Antimicrobial and Antibiofilm Activity of a Recombinant Fragment of β-Thymosin of Sea Urchin Paracentrotus lividus

 ,
, 
 and
and
Abstract
:1. Introduction
2. Results
2.1. Antibacterial Activity of RP1
2.2. Interference with Biofilm Formation
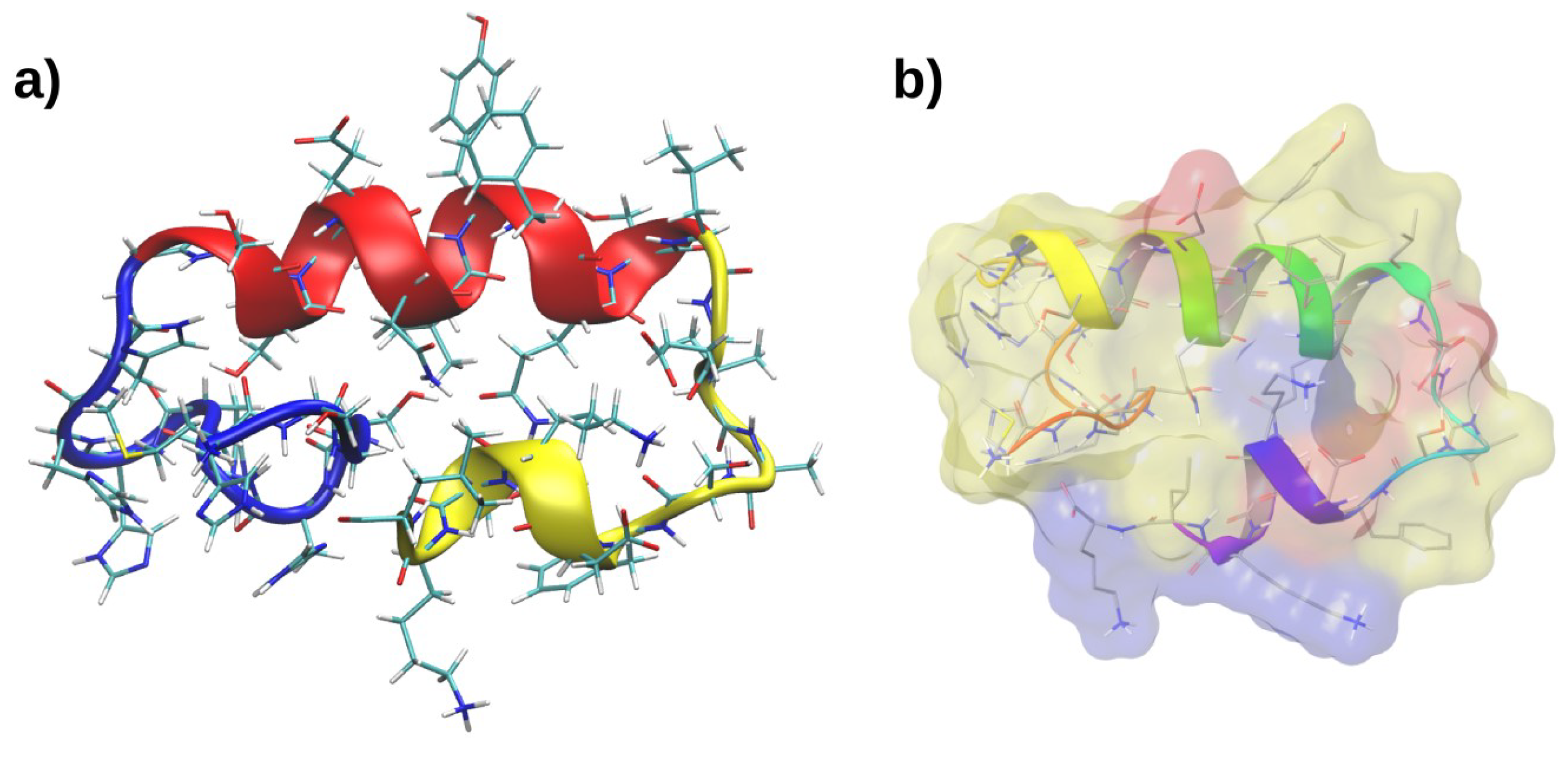
2.3. Molecular Dynamics of RP1
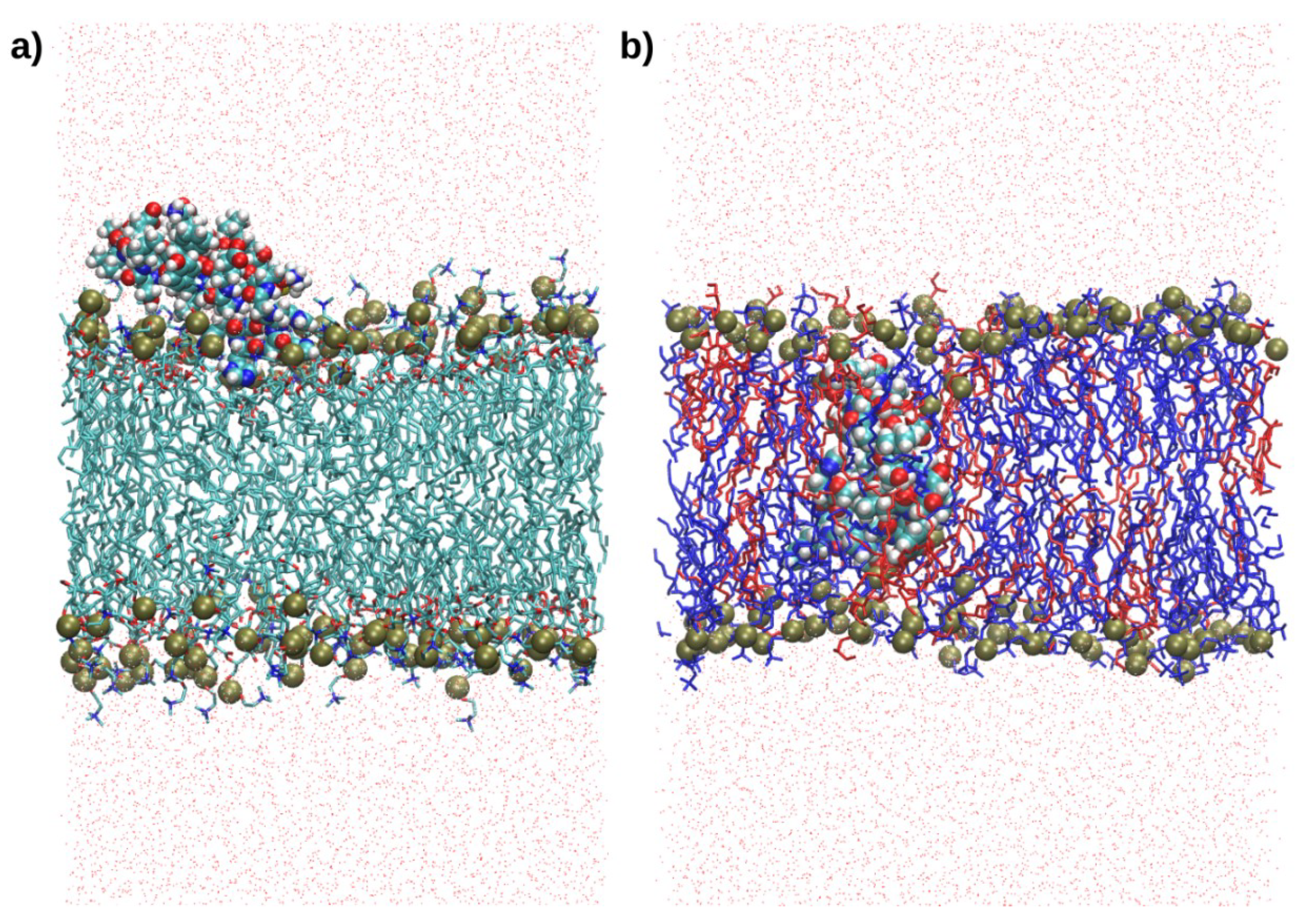
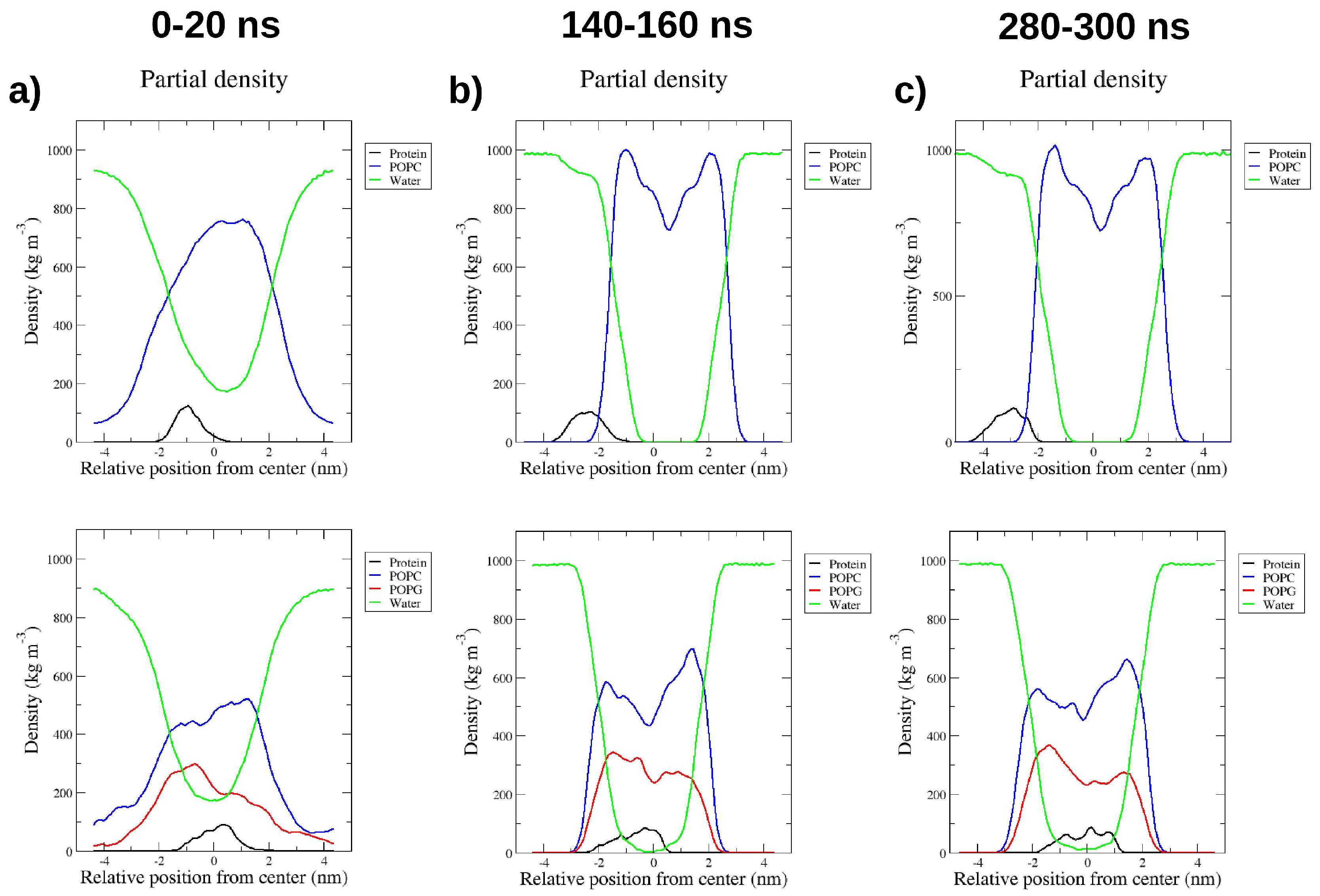
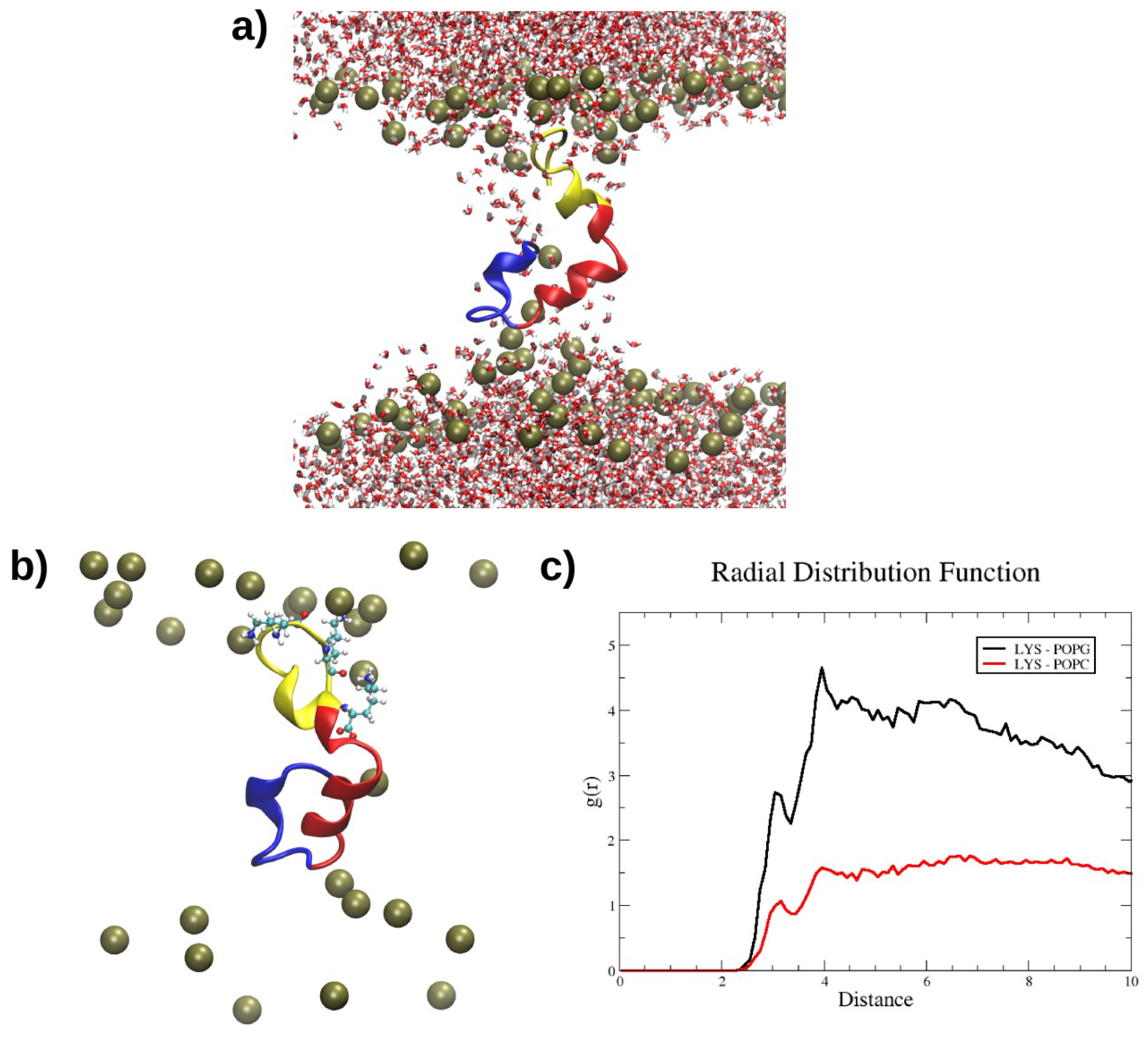
2.4. Interactions with Membrane Models In Silico
3. Discussion
4. Materials and Methods
4.1. Paracentrin 1 Gene Cloning
4.2. Peptide Expression
4.3. Microbial Strains
4.4. Minimum Inhibitory Concentrations (MICs)
4.5. Evaluation of Biofilm Formation and Biofilm Prevention Assay
4.6. Molecular Dynamics Simulations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fair, R.J.; Tor, Y. Antibiotics and bacterial resistance in the 21st century. Perspect. Med. Chem. 2014, 6, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.G.; Bowler, P.G. Clinician perceptions of wound biofilm. Int. Wound J. 2016, 13, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, P.; Allison, D.; McBain, A. Biofilms in vitro and in vivo: Do singular mechanisms imply cross-resistance? J. Appl. Microbiol. 2002, 92, 98–110. [Google Scholar] [CrossRef]
- Cascioferro, S.; Cusimano, M.G.; Schillaci, D. Antiadhesion agents against Gram-positive pathogens. Future Microbiol. 2014, 9, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Projan, S.J.; Youngman, P.J. Antimicrobials: New solutions badly needed. Curr. Opin. Microbiol. 2002, 5, 463–465. [Google Scholar] [CrossRef]
