New 3-Hydroxyquinaldic Acid Derivatives from Cultures of the Marine Derived Actinomycete Streptomyces cyaneofuscatus M-157

 , ,
, , 
Abstract
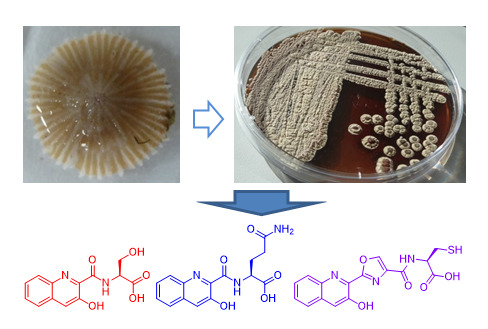
:
1. Introduction
2. Results
2.1. Taxonomy and Phylogenetic Analysis of the Strain M-157
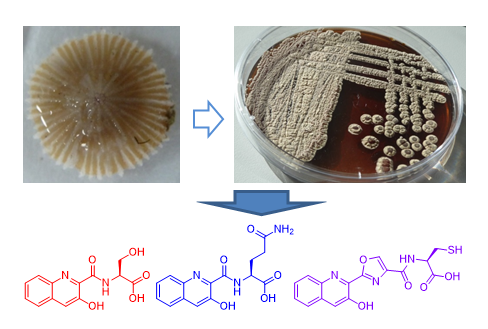
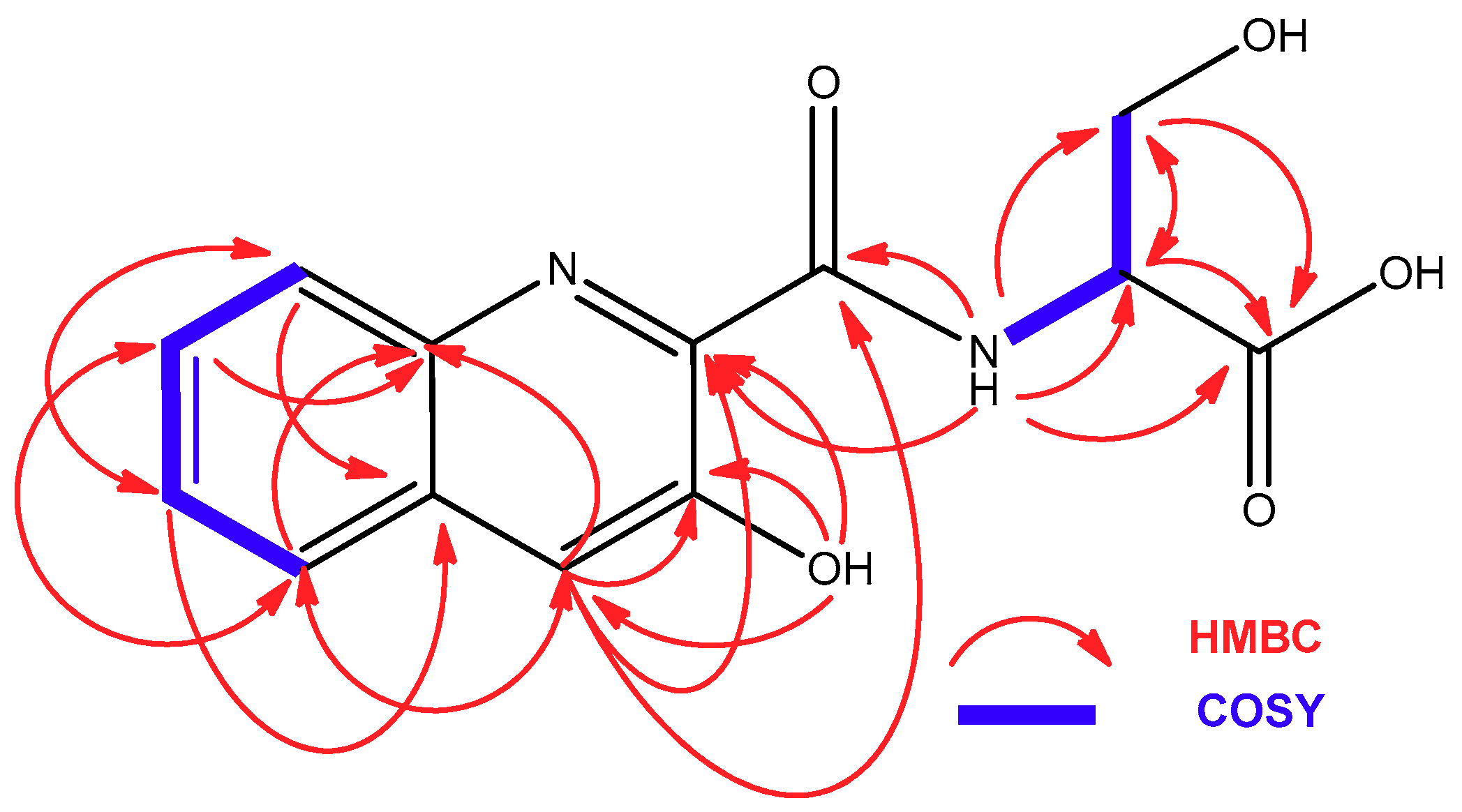
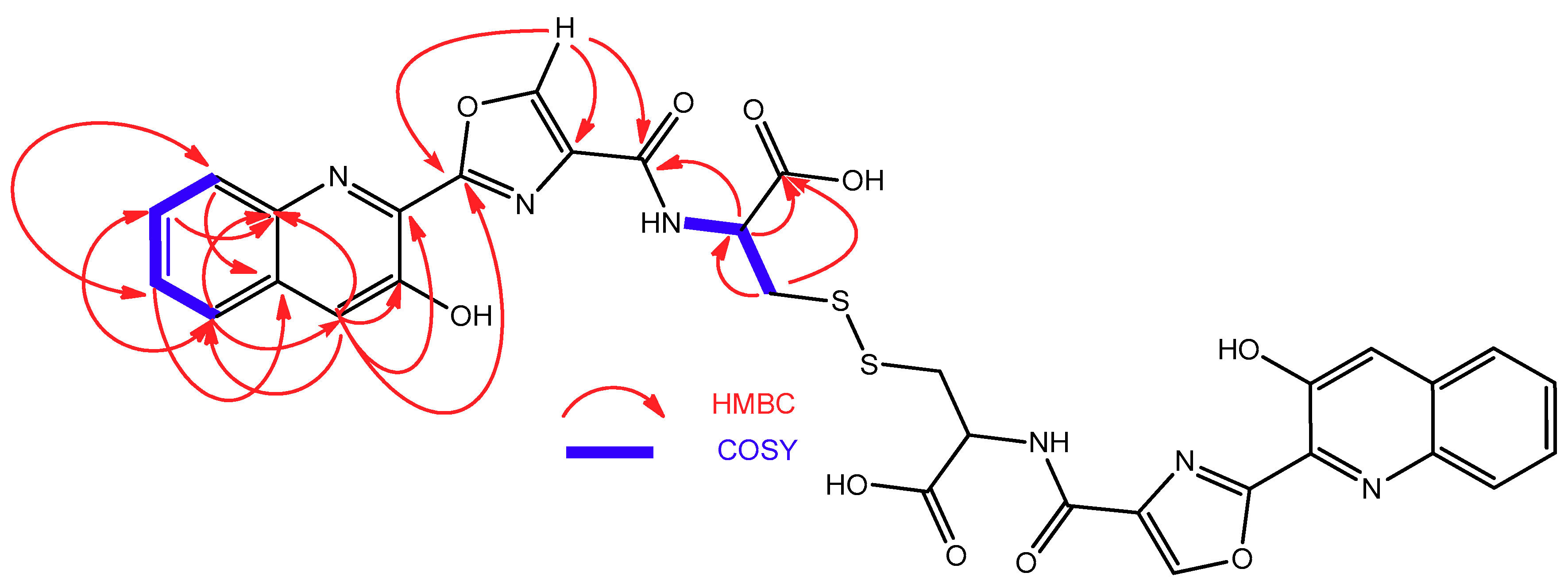
2.2. Isolation and Structural Elucidation of Compounds 1–3
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Taxonomic Identification of the Producing Microorganism
4.3. Fermentation of the Producing Microorganism
4.4. Extraction and Bioassay-Guided Isolation
4.5. Hydrolysis and Marfey’s Analysis of Compounds 1 and 2
4.6. Oxidation of Compound 3 with Performic Acid and Marfey’s Analysis
4.7. Antibacterial Activity Assays
4.8. Cytotoxicity Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Range, M.J.; Ruddock, J.C.; Pacey, M.S.; Cullen, W.P.; Huang, L.H.; Jefferson, M.T.; Whipple, E.B.; Maeda, H.; Tone, J. UK-63,052 complex, new quinomycin antibiotics from Streptomyces braegensis subsp. japonicus; Taxonomy, fermentation, isolation, characterisation and antimicrobial activity. J. Antibiot. 1989, 42, 206–217. [Google Scholar] [CrossRef]
- Lim, C.L.; Nogawa, T.; Uramoto, M.; Okano, A.; Hongo, Y.; Nakamura, T.; Koshino, H.; Takahashi, S.; Ibrahim, D.; Osada, H. RK-1355A and B, novel quinomycin derivatives isolated from a microbial metabolites fraction library based on NPPlot screening. J. Antibiot. 2014, 67, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, H.; Sakai, F.; Nishiyama, Y.; Ohbayashi, M.; Imanishi, H.; Konishi, M.; Miyaki, T.; Koshiyama, H.; Kawaguchi, H. BBM-928, a new antitumor antibiotic complex I. Production, isolation, characterization and antitumor activity. J. Antibiot. 1980, 33, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Hoshino, Y.; Sasahira, T.; Kawaguchi, H. BBM-928, a new antitumor antibiotic complex II. Taxonomic studies on the producing organism. J. Antibiot. 1980, 33, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M.; Ohkuma, H.; Sakai, F.; Tsuno, T.; Koshiyama, H.; Naito, T.; Kawaguchi, H. BBM-928, a new antitumor antibiotic complex. III. Structure determination of BBM-928 A, B and C. J. Antibiot. 