2. Results and Discussion
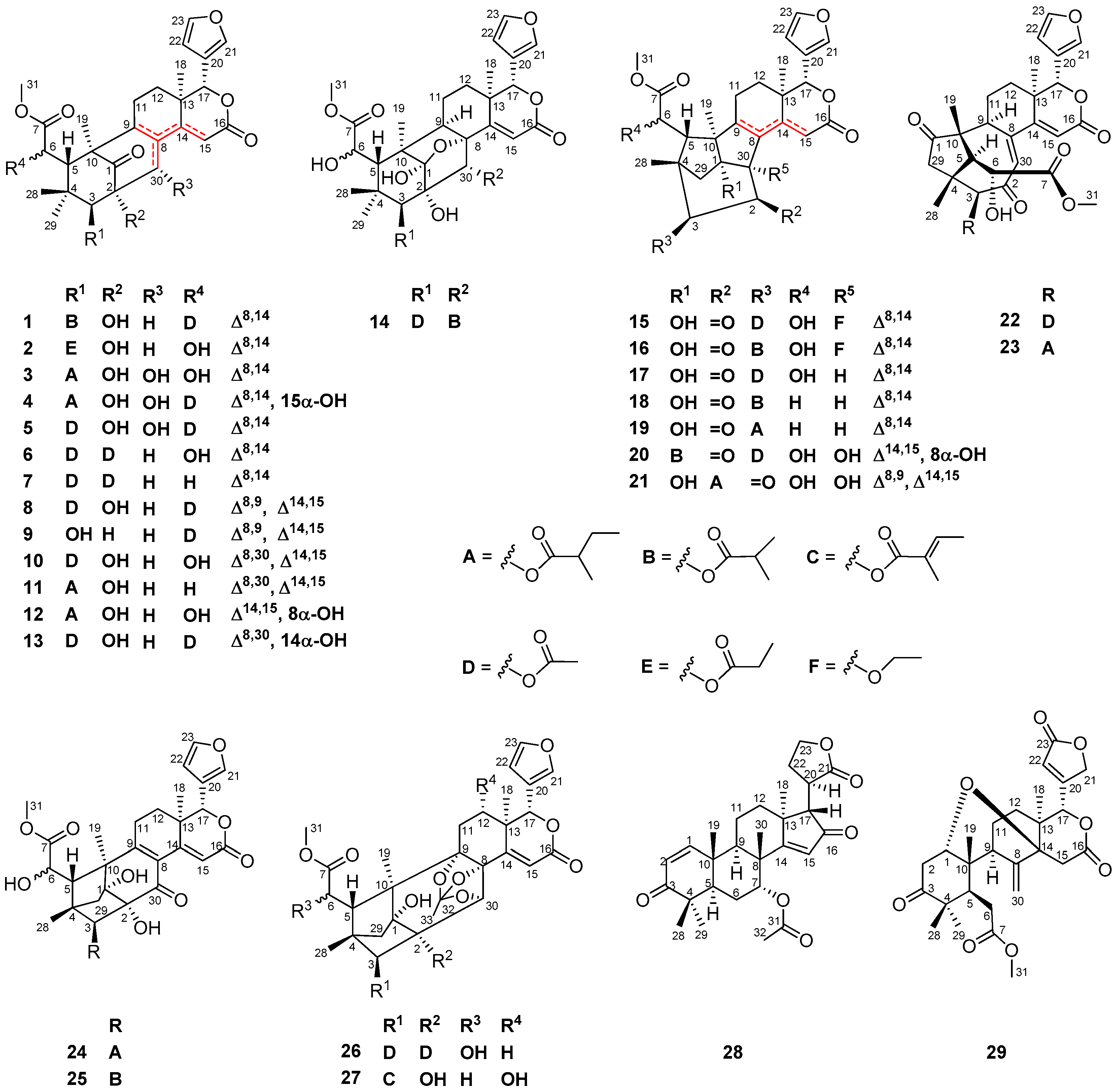
In this paper, 16 compounds (viz. 1, 3–6, 8, 9, 12–14, 20, 21, 24, 26, 27, and 29) were obtained from seeds of the Thai X. moluccensis, whereas 13 compounds (viz. 2, 7, 10, 11, 15–19, 22, 23, 25, and 28) were isolated from those of the Indian X. moluccensis.
Compound
1 was obtained as a colorless crystal. The molecular formula of
1 was established from the positive HRESIMS ion peak at
m/
z 615.2785 (calcd. for [M + H]
+, 615.2800) to be C
33H
42O
11, implying thirteen degrees of unsaturation. According to the
1H and
13C NMR spectroscopic data (
Table 1 and
Table 2), five elements of unsaturation were due to four ester groups, a keto carbonyl function, and three carbon-carbon double bonds; thus, the molecule was pentacyclic. The
1H and
13C NMR spectroscopic data (
Table 1 and
Table 2) showed the presence of a β-substituted furan ring [δ
H 7.52 br d (
J = 0.8 Hz H-21), 6.45 br d (
J = 1.2 Hz H-22), 7.44 t (
J = 1.6 Hz H-23)], four tertiary methyl groups (δ
H 1.27 s H
3-19, 1.04 s H
3-29, 1.05 s H
3-18, 0.84 s H
3-28), a methoxy (δ
H 3.73 s H
3-31), and a keto function (δ
C 216.7 qC), indicating a mexicanolide-type limonoid for
1.
The NMR spectroscopic data of
1 (
Table 1 and
Table 2) were similar to those of khayalenoid H [
19], except for the replacement of the 3-
O-acetyl group in khayalenoid H by an isobutyryloxy group [δ
H 2.64 m 1H, 1.22 d (
J = 7.2 Hz 3H), 1.24 d (
J = 7.2 Hz 3H); δ
C 175.9 qC, 34.5 CH, 19.9 CH
3, 18.4 CH
3] in
1. The presence of the isobutyryloxy group was corroborated by
1H-
1H COSY correlations between H
3-34/H-33 and H
3-35/H-33. The significant HMBC correlation from H-3 (δ
H 4.97 s) to the carbonyl carbon (δ
C 175.9 qC) of the isobutyryloxy group placed it at C-3 (
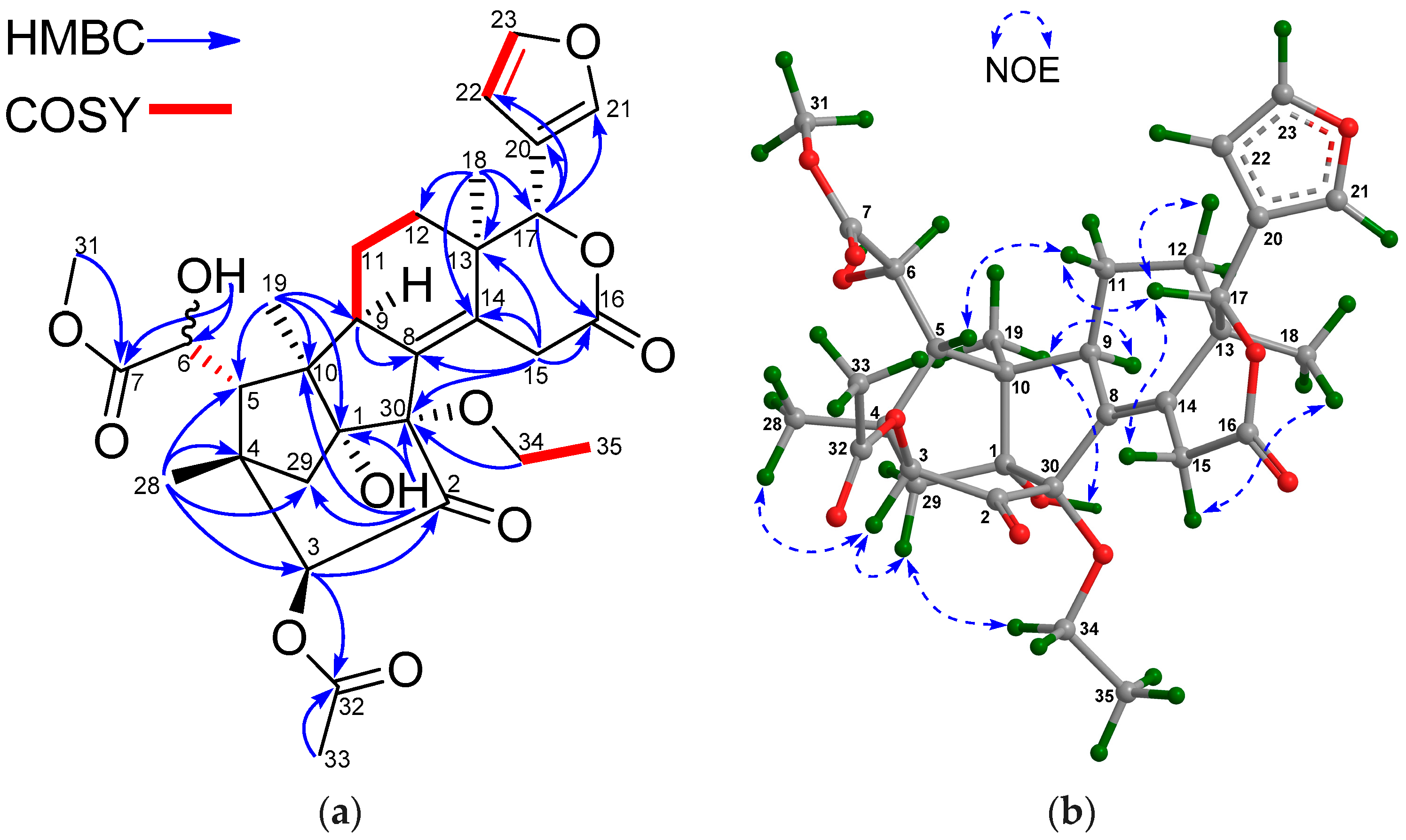
Figure 2a).