- Tincu, J.A.; Taylor, S.W. Antimicrobial peptides from marine invertebrates. Antimicrob. Agents Chemother. 2004, 48, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Blencke, H.M.; Haug, T.; Stensvåg, K. Antimicrobial peptide in Echinoderm host defense. Dev. Comp. Immunol. 2015, 49, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, D.; Cusimano, M.G.; Cunsolo, V.; Saletti, R.; Russo, D.; Vazzana, M.; Vitale, M.; Arizza, V. Immune mediators of sea-cucumber Holothuria tubulosa (Echinodermata) as source of novel antimicrobial and anti-staphylococcal biofilm agents. AMB Express 2013, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schillaci, D.; Arizza, V.; Parrinello, N.; Di Stefano, V.; Fanara, S.; Muccilli, V.; Cunsolo, V.; Haagensen, J.; Molin, S. Antimicrobial and antistaphylococcal biofilm activity from the sea urchin Paracentrotus lividus. J. Appl. Microbiol. 2010, 108, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schillaci, D.; Cusimano, M.G.; Spinello, A.; Barone, G.; Russo, D.; Vitale, M.; Parrinello, D.; Arizza, V. Paracentrin 1, a synthetic antimicrobial peptide from the sea-urchin Paracentrotus lividus, interferes with staphylococcal and Pseudomonas aeruginosa biofilm formation. AMB Express 2014, 4, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schillaci, D.; Vitale, M.; Cusimano, M.G.; Arizza, V. Fragments of beta-thymosin from the sea urchin Paracentrotus lividus as potential antimicrobial peptides against staphylococcal biofilms. Ann. N. Y. Acad. Sci. 2012, 1270, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maupetit, J.