.; Kammerer, B.; Gust, B.; Heide, L. A gene cluster for prenylated naphthoquinone and prenylated phenazine biosynthesis in Streptomyces cinnamonensis DSM 1042. ChemBioChem 2006, 7, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Hayashi, Y.; Kuzuyama, T.; Furihata, K.; Itoh, N.; Seto, H.; Dairi, T. Biosynthesis of a natural polyketide-isoprenoid hybrid compound, furaquinocin A: Identification and heterologous expression of the gene cluster. J. Bacteriol. 2006, 188, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kuzuyama, T.; Furihata, K.; Itoh, N.; Seto, H.; Dairi, T. A Relationship between the Mevaolnate Pathway and Isoprenoid Production in Actinomycetes. J. Antibiot. 2003, 56, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Kuzuyama, T.; Noel, J.P.; Richard, S.B. Structural basis for the promiscuous biosynthetic prenylation of aromatic natural products. Nature 2005, 435, 983–987. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, J.B.; Banskota, A.H.; Charan, R.D.; Schlingmann, G.; Zazopoulos, E.; Piraee, M.; Janso, J.; Bernan, V.S.; Aouidate, M.; Farnet, C.M.; et al. Biosynthesis of Diazepinomicin/ECO-4601, a Micromonospora Secondary Metabolite with a Novel Ring System. J. Nat. Prod. 2008, 71, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Saikia, S.; Nicholson, M.J.; Young, C.; Parker, E.J.; Scott, B. The genetic basis for indole-diterpene chemical diversity in filamentous fungi. Mycol. Res. 2008, 112, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Saleh, O.; Gust, B.; Boll, B.; Fiedler, H.P.; Heide, L. Aromatic prenylation in phenazine biosynthesis: Dihydrophenazine-1-carboxylate dimethylallyltransferase from Streptomyces anulatus. J. Biol. Chem. 2009, 284, 14439–14447. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Moffitt, M.C.; Zazopoulos, E.; McAlpine, J.B.; Dorrestein, P.C.; Moore, B.S. Molecular basis for chloronium-mediated meroterpene cyclization: Cloning, sequencing, and heterologous expression of the napyradiomycin biosynthetic gene cluster. J. Biol. Chem. 2007, 282, 16362–16368. [Google Scholar] [CrossRef] [PubMed]
- Zeyhle, P.; Bauer, J.S.; Kalinowski, J.; Shin-ya, K.; Gross, H.; Heide, L. Genome-based discovery of a novel membrane-bound 1,6-dihydroxyphenazine prenyltransferase from a marine actinomycete. PLoS ONE 2014, 9, e99122. [Google Scholar] [CrossRef] [PubMed]
- Hillwig, M.L.; Zhu, Q.; Liu, X. Biosynthesis of ambiguine indole alkaloids in cyanobacterium Fischerella ambigua. ACS Chem. Biol. 2014, 9, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Steffensky, M.; Mühlenweg, A.; Wange, Z.-X.; Li, S.-M.; Heide, L. Identification of the Novobiocin Biosynthetic Gene Cluster of Streptomyces spheroides NCIB 11891. Antimicrob. Agents Chemother. 2000, 44, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Heide, L. Prenyl transfer to aromatic substrates: Genetics and enzymology. Curr. Opin. Chem. Biol. 2009, 13, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.-H.; Ko, T.-P.; Wang, A.H.-J. Structure, mechanism and function of prenyltransferases. Eur. J. Biochem. 2002, 269, 3339–3354. [Google Scholar] [CrossRef] [PubMed]
- Crouch, N.P.; Adlington, R.M.; Baldwin, J.E.; Lee, M.-H.; MacKinnon, C.H. A Mechanistic Rationalism for the Substrate Specificity of Recombinant Mammalian 4-Hydroxyphenylpyruvate Dioxygenase (4-HPPD). Tetrahedron 1997, 53, 6993–7010. [Google Scholar] [CrossRef]
- Selmer, T.; Andrei, P.I. p-Hydroxyphenylacetate decarboxylase from Clostridium difficile. Eur. J. Biochem. 2001, 268, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Rzhetsky, A.; Hsu, L.C.; Chang, C. Human aldehyde dehydrogenase gene family. Eur. J. Biochem. 1998, 251, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Garrattini, E.; Fratelli, M.; Terao, M. The mammalian aldehyde oxidase gene family. Hum. Genom. 2009, 4, 119–130. [Google Scholar] [CrossRef]