The relative configuration of
1 was established by diagnostic NOE interactions (
Figure 2b). Those between H-17/H-11β, H-17/H-12β, H-17/H-5, and H-5/H
3-28 revealed their cofacial relationship, and were arbitrarily assigned as the β-oriented H-17 and H-5. NOE interactions between H-9/H-12α, H-9/H
3-19, H
3-18/H-12α, H-3/H
3-29, and 2-OH/H
3-29 assigned the α-orientation for H-9, H
3-18, H
3-19, H-3, and 2-OH.
In order to establish the absolute configuration of
1, single-crystal X-ray diffraction analysis with Cu Kα radiation (Flack parameter of −0.06 (10), Flack x of −0.14 (11), and Hooft y of −0.04 (3); suitable crystals of
1 were obtained from acetone/methanol (1:2) at room temperature) was employed, which unequivocally assigned the absolute configuration of
1 as 2
R,3
S,5
S,6
R,9
S,10
R,13
R,17
R. The computer-generated perspective drawing of the X-ray structure of
1 is shown in
Figure 3. Therefore, the absolute configuration of
1 named xylomolin A
1 was assigned as shown (
Figure 2a).
Compound
2 was obtained as an amorphous white powder. The molecular formula was determined to be C
30H
38O
10 by the negative HRESIMS ion peak at
m/
z 593.2143 (calcd. for [M + Cl]
−, 593.2159). The NMR spectroscopic data of
2 resembled those of
1 (
Table 1 and
Table 2), except for the replacement of the 3-
O-isobutyryl moiety and the 6-
O-acetyl group in
1 by a 3-
O-propionyl moiety (δ
H 2.45 m 1H, 2.43 m 1H, 1.20 t (
J = 7.6 Hz 3H); δ
C 173.3 qC, 28.0 CH
2, 9.4 CH
3) and a 6-OH group in
2, respectively. The presence of a 3-
O-propionyl moiety was confirmed by
1H-
1H COSY correlations between H
3-34/H
2-33 and HMBC cross-peaks between H
3-34/C-33, H
3-34/C-32, H
2-33/C-32, and H-3/C-32. The relative configuration of
2 (except for that of C-6) was determined to be the same as that of
1 by NOE interactions between H-17/H-15β, H-15β/H-30β, H-17/H-12β, H-17/H-11β, H-11β/H-5, H-17/H-5, and H-5/H
3-28, and those between H-9/H
3-19, H
3-19/H
3-29, H
3-29/H-3, and H
3-18/H-15α. Thus, the structure of
2—named xylomolin A
2—was assigned as shown in
Figure 1.
Compound
3 gave the molecular formula C
32H
42O
11 as established by the HRESIMS ion peak at
m/
z 625.2620 (calcd. for [M + Na]
+, 625.2619). The NMR spectroscopic data of
3 (
Table 1 and
Table 2) were similar to those of moluccensin S [
20], except for the presence of an additional 30-OH group, which was supported by the downshifted C-30 signal (δ
C 73.0 CH in
3, whereas δ
C 44.6 CH
2 in moluccensin S) and HMBC correlations from the proton of 30-OH to C-30, C-2, and C-8. NOE interactions between H-17/H-12β, H-17/H-11β, H-5/H-12β, H-5/H-17, H-5/H
3-28, and H-5/H-30 revealed the β-orientation for H-5, H-17, H-30, and H
3-28, and the corresponding 30α-OH. Similarly, those between H-3/2-OH, H
3-29/2-OH, H-9/H
3-19, H
3-29/H
3-19, and H
3-18/H-11α assigned the α-orientation for H-3, H-9, H
3-18, H
3-19, H
3-29, and 2-OH. Thus, the structure of
3—named xylomolin A
3—was determined to be 30α-hydroxy-moluccensin S.
Compound
4 provided the molecular formula of C
34H
44O
13 as determined from the positive HRESIMS ion peak at
m/
z 683.2675 (calcd. for [M + Na]
+, 683.2674). The
1H and
13C NMR spectroscopic data of
4 resembled those of
3 (
Table 1 and
Table 2), the difference being the presence of a 15-OH function (δ
H 3.66 br s) and a 6-acetoxy group (δ
H 2.18 s 3H; δ
C 169.8 qC, 21.0 CH
3) in
4. The existence of a 15-OH function was corroborated by the downshifted C-15 signal (δ
C 65.4 CH in
4, whereas δ
C 32.8 CH
2 in
3) and HMBC correlations from the proton of 15-OH to C-14, C-15, and C-16. The HMBC correlation from H-6 to the carbonyl carbon (C-37) of the acetoxy group placed it at C-6. The relative configuration of
4 (except that of C-15) was assigned as the same as that of
3 on the basis of NOE interactions. Those between H-17/H-15 and H-5/H-30 assigned the β-oriented H-15 and H-30, and the corresponding 15α-OH group. Consequently, the structure of
4—named xylomolin A
4—was identified as 15α-hydroxy-6-acetoxy-xylomolin A
3.
The molecular formula of
5 was determined to be C
31H
38O
12 by the positive HRESIMS ion peak at
m/
z 625.2252 (calcd. for [M + Na]
+, 625.2255). The
1H and
13C NMR spectroscopic data of
5 (
Table 1 and
Table 2) were closely related to those of khayalenoid H, the difference being the existence of a 30-OH function, which was supported by the downshifted C-30 signal (δ
C 72.9 CH in
5, whereas δ
C 44.4 CH
2 in khayalenoid H). HMBC correlations from the proton of 30-OH (δ
H 2.67 br s) to C-30 and C-8 demonstrated the above deduction. The NOE interaction between H-5/H-30 assigned the β-oriented H-30 and the corresponding 30α-OH. Thus, the structure of
5—named xylomolin A
5—was assigned as 30α-hydroxy-khayalenoid H.
Compound
6 was isolated as a white and amorphous powder. Its molecular formula was determined to be C
31H
38O
11 from the positive HRESIMS ion peak at
m/
z 609.2311 (calcd. for [M + Na]
+, 609.2306). The
1H and
13C NMR spectroscopic data of
6 (
Table 1 and
Table 2) closely resembled those of khayalenoid H [
19], except for the replacement of the 2-OH function and the 6-
O-acetyl group in khayalenoid H by a 2-
O-acetyl (δ
H 2.13 s 3H; δ
C 169.1 qC, 21.8 CH
3) and a 6-OH group (δ
H 2.80 br s) in
6, respectively. HMBC correlations from the proton of the 6-OH group to C-5, C-6,and C-7 placed it at C-6. The presence of the 2-
O-acetyl group in
6 was corroborated by the downshifted C-2 in
6 (δ
C 86.5 qC in
6, whereas δ
C 77.9 qC in khayalenoid H). HMBC correlations from H
2-30 and H-3 to C-2 confirmed the above deduction. The relative configuration of
6 was determined to be the same as that of khayalenoid H based on diagnostic NOE interactions between H-17/H-15β, H-17/H-12β, H-17/H-5, and H-5/H
3-28 and those between H-9/H
3-19, H
3-19/H
3-29, and H
3-18/H-15α. Therefore, the structure of
6—named xylomolin A
6—was determined to be 2-
O-acetyl-6-
O-deacetyl-khayalenoid H.
The molecular formula of
7 was determined to be C
31H
38O
10 by the positive HRESIMS ion peak at
m/
z 593.2358 (calcd. for [M + Na]
+, 593.2357). The
1H and
13C NMR spectroscopic data of
7 (
Table 1 and
Table 2) were closely related to those of
6, except for the absence of the 6-OH group, which was corroborated by the upshifted CH
2-6 signal (δ
H 2.32 dd (
J = 16.5, 1.6 Hz), 2.42 dd (
J = 16.5, 10.7 Hz), δ
C 33.3) in
7.