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009, 37, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Martín, S.; Salgado, J. Self-assembling of peptide/membrane complexes by atomistic molecular dynamics simulations. Biophys. J. 2007, 92, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.; Faye, I.; Hofsten, P.V.; Kockum, K.; Lee, J.Y.; Xanthopoulos, K.; Bennich, H.; Engström, Å.; Merrifield, R.; Andreu, D. On the primary structures of lysozyme, cecropins and attacins from Hyalophora cecropia. Dev. Comp. Immunol. 1985, 9, 551–558. [Google Scholar] [CrossRef]
- Levashina, E.A.; Ohresser, S.; Bulet, P.; Reichhart, J.M.; Hetru, C.; Hoffmann, J.A. Metchnikowin, a novel immune-inducible proline-rich peptide from Drosophila with antibacterial and antifungal properties. Eur. J. Biochem. 1995, 233, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Segovia, L.J.T.; Ramírez, G.A.T.; Arias, D.C.H.; Duran, J.D.R.; Bedoya, J.P.; Osorio, J.C.C. Identification and characterization of novel cecropins from the Oxysternon conspicillatum neotropic dung beetle. PLoS ONE 2017, 12, 0187914. [Google Scholar] [CrossRef]
- Li, C.; Haug, T.; Moe, M.K.; Styrvold, O.B.; Stensvåg, K. Centrocins: Isolation and characterization of novel dimeric antimicrobial peptides from the green sea urchin, Strongylocentrotus droebachiensis. Dev. Comp. Immunol. 