1981, 34, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Toda, S.; Sugawara, K.; Nlshiyama, Y.; Ohbayashi, M.; Ohkusa, N.; Yamamoto, H.; Konishi, M.; Oki, T. Quinaldopeptin, a novel antibiotic of the quinomycin family. J. Antibiot. 1990, 43, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.A.; Bush, J.A. Sandramycin, a novel antitumor antibiotic produced by a Nocardioides sp. Production, isolation, characterization and biological properties. J. Antibiot. 1989, 42, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.A.; Colson, K.L.; Belofsky, G.N.; Bleiberg, B.B. Sandramycin, a novel antitumor antibiotic produced by a Nocardioides sp. II. Structure determination. J. Antibiot. 1993, 46, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Romero, F.; Espliego, F.; Pérez Baz, J.; García de Quesada, T.; Grávalos, D.; de la Calle, F.; Fernández-Puentes, J.L. Thiocoraline, a New Depsipeptide with Antitumor Activity Produced by a Marine Micromonospora. I. Taxonomy, Fermentation, Isolation, and Biological Activities. J. Antibiot. 1997, 50, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Pérez Baz, J.; Cañedo, L.M.; Fernández Puentes, J.L.; Silva Elipe, M.V. Thiocoraline, a Novel Depsipeptide with Antitumor Activity Produced by a Marine Micromonospora. II. Physico-chemical Properties and Structure Determination. J. Antibiot. 1997, 50, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Wyche, T.P.; Hou, Y.; Braun, D.; Cohen, H.C.; Xiong, M.P.; Bugni, T.S. First Natural Analogs of the Cytotoxic Thiodepsipeptide Thiocoraline A from a Marine Verrucosispora sp. J. Org. Chem. 2011, 76, 6542–6547. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.; Marco, E.; García-Hernández, V.; Domingo, A.; Llamas-Saiz, A.L.; Porto-Sandá, S.; Riguera, R.; Laine, W.; David-Cordonnier, M.-H.; Bailly, C.; et al. Antitumor Activity, X-ray Crystal Structure, and DNA Binding Properties of Thiocoraline A, a Natural Bisintercalating Thiodepsipeptide. J. Med. Chem. 2007, 50, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Vizcaíno, A.; González, V.; Braña, A.F.; Palacios, J.J.; Otero, L.; Fernández, J.; Molina, A.; Kulik, A.; Vázquez, F.; Acuña, J.L.; et