1H-
1H COSY correlations between H
2-6/H-5 and HMBC cross-peaks from H
2-6 and C-5 and C-7 confirmed the above result. The analysis of diagnostic NOE interactions revealed that
7 possessed the same relative configuration as that of
6. Therefore, the structure of
7—named xylomolin A
7—was assigned as 6-dehydroxy-xylomolin A
6.
Compound
8 was isolated as a white and amorphous powder. It had a molecular formula of C
31H
36O
11 as deduced from the positive HRESIMS ion peak at
m/
z 607.2151 (calcd. for [M + Na]
+, 607.2150). The
1H and
13C NMR spectroscopic data of
8 (
Table 3 and
Table 4) were similar to those of heytrijunolide E [
21] with Δ
8,9 and Δ
14,15 conjugated double bonds, except for the absence of the 15-OH group and the presence of an acetoxy group at C-3 in
8. The absence of the 15-OH group was confirmed by the upshifted C-15 signal (δ
C 111.6 qC in
8, whereas δ
C 134.7 qC in heytrijunolide E) and HMBC correlations between H-15/C-14, H-15/C-16, H-15/C-8, and H-15/C-13. The HMBC correlation from H-3 (δ
H 5.51 s) to the carbonyl carbon (δ
C 170.2 qC) of the acetoxy group placed it at C-3. The relative configuration of
8 was established to be the same as that of heytrijunolide E based on diagnostic NOE interactions between H
3-28/H-5, H-5/H-11β, H-12β/H-17, H
3-29/H-3, H-12α/H
3-18, and H-11α/H
3-19. Therefore, the structure of
8—named xylomolin B
1—was assigned as 15-dehydroxy-3β-acetoxy-heytrijunolide E.
Compound
9 provided the molecular formula C
29H
34O
9 as established by the positive HRESIMS ion peak at
m/
z 527.2273 (calcd. for [M + H]
+, 527.2276). The
1H and
13C NMR spectroscopic data of
9 (
Table 3 and
Table 4) were similar to those of heytrijunolide E [
21], the difference being the absence of the 2-OH and 15-OH groups in
9, which was corroborated by the upshifted C-15 (δ
C 110.6 CH in
9, whereas δ
C 134.7 qC in heytrijunolide E) and C-2 (δ
C 50.5 CH in
9, whereas δ
C 78.2 qC in heytrijunolide E) signals. A proton (δ
H 3.02 t (
J = 6.0 Hz)) exhibiting
1H-
1H COSY correlations to H-3 and H-30 and HMBC cross-peaks to C-1, C-3, C-4, and C-30 was assigned as H-2. The existence of H-15 (δ
H 5.89 s) was further confirmed by its HMBC correlations to C-8, C-13, C-14, and C-16. The relative configuration of
9 was determined to be the same as that of heytrijunolide E based on diagnostic NOE interactions between H
3-28/H-5, H-5/H-11β, H-12β/H-17, H
3-29/H-3, H-12α/H
3-18, and H-11α/H
3-19. Thus, the structure of
9—named xylomolin B
2—was assigned as 2,15-dedihydroxy-heytrijunolide E.
Compound
10 had a molecular formula of C
29H
34O
10 as determined from the positive HRESIMS ion peak at
m/
z 543.2238 (calcd. for [M + H]
+, 543.2230). The NMR data of
10 (
Table 3 and
Table 4) closely resembled those of moluccensin U [
20], except for the replacement of the 3-
O-(2-methyl)butyryl group in moluccensin U by an acetoxy group (δ
H 2.22 s 3H; δ
C 169.9 qC, 20.7 CH
3) in
10. The significant HMBC cross-peak from H-3 (δ
H 4.83 s) to the carbonyl carbon of the above acetoxy group placed it at C-3. NOE interactions between H-11β/H-17, H-12β/H-17, H-11β/H-5, H-5/H
3-28, H-9/H
3-18, H-9/H
3-19, and H
3-29/H-3 revealed the same relative configuration of
10 as that of moluccensin U. Therefore, the structure of
10—named xylomolin C
1—was assigned as 3-
O-acetyl-3-de(2-methyl) butyryloxy-moluccensin U.
Compound
11 afforded the molecular formula C
32H
40O
9 as established by the positive HRESIMS ion peak at
m/
z 591.2570 (calcd. for [M + Na]
+, 591.2565). The
1H and
13C NMR spectroscopic data of
11 (
Table 3 and
Table 4) closely resembled those of swietmanin G [
22], the difference being the replacement of the 3-isobutyryloxy group in swietmanin G by a 2-methylbutyryloxy group (δ
H 2.57 m 1H, 1.61 m 1H, 1.81 m 1H, 1.04 t (
J = 7.2 Hz 3H), 1.25 d (
J = 6.8 Hz 3H); δ
C 176.0 qC, 41.4 CH, 26.9 CH
2, 11.9 CH
3, 17.0 CH
3) in
11. The deduction was confirmed by
1H-
1H COSY correlations between H
3-36/H-33, H
3-35/H
2-34, and H
2-34/H-33, and HMBC correlations between H-3/C-32, H
3-35/C-34, H
3-35/C-33, H
3-36/C-33, H
3-36/C-32, and H
3-36/C-34. NOE interactions between H-12β/H-17, H-17/H-11β, H-5/H-11β, H-5/H
3-28, H-9/H
3-18, H-9/H
3-19, and H
3-29/H-3 assigned the α-orientation for H-9, H
3-18, H
3-19, and H-3. Thus, the structure of
11—named xylomolin C
2—was assigned as 3-
O-(2-methyl)butyryl-3-deisobutyryloxy-swietmanin G.
Compound
12 had the molecular formula C
32H
42O
11 as determined from the positive HRESIMS ion peak at
m/
z 625.2612 (calcd. for [M + Na]
+, 625.2619). The
1H and
13C NMR spectroscopic data of
12 (
Table 3 and
Table 4) were similar to those of moluccensin U [
20], except for the absence of the Δ
8,30 double bond and the presence of an 8-OH group. This finding was verified by the upshifted C-8 (δ
C 71.3 qC) and C-30 (δ
C 45.9 CH
2) signals as compared with those (δ
C 134.5 qC and 133.6 CH) of moluccensin U, respectively. The presence of the 8-OH group was confirmed by HMBC correlations from its proton to C-8, C-9, and C-30. NOE interactions between 8-OH/H-9 and 8-OH/H
3-18 assigned the α-orientation for the 8-OH group. The relative configuration of
12 (except that of C-8) was established to be identical to that of moluccensin U based on diagnostic NOE interactions between H-12β/H-17, H-5/H
3-28, H-9/H
3-19, and H
3-29/H-3. Thus, the structure of
12—named xylomolin D—was assigned as 8,30-dihydrogen-8α-hydroxy-moluccensin U.