2010, 34, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Andrew Tincu, J.; Taylor, S.W.; Menzel, L.P.; Waring, A.J. Natural peptide antibiotics from tunicates: Structures, functions and potential uses. Integr. Comp. Biol. 2003, 43, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Yi, K.S.; Matsuzaki, K.; Kim, M.S.; Kim, S.C. Structure–activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: The proline hinge is responsible for the cell-penetrating ability of buforin II. Proc. Natl. Acad. Sci. 2000, 97, 8245–8250. [Google Scholar] [CrossRef] [PubMed]
- Sawai, M.V.; Jia, H.P.; Liu, L.; Aseyev, V.; Wiencek, J.M.; McCray, P.B.; Ganz, T.; Kearney, W.R.; Tack, B.F. The NMR structure of human β-defensin-2 reveals a novel α-helical segment. Biochemistry 2001, 40, 3810–3816. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [PubMed]
- Meister, M.; Lemaitre, B.; Hoffmann, J.A. Antimicrobial peptide defense in Drosophila. Bioessays 1997, 19, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Barra, D.; Simmaco, M. Amphibian skin: A promising resource for antimicrobial peptides. Trends Biotechnol. 1995, 13, 205–209. [Google Scholar] [CrossRef]
- Barra, D.; Simmaco, M.; Boman, H.G. Gene-encoded peptide antibiotics and innate immunity. FEBS lett. 1998, 430, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Simmaco, M.; Mignogna, G.; Barra, D. Antimicrobial peptides from amphibian skin: What do they tell us? Biopolymers 1998, 47, 435–450. [Google Scholar] [CrossRef]
- Canny, G.; Levy, O.; Furuta, G.T.; Narravula-Alipati, S.; Sisson, R.B.; Serhan, C.N.; Colgan, S.P. Lipid mediator-induced expression of bactericidal/permeability-increasing protein (BPI) in human mucosal epithelia. Proc. Natl. Acad. Sci. USA 2002, 99, 3902–3907. [Google Scholar] [CrossRef] [PubMed]
