APETx4, a Novel Sea Anemone Toxin and a Modulator of the Cancer-Relevant Potassium Channel KV10.1

 ,
, 
 ,
,
Abstract
:1. Introduction
2. Results
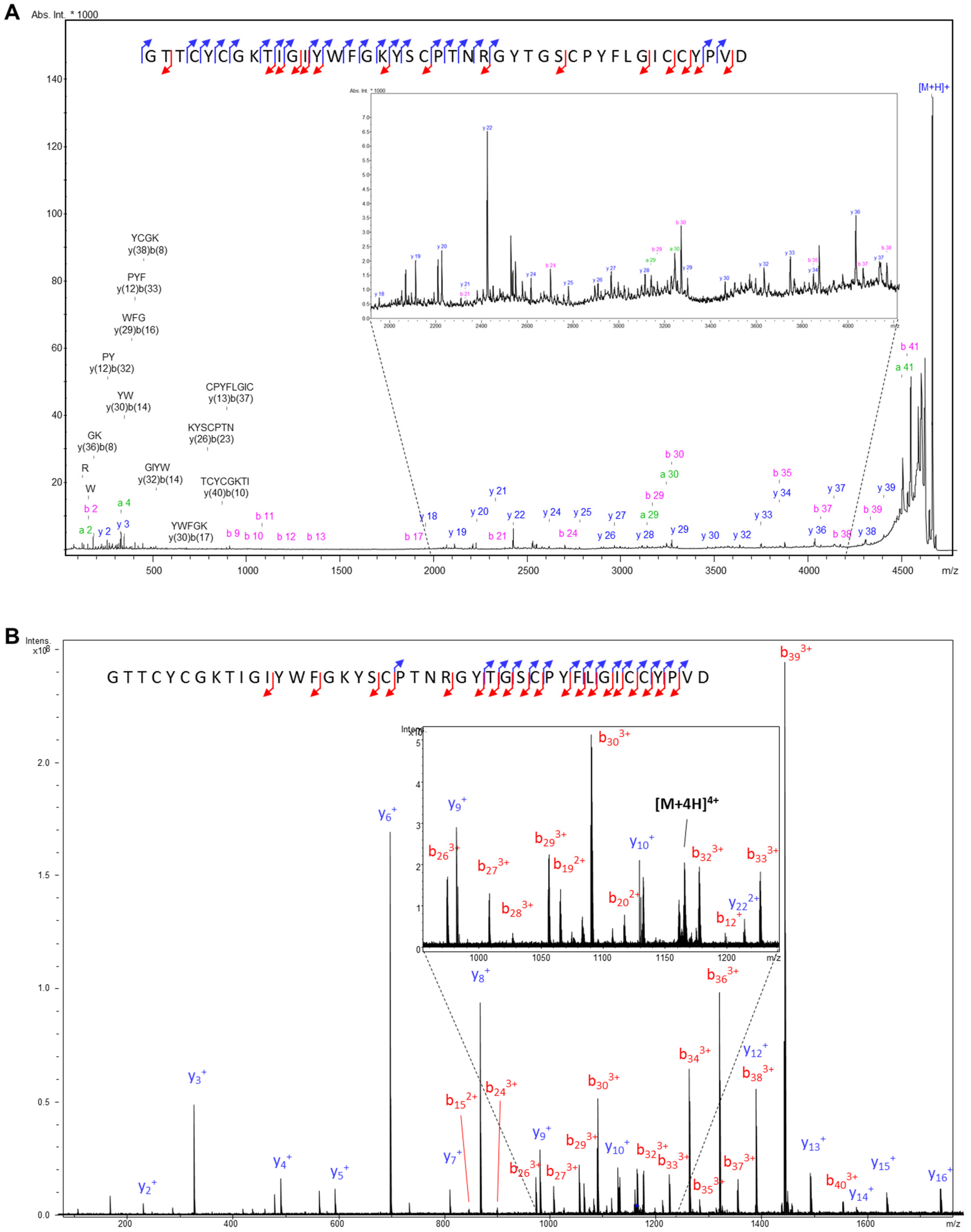
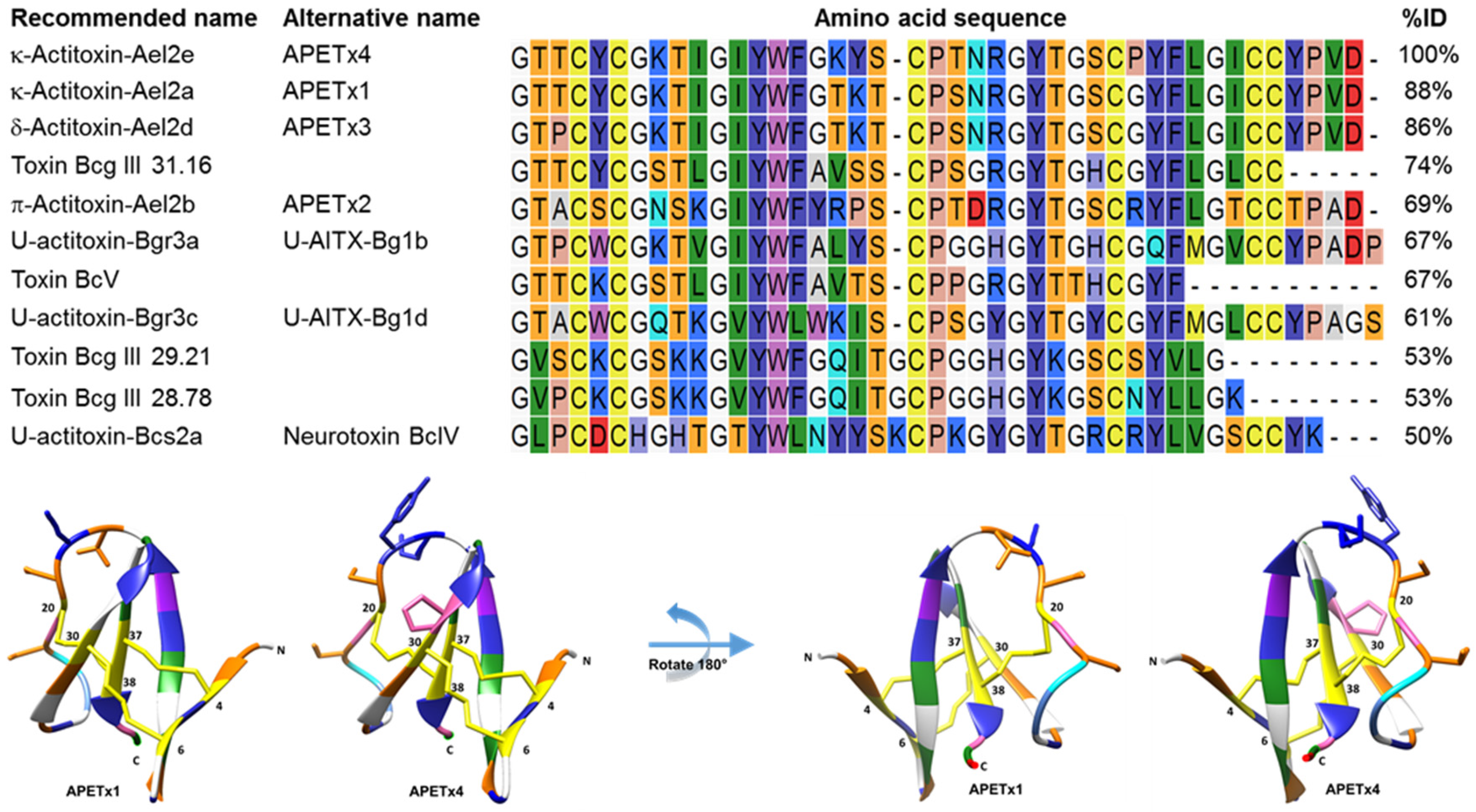
2.1. Activity-Guided Purification and Identification of A Novel KV10.1 Inhibitor