Compound
13 was obtained as an amorphous white power. The molecular formula was determined to be C
31H
38O
12 by the positive HRESIMS ion peak at
m/
z 625.2250 (calcd. for [M + Na]
+, 625.2255). The
1H and
13C NMR spectroscopic data of
13 (
Table 3 and
Table 4) were similar to those of
8, except for the replacement of Δ
8,9 and Δ
14,15 conjugated double bonds in
8 by a Δ
8,30 double bond (δ
C 138.8 qC C-8, 130.2 CH C-30) in
13, and the presence of an additional 14-OH group. HMBC correlations between H
2-15/C-14, H
2-15/C-16, H
2-15/C-13, H-30/C-14, H-30/C-9, and H-9/C-8 demonstrated the above deduction. In order to establish the relative configuration of the 14-OH group in
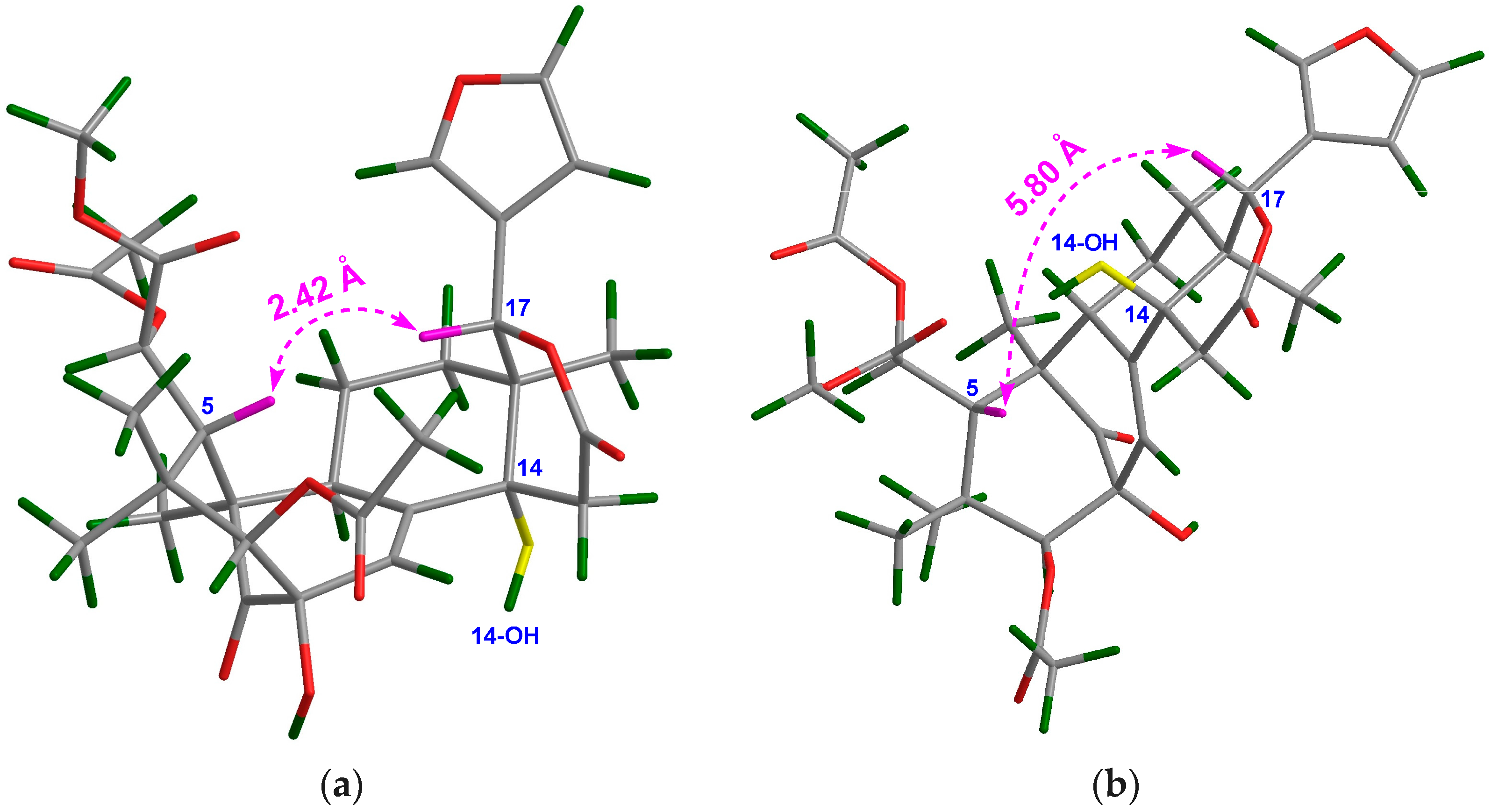
13, two possible 3D structures with a 14α-OH and 14β-OH groups, respectively, were simulated by using the ChemBio3D software (
Figure 4). When the 14-OH group occupies α-orientation, the space distance between H-5/H-17 is around 2.4 Å (
Figure 4a), implying the presence of a strong NOE interaction between these protons. On the contrary, when the 14-OH group occupies the β-orientation, the space distance between H-5/H-17 is around 5.8 Å (
Figure 4b), indicating the absence of a NOE interaction between these protons. Quite evidently, the NOE interaction between H-5/H-17 could be utilized as an effective criterion to resolve the relative configuration of the 14-OH group. Thus, the orientation of the 14-OH group in
13 was assigned as α based on the strong NOE interaction between H-5/H-17. Furthermore, the relative configuration of the whole molecule of
13 (except that of C-14) was determined to be the same as that of
8 on the basis of NOE interactions between H
3-28/H-5, H-5/H-11β, H-12β/H-17, H
3-29/H-3, H-12α/H
3-18, and H-11α/H
3-19. Thus, the structure of
13—named xylomolin E—was assigned as depicted.
Compounds
2–
13 are analogues of
1. From the point of view of biogenetic origins, these mexicanolides should possess the same absolute configurations of carbon skeletons as that of
1. The absolute sterostructures of
2–
13 are shown as in
Figure 1.
The molecular formula of
14 was determined to be C
33H
42O
13 by the positive HRESIMS ion peak at
m/
z 669.2521 (calcd. for [M + Na]
+, 669.2518). The NMR spectroscopic data of
14 (
Table 3 and
Table 4) were similar to those of xylorumphiin H [
23], being a mexicanolide containing a C1-
O-C8 bridge, except for the presence of an additional Δ
14,15 double bond (δ
H 6.08 s 1H; δ
C 158.3 qC, 118.4 CH) and an additional 6-OH function (δ
H 2.93 s) in
14. The existence of the Δ
14,15 double bond was corroborated by HMBC correlations between H
3-18/C-14, H-15/C-8, and H-15/C-16. The downshifted C-6 signal (δ
C 71.7 CH in
14, whereas δ
C 32.3 CH
2 in xylorumphiin H), along with HMBC cross-peaks from H-6 to C-5 and C-7, supported the location of a hydroxy group at C-6. Similar NOE interactions of
14 as those of xylorumphiin H suggested that both mexicanolides possessed the same relative configuration. Thus, the structure of
14—named xylomolin F—was assigned as 6-hydroxy-14,15-dedihydrogen-xylorumphiin H.
The molecular formula of
15 was established by the positive HRESIMS ion peak at
m/
z 587.2494 (calcd. for [M + H]
+, 587.2492) to be C
31H
38O
11, implying thirteen degrees of unsaturation. According to the NMR spectroscopic data of
15 (
Table 5 and
Table 6), seven elements of unsaturation were due to three carbon-carbon double bonds, one carbonyl group, and three ester functionalities; thus,
15 should be hexacyclic. The NMR spectroscopic data of
15 resembled those of thaixylomolin L [
18], being a khayanolide isolated from seeds of the Thai
X. moluccensis, except for the presence of an additional 6-OH group in
15. Strong
3J HMBC correlations from H
2-29 to C-30 further confirmed a khayanolide for
15 instead of a phragmalin, which should exhibit weak
4J HMBC correlations between H
2-29/C-30. HMBC cross-peaks from an active proton (δ
H 2.91 d (
J = 3.4 Hz)) to C-5 (δ
C 45.4, CH), C-6 (δ
C 72.0, CH), and C-7 (δ
C 175.3, qC) (
Figure 5a) revealed the existence of a 6-OH group in
15.
The relative configuration of
15 was assigned by analysis of NOE interactions (
Figure 5b). Those between H-17/H-12β, H-17/H-15β, H-17/H-11β, and H-11β/H-5 revealed their cofacial relationship and were assigned as β-oriented. In turn, NOE interactions between H
3-18/H-15α, H-9/H
3-19, H
3-19/1-OH, and H-34/H
pro-R-29 indicated the α-orientation for H-9, H
3-18, H
3-19, 1-OH, and 30-OEt. The NOE interaction between H-3/H
pro-R-29 established the 3α-H and the corresponding 3β-acetoxy function. Therefore, the relative configuration of
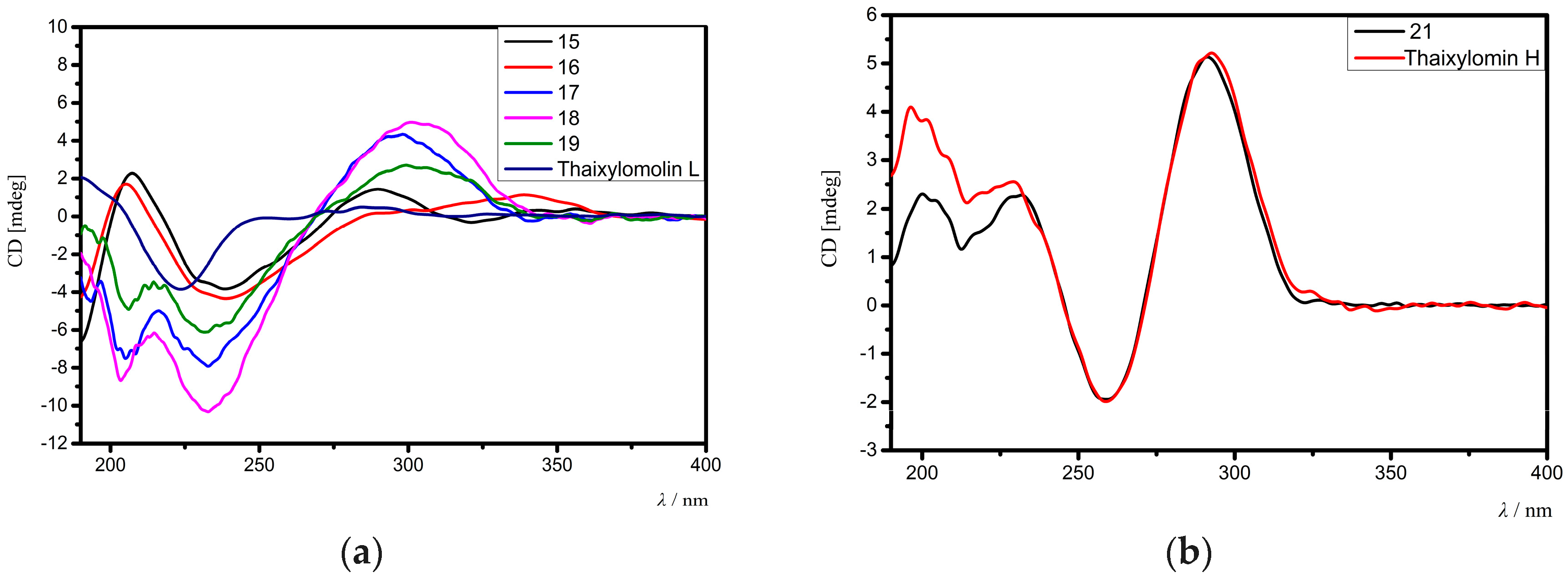
15 was determined. Comparison of the electronic circular dichroism (ECD) spectrum of
15 with that of thaixylomolin L [
18] showed that
15 had the same 1
R,3
S,4
R,5
S,9
R,10
S,13
R,17
R,30
S-absolute configuration as that of thaixylomolin L (
Figure 6a). Thus, the structure of
15—named xylomolin G
1—was assigned as depicted.