- Prescott, A.G.; Lloyd, M.D. The iron(II) and 2-oxoacid-dependent dioxygenases and their role in metabolism (1967 to 1999). Nat. Prod. Rep. 2000, 17, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 monooxygenases: An update on perspectives for synthetic application. Trends. Biotechnol. 2012, 30, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Arias-Barrau, E.; Olivera, E.R.; Luengo, J.M.; Fernandez, C.; Galan, B.; Garcia, J.L.; Diaz, E.; Minambres, B. The homogentisate pathway: A central catabolic pathway involved in the degradation of L-phenylalanine, L-tyrosine, and 3-hydroxyphenylacetate in Pseudomonas putida. J. Bacteriol. 2004, 186, 5062–5077. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.A.; Díaz, E.; García, J.L. Molecular Characterization of the 4-Hydroxyphenylacetate Catabolic Pathway of Escherichia coli W: Engineering a Mobile Aromatic Degradative Cluster. J. Bacteriol. 1996, 178, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Zhang, H. Transition Metal-Catalyzed Nonoxidative Decarboxylation Reactions. Biochemistry 2006, 45, 10408–10411. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Masai, E.; Kitayama, H.; Harada, K.; Katayama, Y.; Fukuda, M. Characterization of the 5-Carboxyvanillate Decarboxylase Gene and Its Role in Lignin-Related Biphenyl Catabolism in Sphingomonas paucimobilis SYK-6. Appl. Environ. Microbiol. 2002, 68, 4407–4415. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Fukuhara, N.; Oikawa, T. Thermophilic, reversible gamma-resorcylate decarboxylase from Rhizobium sp. strain MTP-10005: Purification, molecular characterization, and expression. J. Bacteriol. 2004, 186, 6855–6863. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, M.M.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Tokunaga, K.; Matsuda, Y.; Fujii, I.; Abe, I.; Ebizuka, Y.; Kushiro, T. Reconstitution of a fungal meroterpenoid biosynthesis reveals the involvement of a novel family of terpene cyclases. Nat. Chem. 2010, 2, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Tokunaga, K.; Radhakrishnan, E.K.; Fujii, I.; Abe, I.; Ebizuka, Y.; Kushiro, T. Identification of a key prenyltransferase involved in biosynthesis of the most abundant fungal meroterpenoids derived from 3,5-dimethylorsellinic acid. Chembiochem 2012, 13, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.C.; Entwistle, R.; Guo, C.J.; Ahuja, M.; Szewczyk, E.; Hung, J.H.; Chiang, Y.M.; Oakley, B.R.; Wang, C.C. Two separate gene clusters encode the biosynthetic pathway for the meroterpenoids austinol and dehydroaustinol in Aspergillus nidulans. J. Am. Chem. Soc. 2012, 134, 4709–4720. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Awakawa, T.; Abe, I. Reconstituted biosynthesis of fungal meroterpenoid andrastin A. Tetrahedron 2013, 69, 8199–8204. [Google Scholar] [CrossRef]
- Matsuda, Y.; Wakimoto, T.; Mori, T.; Awakawa, T.; Abe, I. Complete biosynthetic pathway of anditomin: Nature’s sophisticated synthetic route to a complex fungal meroterpenoid. J. Am. Chem. Soc. 2014, 136, 15326–15336. [Google Scholar] [CrossRef] [PubMed]
- Pojer, F.; Li, S.-M.; Heide, L. Molecular cloning and sequence analysis of the clorobiocin biosynthetic gene cluster: New insights into the biosynthesis of the aminocoumarin antibiotics. Microbiology 2002, 148, 3901–3911. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.; Heide, L. Characterization of Polyprenyldiphosphate: 4-Hydroxybenzoate Polyprenyltransferase from Eschericia coli. Biochim. Biophys. Acta 1994, 1212, 93–102. [Google Scholar] [CrossRef]
- Norris, S.R.; Barrette, T.R.; DellaPenna, D. Genetic Dissection of Carotenoid Synthesis in Arabidopsis Defines Plastoquinone as an Essential Component of Phytoene Desaturation. Plant Cell 1995, 7, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Young, L.G.; Leppik, R.A.; Hamilton, J.A.; Gibson, F. Biosynthesis in Escherichia coli K-12: 4-Hydroxybenzoate Octaprenyltransferase. J. Bacteriol. 1972, 110, 18–25. [Google Scholar] [PubMed]