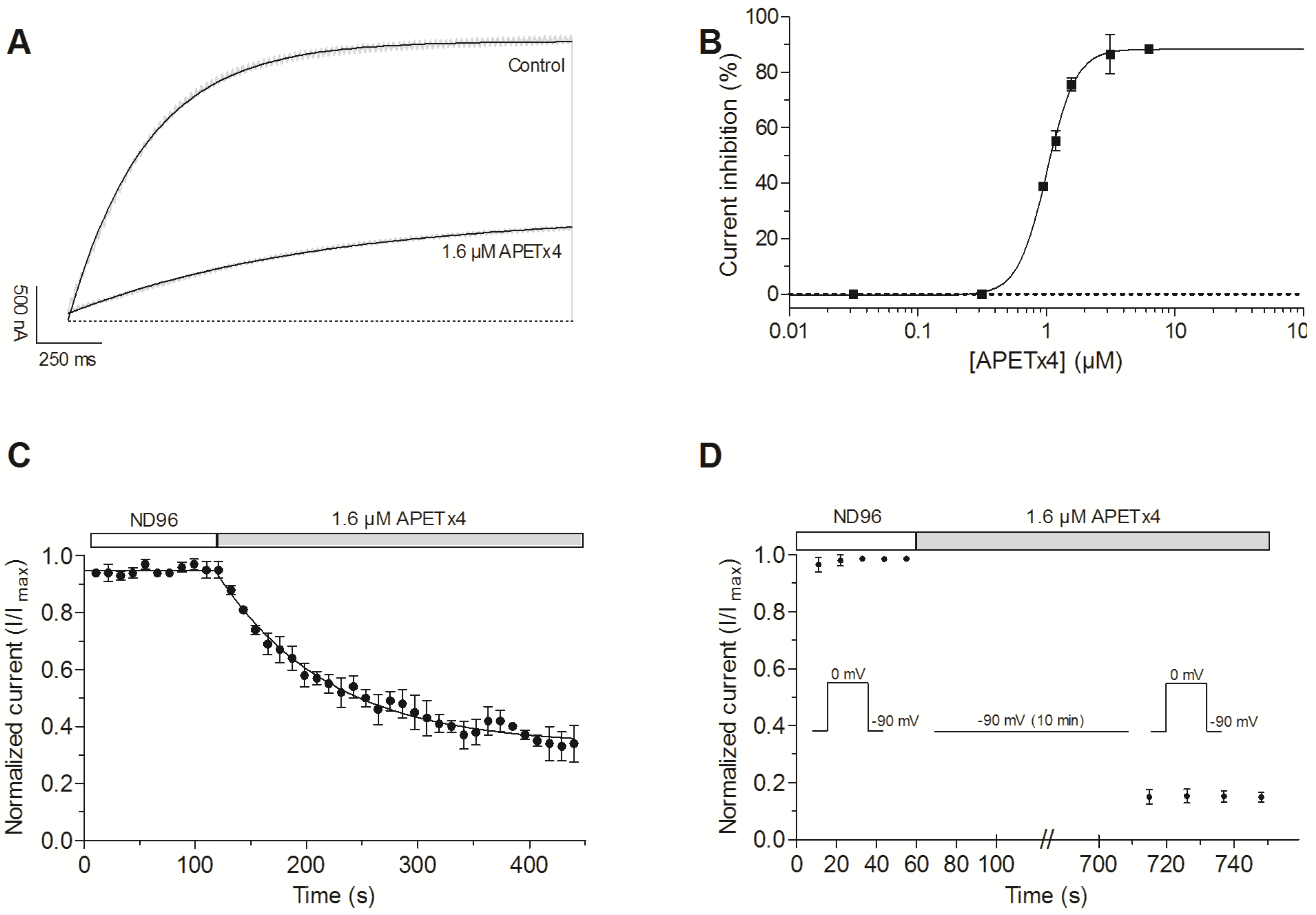
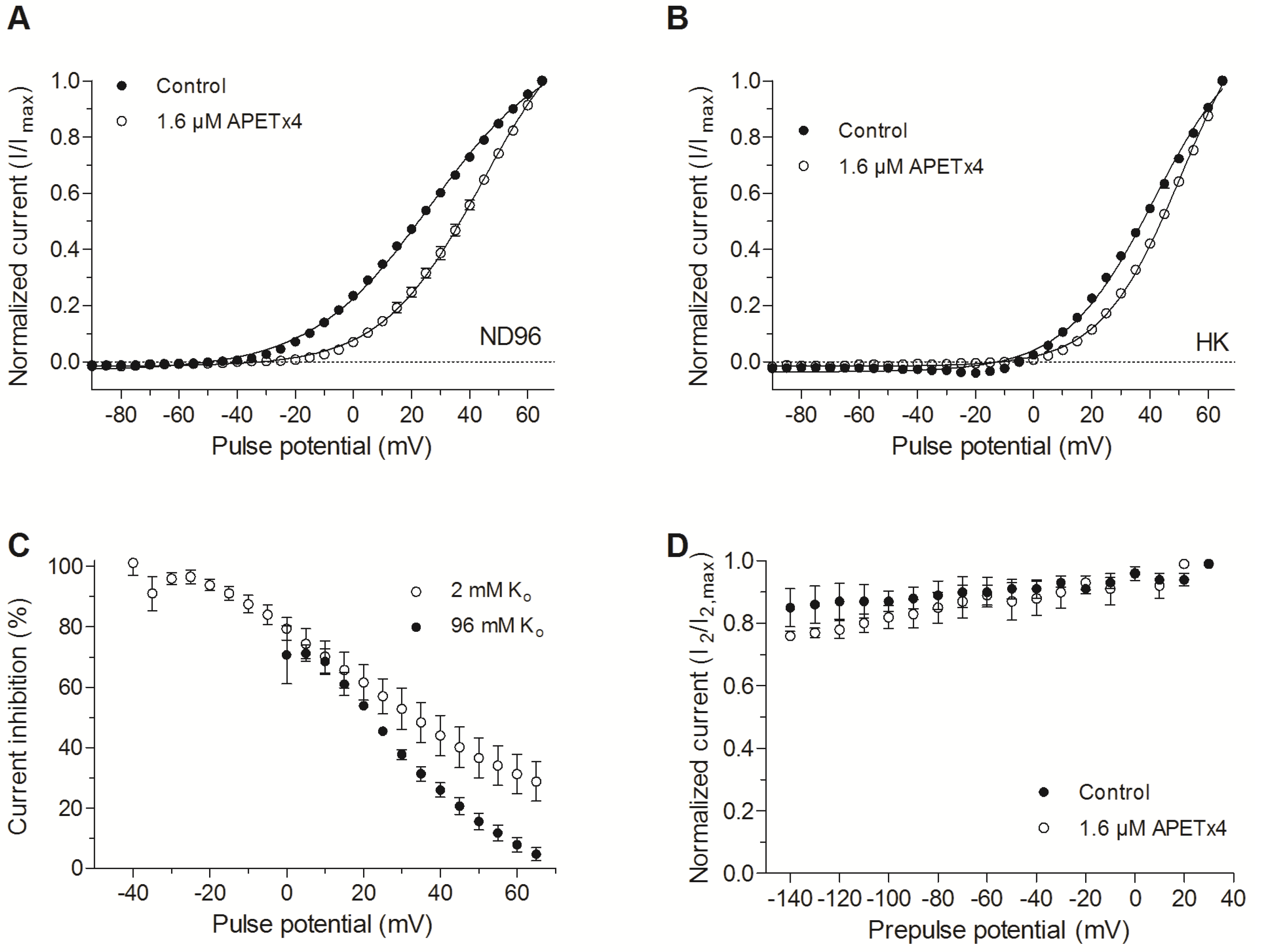
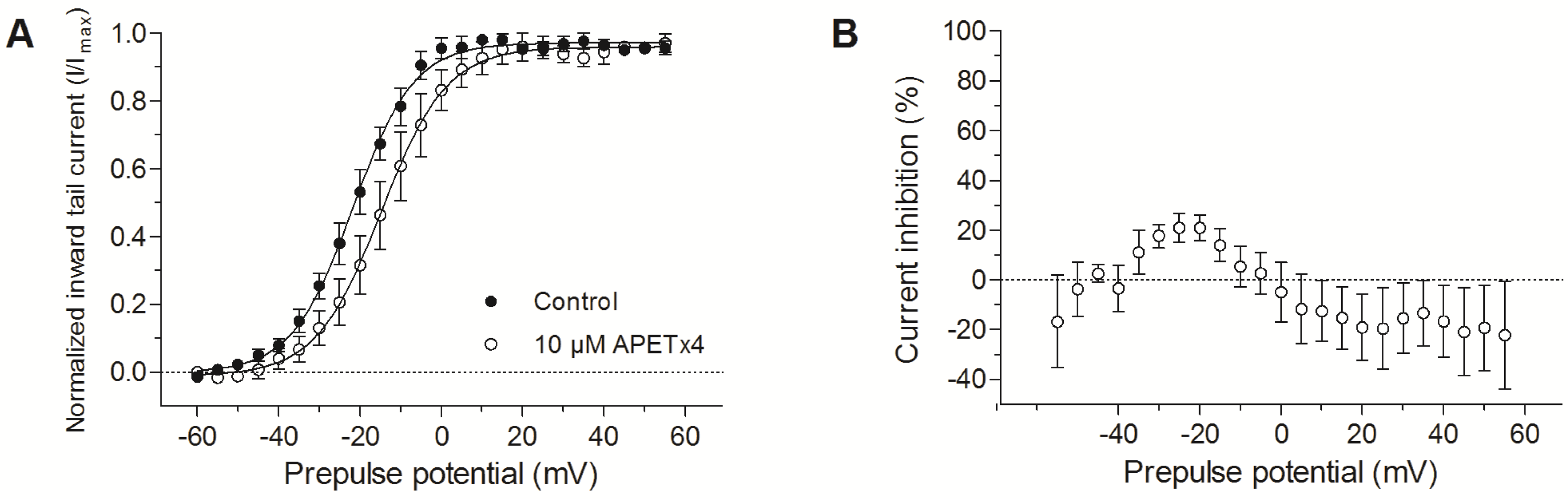
2.2. APETx4 is A Gating Modifier of KV10.1
2.3. Effect of APETx4 on Different Voltage-Gated Ion Channels
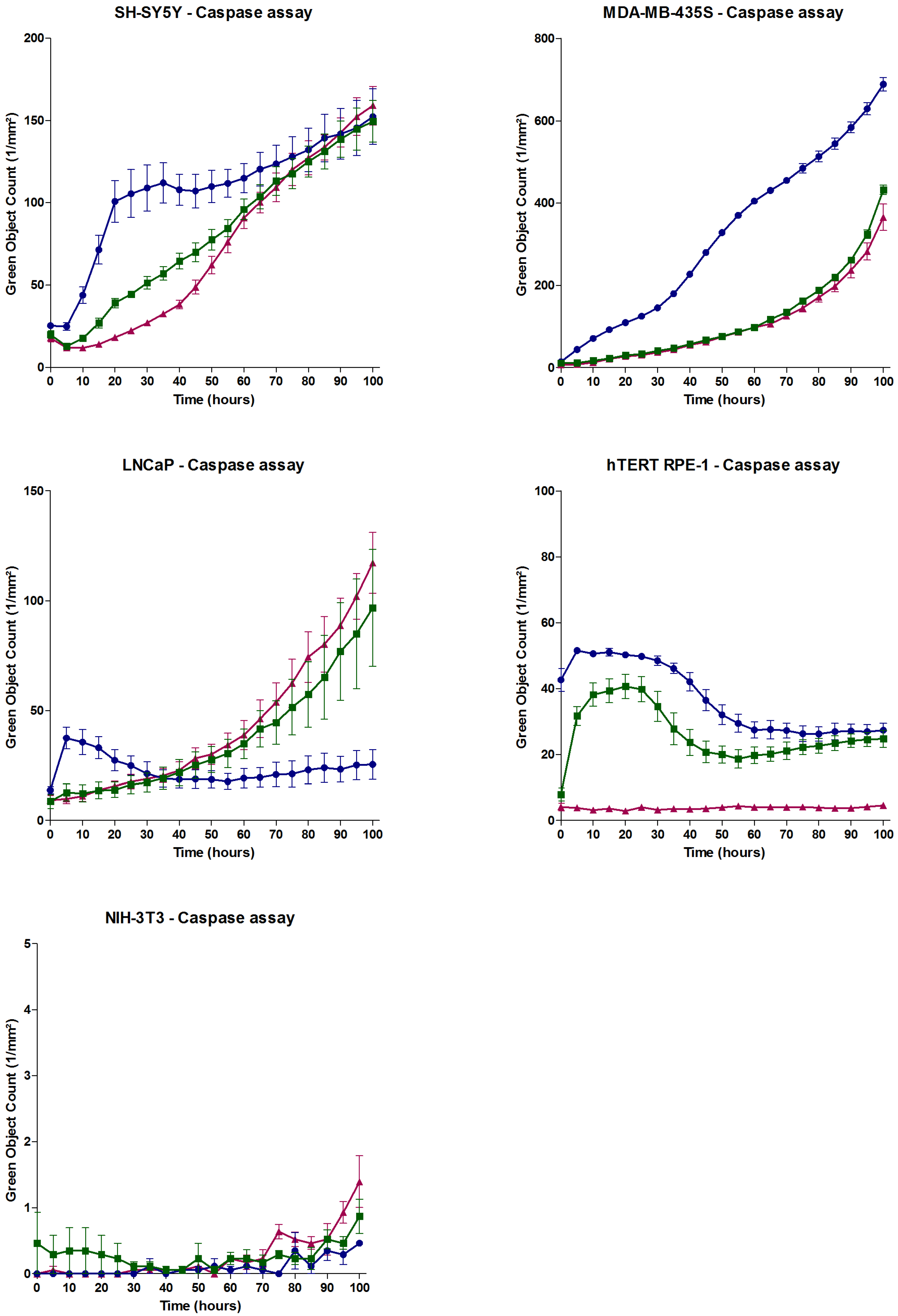