Compound
16 had a molecular formula of C
33H
42O
11 as determined from the positive HRESIMS ion peak at
m/
z 615.2809 (calcd. for [M + H]
+, 615.2805). The NMR spectroscopic data of
16 (
Table 5 and
Table 6) were closely related to those of
15, the difference being the replacement of 3-acetoxy group in
15 by an isobutyryloxy group (δ
H 2.57 m, 1.20 d (
J = 7.2 Hz), 1.22 d (
J = 7.2 Hz); δ
C 176.0 qC, 33.8 CH, 19.0 CH
3, 19.1 CH
3) in
16. The presence of the above isobutyryloxy group was further supported by
1H-
1H COSY correlations between H
3-34/H-33 and H
3-35/H-33, and HMBC correlations between H-33/C-32, H
3-34/C-32, and H
3-35/C-32. The significant HMBC cross-peak from H-3 to the carbonyl carbon (C-32) of the isobutyryloxy group confirmed its location at C-3. NOE interactions between H-17/H-12β, H-17/H-15β, H-17/H-11β, H-11β/H-5, H-17/H-5, H-5/H
3-28, H-3/H
pro-
R-29, H-15α/H
3-18, H
3-18/H-9, H-9/H
3-19, and 1-OH/H-9 indicated the same relative configuration of
16 as that of
15. Comparison of the ECD spectrum of
16 with that of thaixylomolin L concluded that
16 had the same 1
R,3
S,4
R,5
S,9
R,10
S,13
R,17
R,30
S-absolute configuration as that of thaixylomolin L (
Figure 6a). Thus, the structure of
16—named xylomolin G
2—was assigned as 3-
O-isobutyryl-3-deacetoxy-xylomolin G
1.
Compound
17 was isolated as an amorphous yellow solid. Its molecular formula was determined to be C
29H
34O
10 by the positive HRESIMS ion peak at
m/
z 543.2246 (calcd. for [M + H]
+, 543.2230). The similarities between the NMR spectroscopic data of
17 (
Table 5 and
Table 6) and
15 revealed their close structural resemblance, except for the absence of the ethoxyl group at C-30, which was confirmed by the upshifted C-30 signal (δ
C 63.0 CH in
17, whereas δ
C 92.2 qC in
15) and HMBC cross-peaks between H-30/C-1, H-30/C-2, H-30/C-8, and H-30/C-10. The relative configuration of
17 was determined to be identical to that of
15 by analysis of NOE interactions. Comparison of ECD spectra of
17 and
15 concluded that both compounds had the same 1
R,3
S,4
R,5
S,9
R,10
S,13
R,17
R,30
S-absolute configuration (
Figure 6a). Thus, the structure of
17—named xylomolin G
3—was assigned as 30-deethoxyl-xylomolin G
1.
Compound
18 provided the molecular formula C
31H
38O
9 as established by the positive HRESIMS ion peak at
m/
z 555.2593 (calcd. for [M + H]
+, 555.2594). The NMR spectroscopic data of
18 (
Table 5 and
Table 6) were similar to those of
15, the difference being the absence of the 30-ethoxyl group and the 6-OH function in
18. The upshifted C-30 (δ
C 63.4 CH in
18, whereas δ
C 92.2 qC in
15) and C-6 (δ
C 34.2 CH
2 in
18, whereas δ
C 72.1 CH in
15) signals and HMBC cross-peaks between H-30/C-1, H-30/C-2, H-30/C-8, H-30/C-10, H-6/C-5, and H-6/C-7 supported the above deduction. The relative and absolute configurations of
18 were determined to be the same as that of
15 by analysis of their NOE interactions and ECD spectra (
Figure 6a). Thus, the structure of
18—named xylomolin G
4—was concluded to be 30-deethoxyl-6-dehydroxy-xylomolin G
1.
Compound
19 gave the molecular formula C
32H
40O
9 as determined from the positive HRESIMS ion peak at
m/
z 569.2753 (calcd. for [M + H]
+, 569.2751). The NMR data of
19 (
Table 5 and
Table 6) were closely related to those of
18, except for the replacement of the 3-isobutyryloxy group in
18 by a 2-methylbutyryloxy group (δ
H 2.47 m 1H, 1.73 m 1H, 1.52 m 1H, 0.94 t (
J = 7.2 Hz 3H), 1.19 d (
J = 7.2 Hz 3H); δ
C 176.0 qC, 40.8 CH, 26.7 CH
2, 11.5 CH
3, 16.5 CH
3) in
19. The presence of the 2-methylbutyryloxy group was supported by the
1H-
1H COSY correlations between H-33/H-34, H-33/H
3-36, and H-34/H
3-35 and HMBC cross-peaks between H-33/C-32, H
2-34/C-32, H
3-36/C-32, H
3-36/C-34, and H
3-35/C-33. The significant HMBC cross-peak from H-3 to the carbonyl carbon (C-32) of the above 2-methylbutyryloxy group assigned its location at C-3. The relative and absolute configurations of
19 were determined to be the same as those of
18 by analysis of their NOE interactions and ECD spectra (
Figure 6a). Thus, the structure of
19—named xylomolin G
5—was assigned as 3-
O-(2-methyl)butyryl-3-deisobutyryloxy-xylomolin G
4.
Compound
20 was isolated as an amorphous white powder. Its molecular formula was determined to be C
33H
40O
13 by the positive HRESIMS ion peak at
m/
z 667.2359 (calcd. for [M + Na]
+, 667.2361). The NMR spectroscopic data of
20 (
Table 5 and
Table 6) resembled those of
15, except for the presence of an additional 8-OH group (δ
H 5.00 s) and the replacement of the Δ
8,14 double bond, 1-OH function, and 30-ethoxyl group in
15 by a Δ
14,15 double bond (δ
H 5.72 br s H-15; δ
C 160.0 qC C-14, 120.2 CH C-15), a 1-
O-isobutyryl moiety (δ
H 2.55 m H-35, 1.11 d (
J = 7.2 Hz H
3-36), 1.10 d (
J = 7.2 Hz H
3-37); δ
C 176.0 qC C-34, 35.1 CH C-35, 19.4 CH
3 C-36, 19.3 CH
3 C-37), and a 30-OH group (δ
H 4.87 s) in
20, respectively (
Table 5 and
Table 6, recorded in CDCl
3). HMBC correlations between H-15/C-14, H-15/C-16, H-15/C-13, H-15/C-8, 8-OH/C-8, and 30-OH/C-30 confirmed the presence of a Δ
14,15 double bond and the existence of two hydroxy groups at C-8 and C-30, respectively. The presence of the isobutyryloxy group was supported by
1H-
1H COSY cross-peaks between H-35/H
3-36 and H-35/H
3-37 and HMBC correlations between H-35/C-34, H
3-36/C-34, and H
3-37/C-34. Its location at C-1 was corroborated by the downshifted C-1 signal (δ
C 92.6 qC in
20, whereas δ
C 84.5 qC in
15). The relative configuration of
20 was established by NOE interactions, in which those between H-17/H-12β, H-12β/H-5, H-5/H
3-28 assigned the β-orientation for H-17, H-5, H
3-28, whereas those between H-11α/H
3-18, H
3-18/8-OH, 8-OH/H-9, H
pro-R-29/H-3, H-9/H
3-19, and H-3/30-OH concluded the α-orientation for H
3-18, 8-OH, H-9, H-3, H
3-19, and 30-OH. Thus, the structure of
20—named xylomolin H—was assigned as depicted.