- Bräuer, L.; Brandt, W.; Wessjohann, L.A. Modeling the E. coli 4-hydroxybenzoic acid oligoprenyltransferase (ubiA transferase) and characterization of potential active sites. J. Mol. Model. 2004, 10, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Levin, E.J.; Liu, S.; Bai, Y.; Lockless, S.W.; Zhou, M. Structure of a membrane-embedded prenyltransferase homologous to UBIAD1. PLoS Biol. 2014, 12, e1001911. [Google Scholar] [CrossRef] [PubMed]
- El Hachimi, Z.; Samuel, O.; Azerad, R. Biochemical study on ubiquinone biosynthesis in Escherichia coli: I. Specificity of para-hydroxybenzoate polyprenyltransferase. Biochimie 1974, 56, 1239–1247. [Google Scholar] [CrossRef]
- Okada, K.; Suzuki, K.; Kamiya, Y.; Zhu, X.; Fujisaki, S.; Nishimura, Y.; Nishino, T.; Nakagawa, T.; Kawamukai, M.; Matsuda, H. Polyprenyl diphosphate synthase essentially defines the length of the side chain of ubiquinone. Biochim. Biophys. Acta 1996, 1302, 217–223. [Google Scholar] [CrossRef]
- Wessjohann, L.; Sontag, B. Prenylation of Benzoic Acid Derivatives Catalyzed by a Transferase from Escherichia coli Overproduction: Method Developments and Substrate Specificity. Angew. Chem. Int. Ed. 1996, 35, 1697–1699. [Google Scholar] [CrossRef]
- Li, W. Bringing Bioactive Compounds into Membranes: The UbiA Superfamily of Intramembrane Aromatic Prenyltransferases. Trends. Biochem. Sci. 2016, 41, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Saleh, O.; Haagen, Y.; Seeger, K.; Heide, L. Prenyl transfer to aromatic substrates in the biosynthesis of aminocoumarins, meroterpenoids and phenazines: The ABBA prenyltransferase family. Phytochemistry 2009, 70, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Tello, M.; Kuzuyama, T.; Heide, L.; Noel, J.P.; Richard, S.B. The ABBA family of aromatic prenyltransferases: Broadening natural product diversity. Cell. Mol. Life Sci. 2008, 65, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Abe, I.; Rohmer, M.; Prestwich, G.D. Enzymatic Cyclization of Squalene and Oxidosqualene to Sterols and Triterpenes. Chem. Rev. 1993, 93, 2189–2208. [Google Scholar] [CrossRef]
- Abe, I.; Tanaka, H.; Noguchi, H. Enzymatic Formation of an Unnatural Hexacyclic C35 Polyprenoid by Bacterial Squalene Cyclase. J. Am. Chem. Soc. 2002, 124, 14514–14515. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.C.; Dominicus, J.M.; Syrén, P.-O.; Nestl, B.M.; Hauer, B. Stereoselective Friedel–Crafts alkylation catalyzed by squalene hopene cyclases. Tetrahedron 2012, 68, 7624–7629. [Google Scholar] [CrossRef]
- Lodeiro, S.; Xiong, Q.; Wilson, W.K.; Kolesnikova, M.D.; Onak, C.S.; Matsuda, S.P. An Oxidosqualene Cyclase Makes Numerous Products by Diverse Mechanisms: A Challenge to Prevailing Concepts of Triterpene Biosynthesis. J. Am. Chem. Soc. 2007, 129, 11213–11222. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Noguchi, H.; Abe, I. Enzymatic Formation of Indole-Containing Unnatural Cyclic Polyprenoids by Bacterial Squalene: Hopene Cyclase. Org. Lett. 2005, 7, 5873–5876. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Zhu, X.; Wilson, W.K.; Ganesan, A.; Matsuda, S.P. Enzymatic Synthesis of an Indole Diterpene by an Oxidosqualene Cyclase: Mechanistic, Biosynthetic, and Phylogenetic Implications. J. Am. Chem. Soc. 2003, 125, 9002–9003. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, Y.; Ohyama, T.; Hoshino, T. Chemo-enzymatic syntheses of drimane-type sesquiterpenes and the fundamental core of hongoquercin meroterpenoid by recombinant squalene-hopene cyclase. Org. Biomol. Chem. 2012, 10, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hoshino, H.; Yoshida, S.; Nakajima, M.; Hoshino, T. Bifunctional triterpene/sesquarterpene cyclase: Tetraprenyl-beta-curcumene cyclase is also squalene cyclase in Bacillus megaterium. J. Am. Chem. Soc. 2011, 133, 17540–17543. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Hoshino, T. Enzymatic cyclizations of squalene analogs with threo- and erythro-diols at the 6,7- or 10,11-positions by recombinant squalene cyclase. Trapping of carbocation intermediates and mechanistic insights into the product and substrate specificities. Org. Biomol. Chem. 2005, 3, 3127–3139. [Google Scholar] [CrossRef] [PubMed]