- Levy, O. Antibiotic proteins of polymorphonuclear leukocytes. Eur. J. Haematol. 1996, 56, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Nissen-Meyer, J.; Nes, I.F. Ribosomally synthesized antimicrobial peptides: Their function, structure, biogenesis, and mechanism of action. Arch. Microbiol. 1997, 167, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, D.; Petruso, S.; Raimondi, M.V.; Cusimano, M.G.; Cascioferro, S.; Scalisi, M.; La Giglia, M.A.; Vitale, M. Pyrrolomycins as potential anti-staphylococcal biofilms agents. Biofouling 2010, 26, 433–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilmann, C.; Gerke, C.; Perdreau-Remington, F.; Götz, F. Characterization of Tn917 insertion mutants of Staphylococcus epidermidis affected in biofilm formation. Infect. Immun. 1996, 64, 277–282. [Google Scholar] [PubMed]
- Spinello, A.; de Almeida, A.; Casini, A.; Barone, G. The inhibition of glycerol permeation through aquaglyceroporin-3 induced by mercury (II): A molecular dynamics study. J. Inorg. Biochem. 2016, 160, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Jambeck, J.P.; Lyubartsev, A.P. Another piece of the membrane puzzle: Extending slipids further. J. Chem. Theory Comput. 2012, 9, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Jämbeck, J.P.; Lyubartsev, A.P. An extension and further validation of an all-atomistic force field for biological membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Farrotti, A.; Bocchinfuso, G.; Palleschi, A.; Rosato, N.; Salnikov, E.; Voievoda, N.; Bechinger, B.; Stella, L. Molecular dynamics methods to predict peptide locations in membranes: LAH4 as a stringent test case. BBA-Biomembr. 2015, 1848, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Strains | MIC (µg/mL) | |