al. Pharmacological Potential of Phylogenetically Diverse Actinobacteria Isolated from Deep-Sea Coral Ecosystems of the Submarine Avilés Canyon in the Cantabrian Sea. Microb. Ecol. 2017, 73, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Braña, A.; Sarmiento-Vizcaíno, A.; Osset, M.; Pérez-Victoria, I.; Martín, J.; de Pedro, N.; de la Cruz, M.; Díaz, C.; Vicente, F.; Reyes, F.; et al. Lobophorin K, a New Natural Product with Cytotoxic Activity Produced by Streptomyces sp. M-207 Associated with the Deep-Sea Coral Lophelia pertusa. Mar. Drugs 2017, 15, 144. [Google Scholar] [CrossRef] [PubMed]
- Braña, A.F.; Sarmiento-Vizcaíno, A.; Pérez-Victoria, I.; Otero, L.; Fernández, J.; Palacios, J.J.; Martín, J.; de la Cruz, M.; Díaz, C.; Vicente, F.; et al. Branimycins B and C, Antibiotics Produced by the Abyssal Actinobacterium Pseudonocardia carboxydivorans M-227. J. Nat. Prod. 2017, 80, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Vizcaíno, A.; Braña, A.; Pérez-Victoria, I.; Martín, J.; de Pedro, N.; Cruz, M.; Díaz, C.; Vicente, F.; Acuña, J.; Reyes, F.; et al. Paulomycin G, a New Natural Product with Cytotoxic Activity against Tumor Cell Lines Produced by Deep-Sea Sediment Derived Micromonospora matsumotoense M-412 from the Avilés Canyon in the Cantabrian Sea. Mar. Drugs 2017, 15, 271. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Victoria, I.; Martín, J.; Reyes, F. Combined LC/UV/MS and NMR Strategies for the Dereplication of Marine Natural Products. Planta Med 2016, 82, 857–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riego, E.; Bayó, N.; Cuevas, C.; Albericio, F.; Álvarez, M. A new approach to 3-hydroxyquinoline-2-carboxylic acid. Tetrahedron 2005, 61, 1407–1411. [Google Scholar] [CrossRef]
- Shaaban, K.A.; Shepherd, M.D.; Ahmed, T.A.; Nybo, S.E.; Leggas, M.; Rohr, J. Pyramidamycins A–D and 3-hydroxyquinoline-2-carboxamide; cytotoxic benzamides from Streptomyces sp. DGC1. J. Antibiot. 2012, 65, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ikai, Y.; Oka, H.; Suzuki, M.; Harada, K.-I. A Nonempirical Method Using LC/MS for Determination of the Absolute Configuration of Constituent Amino Acids in a Peptide: Combination of Marfey’s Method with Mass Spectrometry and Its Practical Application. Anal. Chem. 1997, 69, 5146–5151. [Google Scholar] [CrossRef]