Compound
21 afforded the molecular formula C
32H
38O
11 as deduced from the positive HRESIMS ion peak at
m/
z 621.2304 (calcd. for [M + Na]
+, 621.2306). The NMR spectroscopic data of
21 (
Table 5 and
Table 6) were similar to those of thaixylomolin H [
18], except for the presence of an additional 6-OH group (δ
H 3.19 s) and the replacement of the 2-acetoxy group by a 2-methylbutyryloxy moiety (δ
H 2.42 m 1H, 1.73 m 1H, 1.48 m 1H, 0.97 t (
J = 7.6 Hz 3H), 1.22 d (
J = 7.2 Hz 3H); δ
C 176.0 qC, 41.3 CH, 26.5 CH
2, 11.7 CH
3, 17.2 CH
3) in
21. The presence of the 6-OH group was supported by the downshifted C-6 signal (δ
C 71.0 CH in
21, whereas δ
C 31.6 CH
2 in thaixylomolin H), the
1H-
1H COSY cross-peak between H-5/H-6, and HMBC correlations between H-6/C-5 and H-6/C-7. The existence of the 2-methylbutyryloxy group was corroborated by
1H-
1H COSY cross-peaks between H-33/H
3-36, H-33/H-34, and H
2-34/H
3-35 and HMBC correlations between H-33/C-32, H-34/C-32, H
3-36/C-32, H-33/C-34, H
3-36/C-34, H
3-35/C-34, H-34/C-33, H
3-35/C-33, and H
3-36/C-33. The significant HMBC correlation from H-2 to the carbonyl carbon (C-32) of the 2-methylbutyryloxy group placed it at C-2. NOE interactions between H-17/H-12β, H-11α/H
3-19, H-11α/H
3-18, H-11β/H-5, H-5/H
3-28, H
3-19/1-OH, H
pro-R-29/H-2, and H-2/30-OH assigned the β-orientation for H-17, H
3-28, and H-5, and the α-orientation for H-2, H
3-18, H
3-19, 30-OH, and 1-OH. The ECD spectrum of
21 was identical to that of thaixylomolin H (
Figure 6b), concluding that
21 had the same 1
R,2
R,4
R,5
R,10
S,13
R,17
R,30
R-absolute configuration as that of thaixylomolin H. Thus, the structure of
21—named xylomolin I—was identified as 6-hydroxy-2-
O-(2-methyl)butyryl-2-deacetoxy-thaixylomolin H.
Compound
22 had the molecular formula C
29H
32O
10 as determined from the positive HRESIMS ion peak at
m/
z 541.2077 (calcd. for [M + H]
+, 541.2074). The similarities between the NMR spectroscopic data of
22 (
Table 7 and
Table 8) and those of trangmolin F [
16], containing a (
Z)-bicyclo[5.2.1]dec-3-en-8-one substructure, revealed their close structural resemblance. However, the 3-
O-isobutyryl function in trangmolin F was replaced by an acetoxy group (δ
H 2.17 s 3H; δ
C 170.4 qC, 20.6 CH
3) in
22, being unambiguously confirmed by HMBC cross-peaks between H-3/C-32 and H
3-33/C-32 (
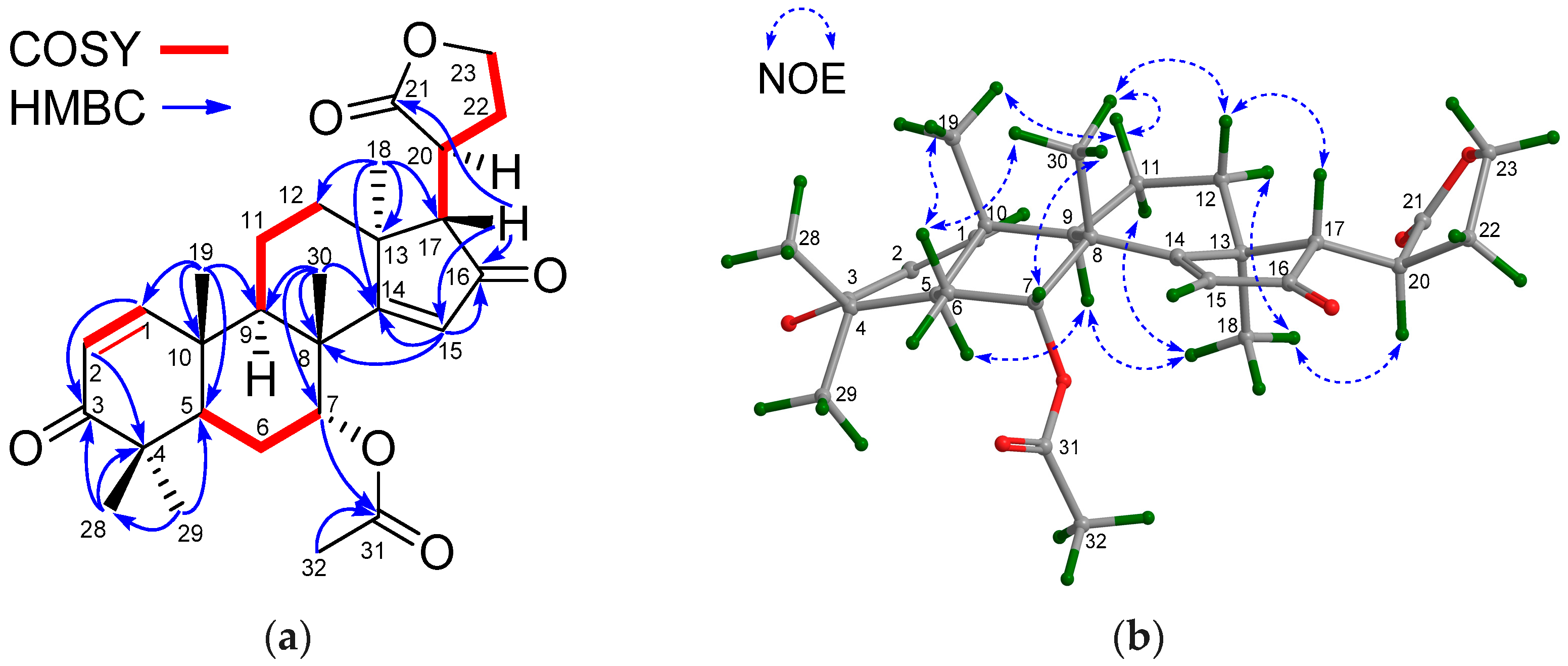
Figure 7a). The relative configuration of
22 was assigned by NOE interactions (
Figure 7b). Those between H-17/H-12β, H-12β/H
3-19, and H
3-19/H-5 revealed their cofacial relationship and were determined as β-oriented, whereas those between H-3/H-9, H
pro-R-29/H-3, and H-12α/H
3-18 indicated the α-orientation for H-3, H-9, and H
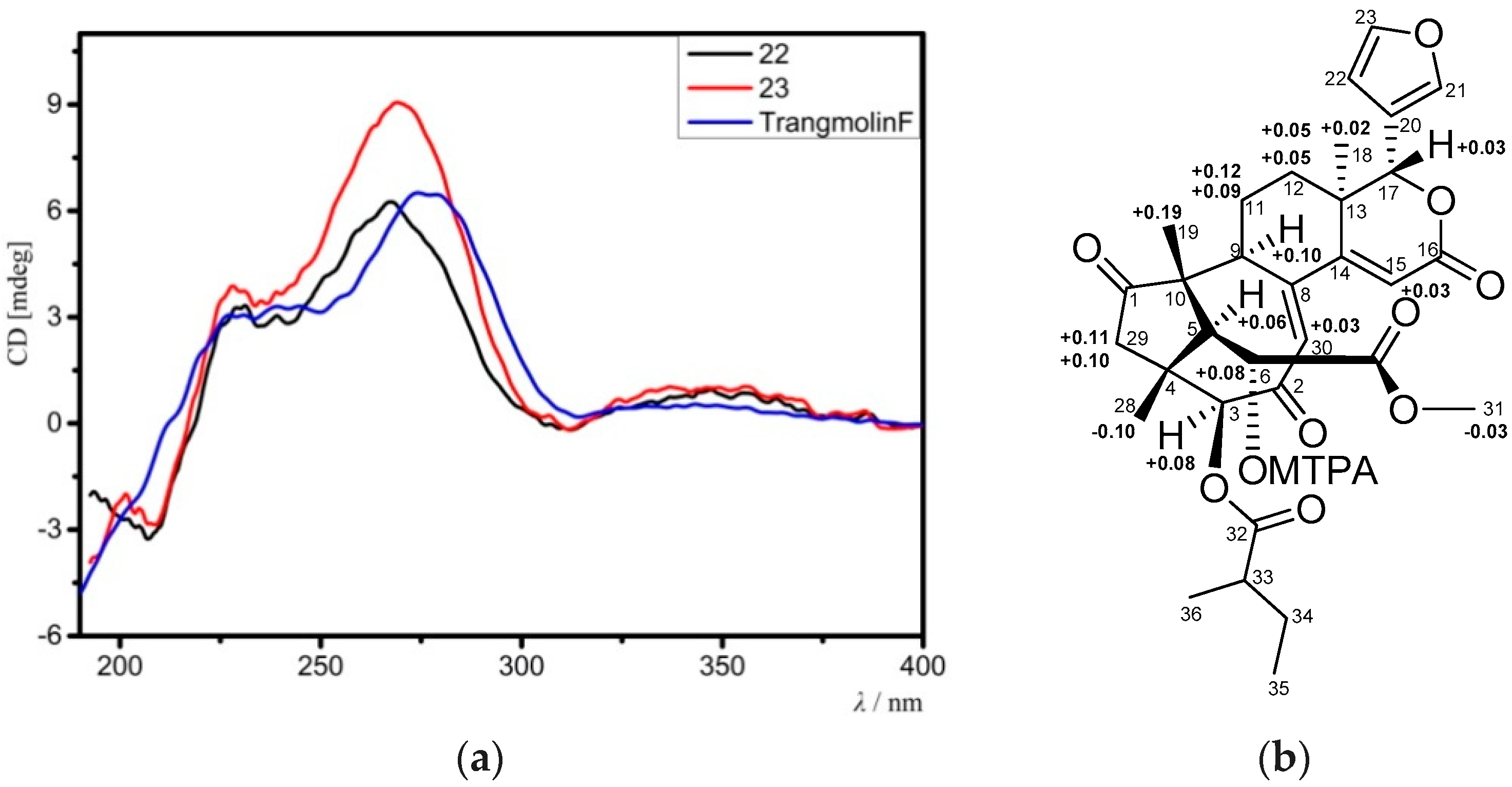
3-18, and the corresponding 3β-acetoxy function. The ECD spectrum of
22 was nicely matched with that of trangmolin F (
Figure 8a), concluding that the absolute configuration of
22 was the same as that of trangmolin F. Thus, the structure of
22—named xylomolin J
1—was assigned as 3-
O-acetyl-3-deisobutyryloxy-trangmolin F.
Compound
23 provided the molecular formula of C
32H
38O
10 as established by the positive HRESIMS ion peak at
m/
z 583.2548 (calcd. for [M + H]
+, 583.2543). The NMR spectroscopic data of
23 (
Table 7 and
Table 8) resembled those of
22, except for the replacement of the 3-acetoxy group in