|---|---|---|
| RP1 | LL-37 | |
| S. aureus ATCC 25923 | 50 | 50 |
| P. aeruginosa ATCC 15442 | 50 | 50 |
| Reference Strains | BIC (µg/mL) | |
|---|---|---|
| RP1 | LL-37 | |
| S. aureus ATCC 25923 | 5.0 ± 0.3 | 1.6 ± 0.04 |
| P. aeruginosa ATCC 15442 | 10.7 ± 0.7 | 11.9 ± 0.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinello, A.; Cusimano, M.G.; Schillaci, D.; Inguglia, L.; Barone, G.; Arizza, V. Antimicrobial and Antibiofilm Activity of a Recombinant Fragment of β-Thymosin of Sea Urchin Paracentrotus lividus. Mar. Drugs 2018, 16, 366. https://doi.org/10.3390/md16100366
Spinello A, Cusimano MG, Schillaci D, Inguglia L, Barone G, Arizza V. Antimicrobial and Antibiofilm Activity of a Recombinant Fragment of β-Thymosin of Sea Urchin Paracentrotus lividus. Marine Drugs. 2018; 16(10):366. https://doi.org/10.3390/md16100366
Chicago/Turabian StyleSpinello, Angelo, Maria Grazia Cusimano, Domenico Schillaci, Luigi Inguglia, Giampaolo Barone, and Vincenzo Arizza. 2018. "Antimicrobial and Antibiofilm Activity of a Recombinant Fragment of β-Thymosin of Sea Urchin Paracentrotus lividus" Marine Drugs 16, no. 10: 366. https://doi.org/10.3390/md16100366
APA StyleSpinello, A., Cusimano, M. G., Schillaci, D., Inguglia, L., Barone, G., & Arizza, V. (2018). Antimicrobial and Antibiofilm Activity of a Recombinant Fragment of β-Thymosin of Sea Urchin Paracentrotus lividus. Marine Drugs, 16(10), 366. https://doi.org/10.3390/md16100366