2.4. Effect of APETx4 on Cancerous and Non-Cancerous Cell Lines
3. Discussion
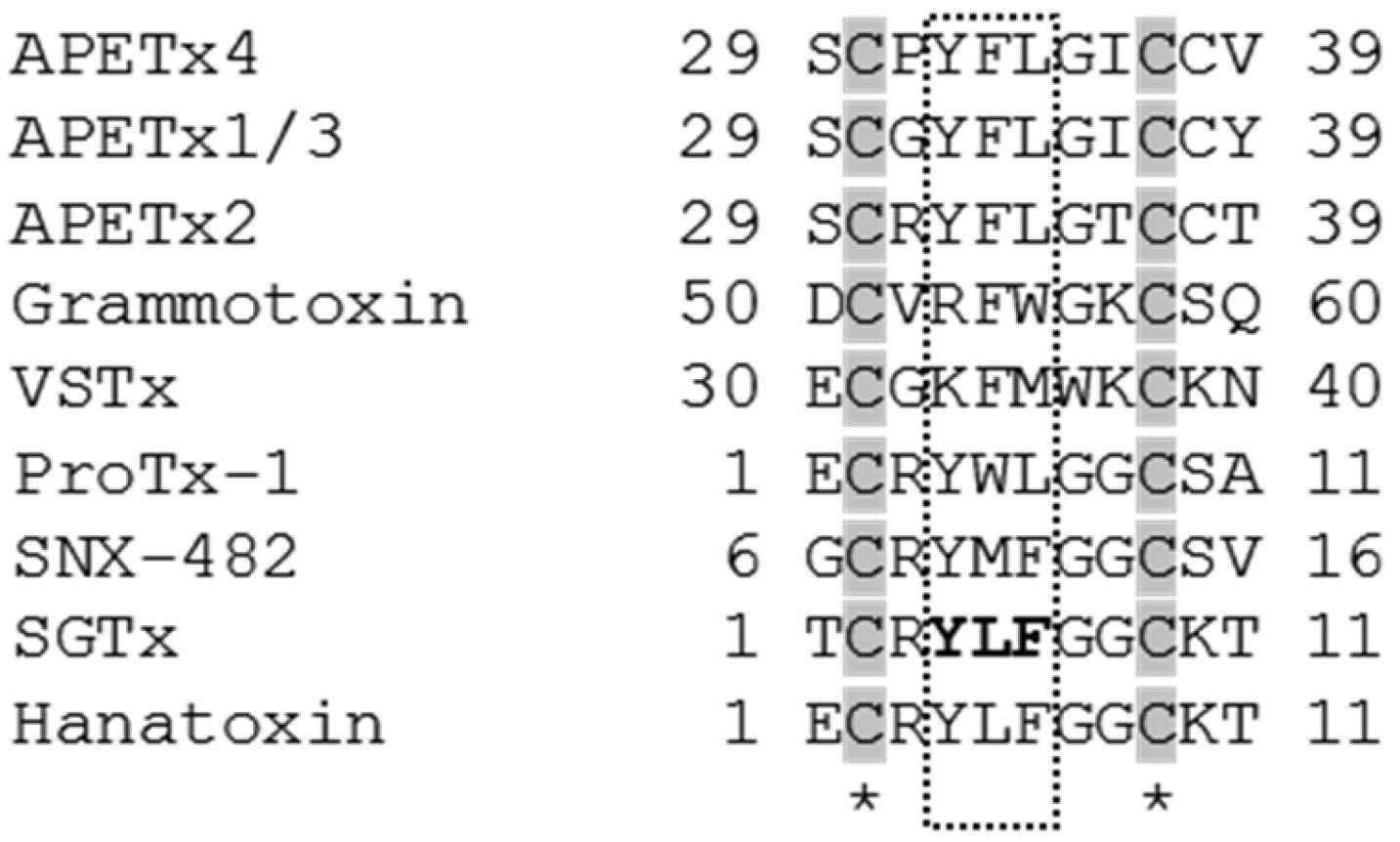
3.1. APETx4 is a Gating Modifier that Presumably Binds to the Voltage Sensor Paddle of KV10.1
3.2. APETx4 is able to Distinguish the Oncogenic KV10.1 Channel from the Cardiac hERG Channel
3.3. The in Vitro Cytotoxic Effect of APETx4 Does not Only Result from Its Effect on KV10.1
4. Materials and Methods
4.1. Purification of APETx4
4.2. Biochemical Characterization and Sequence Analysis
4.3. Expression of Voltage-Gated Ion Channels in Xenopus Laevis Oocytes
4.4. Electrophysiological Recordings
4.5. Live Cell Imaging
4.5.1. Cell Cultures
4.5.2. Proliferation, Cytotoxicity and Apoptosis Assays
4.6. Data Analysis and Molecular Modelling
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Cancer: Fact Sheet. February 2017. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 14 June 2017).