22 by a 2-methylbutyryloxy moiety (δ
H 2.51 m 1H, 1.53 m 1H, 1.74 m 1H, 0.96 t (
J = 7.2 Hz, 3H), 1.18 d (
J = 7.2 Hz, 3H); δ
C 176.2 qC, 40.7 CH, 26.6 CH
2, 11.5 CH
3, 16.4 CH
3) in
23. The presence of the 2-methylbutyryloxy group was further evidenced by
1H-
1H COSY cross-peaks between H-33/H
3-36, H-33/H
2-34, and H
2-34/H
3-35 and HMBC correlations between H-33/C-32, H-34/C-32, and H
3-36/C-32. The HMBC correlation from H-3 to the carbonyl carbon (C-32) of the above 2-methylbutyryloxy group placed it at C-3. The relative configuration of
23 was confirmed to be the same as that of
22 by analysis of NOE interactions. Comparison of ECD spectra of compounds
23,
22, and trangmolin F (
Figure 8a) revealed that these compounds had the same absolute configuration. The absolute configuration of C-6 was further determined by the modified Mosher α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) ester method [
24]. The ∆δ values of H-5, H
3-19, and H
3-29 were positive, while that of H
3-31 was negative (
Figure 8b). This regular arrangement concluded the
R-absolute configuration for C-6. Finally, the absolute configuration of
23—named xylomolin J
2—was unequivocally established as 3
S,4
R,5
S,6
R,9
S,10
R,13
R,17
R.Compound
24 had the molecular formula C
32H
38O
11 as determined from the positive HRESIMS ion peak at
m/
z 621.2306 (calcd. for [M + Na]
+, 621.2306). The NMR spectroscopic data of
24 (
Table 7 and
Table 8) were similar to those of moluccensin I [
25], except for the presence of an additional 6-OH group (δ
H 3.12 br s) and the replacement of the 1-
O-isobutyryl group in moluccensin I by a 1-OH function (δ
H 2.81 s) in
24. The downshifted C-6 signal (δ
C 71.5 CH in
24, whereas δ
C 33.2 CH
2 in moluccensin I) and HMBC correlations from the active proton (δ
H 3.12 br s) to C-5, C-6, and C-7 revealed the presence of the 6-OH group (
Figure 9a). The existence of the 1-OH function was confirmed by the upshifted C-1 signal (δ
C 85.8 qC in
24, whereas δ
C 90.8 qC in moluccensin I) and strong HMBC cross-peaks from the active proton (δ
H 2.81 s) to C-1, C-2, and C-10 (
Figure 9a). The relative configuration of
24 was identified as the same as that of moluccensin I based on NOE interactions between H-17/H-12β, H-5/H-11β, H-5/H-28, H
3-18/H-11α, H
3-19/H
pro-S-29, H-3/H
pro-R-29, and 2-OH/H
pro-R-29 (
Figure 9b). Therefore, the structure of
24—named xylomolin K
1—was assigned as 6-hydroxy-1-
O-deisobutyryl-moluccensin I.
Compound
25 provided the molecular formula C
31H
36O
11 as determined from the positive HRESIMS ion peak at
m/
z 607.2164 (calcd. for [M + Na]
+, 607.2150). The NMR spectroscopic data of
25 (
Table 7 and
Table 8) were similar to those of
24, except for the replacement of 3-
O-(2-methyl)butyryl in
24 by an isobutyryloxy group (δ
H 2.45 m 1H, 1.11 d (
J = 6.8 Hz, 3H), 1.11 d (
J = 6.8 Hz, 3H); δ
C 174.8 qC, 34.2 CH, 18.8 CH
3, 19.0 CH
3) in
25. The HMBC correlation from H-3 to the carbonyl carbon (C-32) of the isobutyryloxy group placed it at C-3. The relative configuration of
25 was determined to be the same as that of
24 based on NOE interactions between H-17/H-12β, H-11β/H-5, H-5/H
3-28, H-12α/H
3-18, H
3-18/H-11α, H
pro-R-29/H-3, and H
pro-S-29/H
3-19. Thus, the structure of
25—named xylomolin K
2—was identified as 3-
O-isobutyryl-3-de(2-methyl)butyryloxy-xylomolin K
1.