- Abe, I.; Rohmer, M. Enzymic Cyclization of 2.3-Dihydrosqualene and Squalene 2.3-Epoxide by Squalene Cyclases: From Pentacyclic to Tetracyclic Triterpenes. J. Chem. Soc. Perkin Trans. 1 1994, 7, 783–791. [Google Scholar] [CrossRef]
- Baunach, M.; Franke, J.; Hertweck, C. Terpenoid Biosynthesis Off the Beaten Track: Unconventional Cyclases and Their Impact on Biomimetic Synthesis. Angew. Chem. Int. Ed. 2015, 54, 2604–2626. [Google Scholar] [CrossRef] [PubMed]
- Tagami, K.; Liu, C.; Minami, A.; Noike, M.; Isaka, T.; Fueki, S.; Shichijo, Y.; Toshima, H.; Gomi, K.; Dairi, T.; et al. Reconstitution of Biosynthetic Machinery from Indole-Diterpene Paxilline in Aspergillus oryzae. J. Am. Chem. Soc. 2013, 135, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Laden, B.P.; Tang, Y.; Porter, T.D. Cloning, heterologous expression, and enzymological characterization of human squalene monooxygenase. Arch. Biochem. Biophys. 2000, 374, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Porter, T.D. Hepatic cytochrome P450 reductase-null mice reveal a second microsomal reductase for squalene monooxygenase. Arch. Biochem. Biophys. 2007, 461, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Dürr, C.; Schnell, H.J.; Luzhetskyy, A.; Murillo, R.; Weber, M.; Welzel, K.; Vente, A.; Bechthold, A. Biosynthesis of the terpene phenalinolactone in Streptomyces sp. Tu6071: Analysis of the gene cluster and generation of derivatives. Chem. Biol. 2006, 13, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Baunach, M.; Ding, L.; Hertweck, C. Bacterial Synthesis of Diverse Indole Terpene Alkaloids by an Unparalleled Cyclization Sequence. Angew. Chem. Int. Ed. 2012, 51, 10293–10297. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 systems-biological variations of electron transport chains. Biochim. Biophys. Acta 2007, 1770, 330–344. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.G.; Eisman, E.B.; Kulkarni, S.; Gerwick, L.; Gerwick, W.H.; Wipf, P.; Sherman, D.H.; Smith, J.L. Structural basis of functional group activation by sulfotransferases in complex metabolic pathways. ACS Chem. Biol. 2012, 7, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Sakowicz, R.; Finer, J.T.; Beraud, C.; Crompton, A.; Lewis, E.; Fritsch, A.; Lee, Y.; Mak, J.; Moody, R.; Turincio, R.; Chabala, J.C.; Gonzales, P.; Roth, S.; Weitman, S.; Wood, K.W. Antitumor activity of a kinesin inhibitor. Cancer Res. 2004, 64, 3276–3280. [Google Scholar] [CrossRef] [PubMed]
- Reddie, K.G.; Roberts, D.R.; Dore, T.M. Inhibition of kinesin motor proteins by adociasulfate-2. J. Med. Chem. 2006, 49, 4857–4860. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, P.; Ueoka, R.; Grauso, L.; Mangoni, A.; Morinaka, B.I.; Gugger, M.; Piel, J. Cyanobacterial ent-Sterol-Like Natural Products from a Deviated Ubiquinone Pathway. Angew. Chem. Int. Ed. 2017, 129, 1–5. [Google Scholar] [CrossRef]
- Müller, W.E.G.; Böhm, M.; Batel, R.; De Rosa, S.; Tommonaro, G.; Müller, I.M.; Schröder, H.C. Application of Cell Culture for the Production of Bioactive Compounds from Sponges: Synthesis of Avarol by Primmorphs from Dysidea avara. J. Nat. Prod. 2000, 63, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Uriz, M.J.; Turon, X.; Galera, J.; Tur, J.M. New light on the cell location of avarol within the sponge Dysidea avara (Dendroceratida). Cell. Tissue Res. 1996, 285, 519–527. [Google Scholar] [CrossRef]










© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, T.E. Biogenetic Relationships of Bioactive Sponge Merotriterpenoids. Mar. Drugs 2017, 15, 285. https://doi.org/10.3390/md15090285
Smith TE. Biogenetic Relationships of Bioactive Sponge Merotriterpenoids. Marine Drugs. 2017; 15(9):285. https://doi.org/10.3390/md15090285
Chicago/Turabian StyleSmith, Thomas E. 2017. "Biogenetic Relationships of Bioactive Sponge Merotriterpenoids" Marine Drugs 15, no. 9: 285. https://doi.org/10.3390/md15090285
APA StyleSmith, T. E. (2017). Biogenetic Relationships of Bioactive Sponge Merotriterpenoids. Marine Drugs, 15(9), 285. https://doi.org/10.3390/md15090285