- American Cancer Society Global Cancer Facts & Figures 3rd Edition. Am. Cancer Soc. 2015. [CrossRef]
- Mello de Queiroz, F.; Suarez-Kurtz, G.; Stühmer, W.; Pardo, L.A. Ether à go-go potassium channel expression in soft tissue sarcoma patients. Mol. Cancer 2006, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.R.; Griesinger, F.; Stühmer, W.; Pardo, L.A. The potassium channel Ether à go-go is a novel prognostic factor with functional relevance in acute myeloid leukemia. Mol. Cancer 2010, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Hemmerlein, B.; Weseloh, R.M.; Mello de Queiroz, F.; Knötgen, H.; Sánchez, A.; Rubio, M.E.; Martin, S.; Schliephacke, T.; Jenke, M.; Stühmer, W.; et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol. Cancer 2006, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Asher, V.; Khan, R.; Warren, A.; Shaw, R.; Schalkwyk, G.V.; Bali, A.; Sowter, H.M. The Eag potassium channel as a new prognostic marker in ovarian cancer. Diagn. Pathol. 2010, 5, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Luo, H.; Jin, X.; Yan, J.; Ai, Y. Aberrant expression of Eag1 potassium channels in gastric cancer patients and cell lines. Med. Oncol. 2007, 24, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.-W.; Wang, X.-G.; Luo, H.-S.; Tan, S.-Y.; Gao, S.; Luo, B.; Jiang, H. Expression and prognostic roles of Eag1 in resected esophageal squamous cell carcinomas. Dig. Dis. Sci. 2008, 53, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.-W.; Yan, J.-J.; An, P.; Lü, P.; Luo, H.-S. Aberrant expression of ether à go-go potassium channel in colorectal cancer patients and cell lines. World J. Gastroenterol. 2007, 13, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Lino de Oliveira, C.; Mello de Queiroz, F.; Pardo, L.A.; Stühmer, W.; Del Bel, E. Eag1 potassium channel immunohistochemistry in the CNS of adult rat and selected regions of human brain. Neuroscience 2008, 155, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Saganich, M.J.; Machado, E.; Rudy, B. Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J. Neurosci. 2001, 21, 4609–4624. [Google Scholar] [PubMed]
- Pardo, L.A.; Sühmer, W. Eag1 as a cancer target. Expert Opin. Ther. Targets 2008, 12, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A.; del Camino, D.; Sánchez, A.; Alves, F.; Brüggemann, A.; Beckh, S.; Stühmer, W. Oncogenic potential of EAG K(+) channels. EMBO J. 1999, 18, 5540–5547. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Mello de Queiroz, F.; Downie, B.R.; Suckow, A.; Stühmer, W.; Pardo, L.A. Silencing the activity and proliferative properties of the human EagI Potassium Channel by RNA Interference. J. Biol. Chem. 2006, 281, 13030–13037. [Google Scholar] [CrossRef] [PubMed]
- García-Ferreiro, R.E.; Kerschensteiner, D.; Major, F.; Monje, F.; Stühmer, W.; Pardo, L.A. Mechanism of block of hEag1 K+ channels by imipramine and astemizole. J. Gen. Physiol. 2004, 124, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lagunas, F.; Carrillo, E.; Pardo, L.A.; Stühmer, W. Gating Modulation of the Tumor-Related Kv10.1 Channel by Mibefradil. J. Cell. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Stary-Weinzinger, A.; Sanguinetti, M.C. ICA-105574 interacts with a common binding site to elicit opposite effects on inactivation gating of EAG and ERG potassium channels. Mol. Pharmacol. 2013, 83, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Varela, D.; Zwick-Wallasch, E.; Knötgen, H.; Sánchez, A.; Hettmann, T.; Ossipov, D.; Weseloh, R.; Contreras-Jurado, C.; Rothe, M.; Stühmer, W.; et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007, 67, 7343–7349. [Google Scholar] [CrossRef] [PubMed]
- Hartung, F.; Stühmer, W.; Pardo, L.A. Tumor cell-selective apoptosis induction through targeting of K(V)10.1 via bifunctional TRAIL antibody. Mol. Cancer 2011, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A.; Gómez-Varela, D.; Major, F.; Sansuk, K.; Leurs, R.; Downie, B.R.; Tietze, L.F.; Stuhmer, W. Approaches targeting Kv10.1 open a novel window for cancer diagnosis and therapy. Curr. Med. Chem. 2012, 19, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Moreels, L.; Peigneur, S.; Yamaguchi, Y.; Vriens, K.; Waelkens, E.; Zhu, S.; Thevissen, K.; Cammue, B.P.A.; Sato, K.; Tytgat, J. Expanding the pharmacological profile of κ-hefutoxin 1 and analogues: A focus on the inhibitory effect on the oncogenic channel Kv10.1. Peptides 2016. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.N.; Sivaraja, V.; Huys, I.; Sasaki, T.; Cheng, B.; Kumar, T.K.S.; Sato, K.; Tytgat, J.; Yu, C.; San, B.C.C.; et al. kappa-Hefutoxin1, a novel toxin from the scorpion Heterometrus fulvipes with unique structure and function. Importance of the functional diad in potassium channel selectivity. J. Biol. Chem. 2002, 277, 30040–30047. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. Gold from the sea: Marine compounds as inhibitors of the hallmarks of cancer. Biotechnol. Adv. 2011, 29, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, P.; D’Auria, M.V.; D.Muller, C.; Tammela, P.; Vuorela, H.; Yli-Kauhaluoma, J. Exploring Marine Resources for Bioactive Compounds. Plant. Med. 2014, 80, 1234–1246. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.-H.; Wang, Y.-J.; Sheng, J.; Wang, F.; Zheng, Y.; Lin, X.-K.; Sun, M. Antitumor Peptides from Marine Organisms. Mar. Drugs 2011, 9, 1840–1859. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Jimenez, G.-M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.-M. Bioactive Peptides and Depsipeptides with Anticancer Potential: Sources from Marine Animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.; Honecker, F. Marine Compounds and Cancer: Where Do We Stand? Mar. Drugs 2015, 13, 5657–5665. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kalimuthu, S. Introduction to Anticancer Drugs from Marine Origin. In Handbook of Anticancer Drugs from Marine Origin; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–13. ISBN 9783319071459. [Google Scholar]
- Rocha, J.; Peixe, L.; Gomes, N.C.M.; Calado, R. Cnidarians as a source of new marine bioactive compounds—An overview of the last decade and future steps for bioprospecting. Mar. Drugs 2011, 9, 1860–1886. [Google Scholar] [CrossRef] [PubMed]
- Frazão, B.; Vasconcelos, V.; Antunes, A. Sea anemone (cnidaria, anthozoa, actiniaria) toxins: An overview. Mar. Drugs 2012, 10, 1812–1851. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Chang, S.C.; Chauhan, S.; Huq, R.; Tajhya, R.B.; Chhabra, S.; Norton, R.S.; Beeton, C. Development of Highly Selective Kv1.3-Blocking Peptides Based on the Sea Anemone Peptide ShK. Mar. Drugs 2015, 13, 529–542. [Google Scholar] [CrossRef] [PubMed]
- A 4 Week Study of the Safety, Tolerability, and Pharmacodynamics of ShK-186 (Dalazatide) in Active Plaque Psoriasis. 2015. Available online: http://clinicaltrials.gov (accessed on 12 September 2017).
- Multiple Ascending Dose Safety Study of ShK-186 (Dalazatide) in Healthy Volunteers (2015). Available online: http://clinicaltrials.gov (accessed on 12 September 2017).
- Bruhn, T.; Schaller, C.; Schulze, C.; Sanchez-Rodriguez, J.; Dannmeier, C.; Ravens, U.; Heubach, J.F.; Eckhardt, K.; Schmidtmayer, J.; Schmidt, H.; Aneiros, a.; Wachter, E.; Béress, L. Isolation and characterisation of five neurotoxic and cardiotoxic polypeptides from the sea anemone Anthopleura elegantissima. Toxicon 2001, 39, 693–702. [Google Scholar] [CrossRef]
- McCommas, S.A. Relationships within the family Actiniidae (Cnidaria, Actiniaria) based on molecular characters. Hydrobiologia 1991, 216–217, 509–512. [Google Scholar] [CrossRef]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, J.S.; Fuentes-Silva, D.; King, G.F. Development of a rational nomenclature for naming peptide and protein toxins from sea anemones. Toxicon 2012, 60, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Béress, L.; Möller, C. A natural point mutation changes both target selectivity and mechanism of action of sea anemone toxins. FASEB J. 2012, 26, 5141–5151. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Loret, E.; Bruhn, T.; Béress, L.; Lazdunski, M. APETx1, a new toxin from the sea anemone Anthopleura elegantissima, blocks voltage-gated human ether-a-go-go-related gene potassium channels. Mol. Pharmacol. 2003, 64, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Salata, J.J.; Bennett, P.B. Saxitoxin is a gating modifier of HERG K+ channels. J. Gen. Physiol. 2003, 121, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, X.-S.X.; Diochot, S.; Lazdunski, M.; Tseng, G.-N.N. APETx1 from sea anemone Anthopleura elegantissima is a gating modifier peptide toxin of the human ether-a-go-go-related potassium channel. Mol. Pharmacol. 2007, 72, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartz, K.J.; MacKinnon, R. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron 1997, 18, 675–682. [Google Scholar] [CrossRef]