Compound
26 was obtained as an amorphous white powder. Its molecular formula was determined to be C
33H
38O
14 by the positive HRESIMS ion peak at
m/
z 681.2149 (calcd. for [M + Na]
+, 681.2154). The NMR spectroscopic data of
26 (
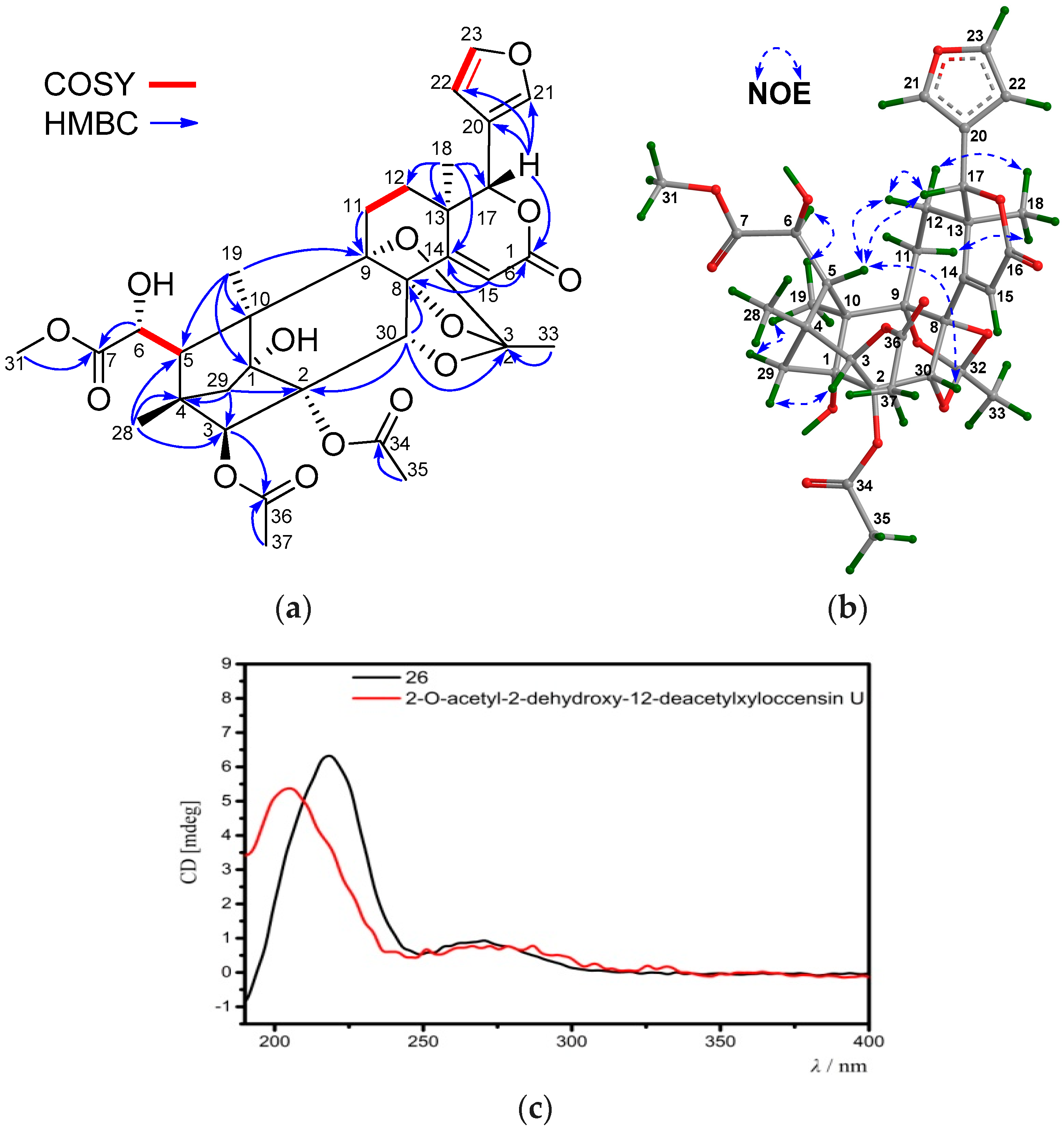
Table 7 and
Table 8) resembled those of 2-
O-acetyl-2-dehydroxy-12-deacetylxyloccensin U [
18], being a phragmalin 8,9,30-
ortho ester isolated from
X. moluccensis, except for the presence of an additional 6-OH group and the absence of the 12-OH group in
26. The presence of the 6-OH group was supported by the downshifted C-6 signal (δ
C 71.5 CH in
26, whereas δ
C 33.7 CH
2 in 2-
O-acetyl-2-dehydroxy-12-deacetylxyloccensin U), the
1H-
1H COSY cross-peak between H-5/H-6 and HMBC correlations between H-6/C-5 and H-6/C-7. The upshifted C-12 signal (δ
C 29.5, CH
2 in
26, whereas δ
C 66.6 CH in 2-
O-acetyl-2-dehydroxy-12-deacetylxyloccensin U),
1H-
1H COSY cross-peaks between H
2-11/H
2-12, and HMBC correlations between H
3-18/C-12 (
Figure 10a) confirmed the absence of the 12-OH group in
26. NOE correlations between H-17/H-12β, H-12β/H-5, H-17/H-5, H-5/H-30, H
3-18/H-12α, H
3-18/H-11α, H
3-19/H
pro-S-29, and H-3/H
pro-R-29 revealed the same relative configuration of
26 as that of 2-
O-acetyl-2-dehydroxy-12-deacetylxyloccensin U (
Figure 10b). The ECD spectrum of
26 was nicely matched with that of 2-
O-acetyl-2-dehydroxy-12-deacetylxyloccensin U (
Figure 10c), revealing the same absolute configuration for the two compounds. Thus, the structure of
26—named xylomolin L
1—was assigned as 6-hydroxy-12-dehydroxy-2-
O-acetyl-2-dehydroxy-12-deacetylxyloccensin U.
The molecular formula of
27 was determined to be C
34H
40O
13 by the positive HRESIMS ion peak at
m/
z 679.2374 (calcd. for [M + Na]
+, 679.2361). The NMR spectroscopic data of
27 (
Table 7 and
Table 8) closely resembled those of swietephragmin G [
26], being a phragmalin 8,9,30-
ortho ester, except for the presence of an additional 12-OH group, which was supported by the downshifted C-12 signal (δ
C 66.5 CH in
27, whereas δ
C 29.2 CH
2 in swietephragmin G),
1H-
1H COSY cross-peaks between H
2-11/H-12, and HMBC correlations between H
3-18/C-12. The strong NOE interaction between H-17/H-12 assigned the β-oriented H-12 and the corresponding α-orientation for the 12-OH group. The NOE interaction between H
3-37/H
3-38 assigned the
E configuration for the double bond of 3-tigloyloxy group. Therefore, the structure of
27—named xylomolin L
2—was identified as 12α-hydroxy-swietephragmin G.
Compound
28 had the molecular formula C
28H
37O
6 as determined from the positive HRESIMS ion peak at
m/
z 469.2603 (calcd. for [M + H]
+, 469.2585). The NMR spectroscopic data of
28 (
Table 7 and
Table 8) were closely related to those of andirolide Q [
27], except for the different positions of the ester carbonyl carbon of the C-17 attached five-membered γ-lactone ring,
viz. C-21 in
28 instead of C-23 in andirolide Q. Significant
1H-
1H COSY correlations between H-17/H-20, H-20/H
2-22, H
2-22/H
2-23 and the HMBC correlation between H-17/C-21 confirmed the above deduction (
Figure 11a). NOE interactions between H-17/H-12β, H-12β/H
3-30, H
3-30/H-7, H
3-30/H-11β, H-11β/H
3-19, and H
3-18/H-11α, H
3-18/H-12α, H
3-18/H-20, H
3-18/H-9, and H-9/H-5 indicated the β-orientation for H-7, H-17, H
3-19, and H
3-30, and the α-orientation for H-5, H-9, H
3-18, and H-20 (
Figure 11b). Hence, the relative configuration of
28—named xylomolin M—was assigned as depicted.
Compound
29 afforded the molecular formula C
27H
34O
8 as deduced from the positive HRESIMS ion peak at
m/
z 509.2147 (calcd. for [M + Na]
+, 509.2146). The NMR spectroscopic data of
29 (
Table 7 and
Table 8) closely resembled those of moluccensin O [
25], except for the absence of the 21-OH group, which was corroborated by the upshifted C-21 signal (δc 72.4 CH
2 in
29, δc 98.1 CH in moluccensin O) and HMBC correlations from H
2-21 (δ
H 4.85 br d (
J = 18.3 Hz), 5.03 dd (
J = 18.3, 2.0 Hz)) to C-20 and C-22. The relative configuration of
29 was assigned as the same as that of moluccensin O based on NOE correlations. Thus, the structure of compound
29—named xylomolin N—was assigned as 21-dehydroxy-moluccensin O.
The antitumor activities of
1,
3,
8,
10,
11,
14–
16,
20,
23,
25, and
27 were tested by the MTT cytotoxity assay against five human tumor cell lines, including human colorectal HCT-8 and HCT-8/T, human ovarian A2780 and A2780/T, and human breast MD-MBA-231 (
Table S1) [
28]. Cisplatin was used as the positive control. Compounds
11 and
23 showed weak activities against the tested cancer cell lines, whereas the other ten compounds were inactive at 100 μM. Compound
23 exhibited selective antitumor activity against human breast MD-MBA-231 cancer cells with an IC
50 value of 37.7 μM.
Anti-HIV activities of
1,
3,
8,
10,
11,
14,
20,
23–
25, and
27 were tested in vitro with the HIV-1 virus transfected 293 T cells [
29]. At the concentration of 20 μM,
1,
11,
23, and
24 showed inhibitory rates of 17.49 ± 6.93%, 24.47 ± 5.04%, 14.77 ± 5.91%, and 14.34 ± 3.92%, respectively (
Table S2). Efavirenz was used as the positive control with an inhibitory rate of 88.54 ± 0.45% at the same concentration.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}