- Takahashi, H.; Kim, J., II; Min, H.J.; Sato, K.; Swartz, K.J.; Shimada, I. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K(+) channels: common surface features of gating modifier toxins. J. Mol. Biol. 2000, 297, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Roh, S.H.; Kim, S.; Lee, C.W.; Kim, J., II; Swartz, K.J. Molecular surface of tarantula toxins interacting with voltage sensors in K(v) channels. J. Gen. Physiol. 2004, 123, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.-D.; Wu, K.-C.; Guo, X.-Z.; Xie, M.-J.; Zhang, J.; Fan, D.-M. Expression and significance of HERG protein in gastric cancer. Cancer Biol. Ther. 2008, 7, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Asher, V.; Warren, A.; Shaw, R.; Sowter, H.; Bali, A.; Khan, R. The role of Eag and HERG channels in cell proliferation and apoptotic cell death in SK-OV-3 ovarian cancer cell line. Cancer Cell Int. 2011, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, V.; Sowter, H.; Shaw, R.; Bali, A.; Khan, R. Eag and HERG potassium channels as novel therapeutic targets in cancer. World J. Surg. Oncol. 2010, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Baron, A.; Rash, L.D.; Deval, E.; Escoubas, P.; Scarzello, S.; Salinas, M.; Lazdunski, M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004, 23, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.E.; Cristofori-Armstrong, B.; Anangi, R.; Rosengren, K.J.; Lau, C.H.Y.; Mobli, M.; Brust, A.; Alewood, P.F.; King, G.F.; Rash, L.D. Understanding the molecular basis of toxin promiscuity: The analgesic sea anemone peptide APETx2 interacts with acid-sensing ion channel 3 and hERG channels via overlapping pharmacophores. J. Med. Chem. 2014, 57, 9195–9203. [Google Scholar] [CrossRef] [PubMed]
- Diana Urrego; Naira Movsisyan; Roser Ufartes; Luis A. Pardo. Periodic expression of Kv10.1 contributes to G2/M progression of cancer and non- transformed cells. Cell Cycle 2016, 15, 99–811. [Google Scholar] [CrossRef]
- Kourie, J.I.; Shorthouse, A.A. Properties of cytotoxic peptide-formed ion channels. Am. J. Physiol. Cell Physiol. 2000, 278, C1063–C1087. [Google Scholar] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinformatics 2016, 54, 5.6.1–5.6.37. [Google Scholar] [PubMed]
- Goujon, M.; Mcwilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL–EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Description | KV10.1 Expression Level | Ref. |
|---|---|---|---|
| SH-SY5Y | Human neuroblastoma cell line | High | [20] |
| LNCaP | Human prostate cancer cell line | Undetectable | [20] |
| NIH-3T3 | Mouse embryonic fibroblast cell line | Undetectable | [14] |
| MDA-MB-435S | Human melanoma cell line | Moderate | [20] |
| hTERT RPE-1 | Human epithelial cell line | Moderate | [20] |
| Cell Line | Description | Medium |
|---|---|---|
| SH-SY5Y | Human neuroblastoma cell line | RPMI + 15% FCS |
| LNCAP | Human prostate cancer cell line | RPMI + 15% FCS |
| NIH-3T3 | Mouse embryonic fibroblast cell line | DMEM + 10% FCS |
| MDA-MB-435S | Human melanoma cell line | RPMI + 10% FCS |
| hTERT RPE-1 | Human epithelial cell line | DMEM:F12 + 10% FCS |
| + 10 µg/mL Hygromycin B |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreels, L.; Peigneur, S.; Galan, D.T.; De Pauw, E.; Béress, L.; Waelkens, E.; Pardo, L.A.; Quinton, L.; Tytgat, J. APETx4, a Novel Sea Anemone Toxin and a Modulator of the Cancer-Relevant Potassium Channel KV10.1. Mar. Drugs 2017, 15, 287. https://doi.org/10.3390/md15090287
Moreels L, Peigneur S, Galan DT, De Pauw E, Béress L, Waelkens E, Pardo LA, Quinton L, Tytgat J. APETx4, a Novel Sea Anemone Toxin and a Modulator of the Cancer-Relevant Potassium Channel KV10.1. Marine Drugs. 2017; 15(9):287. https://doi.org/10.3390/md15090287
Chicago/Turabian StyleMoreels, Lien, Steve Peigneur, Diogo T. Galan, Edwin De Pauw, Lászlo Béress, Etienne Waelkens, Luis A. Pardo, Loïc Quinton, and Jan Tytgat. 2017. "APETx4, a Novel Sea Anemone Toxin and a Modulator of the Cancer-Relevant Potassium Channel KV10.1" Marine Drugs 15, no. 9: 287. https://doi.org/10.3390/md15090287
APA StyleMoreels, L., Peigneur, S., Galan, D. T., De Pauw, E., Béress, L., Waelkens, E., Pardo, L. A., Quinton, L., & Tytgat, J. (2017). APETx4, a Novel Sea Anemone Toxin and a Modulator of the Cancer-Relevant Potassium Channel KV10.1. Marine Drugs, 15(9), 287. https://doi.org/10.3390/md15090287