- Varga-Visi, E.; Terlaky-Balla, E.; Pohn, G.; Kamether, L.; Csapó, J. RPHPLC determination of l- and d-cystine and cysteine as cysteic acid. Chromatographia 2000, 51, S325–S327. [Google Scholar] [CrossRef]
- Walsh, C.T.; Malcolmson, S.J.; Young, T.S. Three Ring Posttranslational Circuses: Insertion of Oxazoles, Thiazoles, and Pyridines into Protein-Derived Frameworks. ACS Chem. Biol. 2012, 7, 429–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D.W. Extending the “One Strain Many Compounds” (OSMAC) Principle to Marine Microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.; Crespo, G.; González-Menéndez, V.; Pérez-Moreno, G.; Sánchez-Carrasco, P.; Pérez-Victoria, I.; Ruiz-Pérez, L.M.; González-Pacanowska, D.; Vicente, F.; Genilloud, O.; et al. MDN-0104, an Antiplasmodial Betaine Lipid from Heterospora chenopodii. J. Nat. Prod. 2014, 77, 2118–2123. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.; Weissbach, U.; Reillo, C.S.; Braña, A.F.; Méndez, C.; Rohr, J.; Salas, J.A. Identification of two genes from Streptomyces argillaceus encoding two glycosyltransferases involved in the transfer of a disaccharide during the biosynthesis of the antitrumor drug mithramycin. J. Bacteriol. 1998, 180, 4929–4937. [Google Scholar] [PubMed]
- Audoin, C.; Bonhomme, D.; Ivanisevic, J.; Cruz, M.; Cautain, B.; Monteiro, M.; Reyes, F.; Rios, L.; Perez, T.; Thomas, O. Balibalosides, an Original Family of Glucosylated Sesterterpenes Produced by the Mediterranean Sponge Oscarella balibaloi. Mar. Drugs 2013, 11, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Cautain, B.; de Pedro, N.; Schulz, C.; Pascual, J.; da S Sousa, T.; Martin, J.; Pérez-Victoria, I.; Asensio, F.; González, I.; Bills, G.F.; et al. Identification of the Lipodepsipeptide MDN-0066, a Novel Inhibitor of VHL/HIF Pathway Produced by a New Pseudomonas Species. PLoS ONE 2015, 10, e0125221. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type * | δH (J in Hz) | |
| 1 | 167.8, C | 168.7, C | ||
| 2 | 134.6, C | 135.4, C | ||
| 3 | 153.1, C | 153.4, C | ||
| 4 | 120.3, CH | 7.89, s | 120.4, CH | 7.87, s |
| 5 | 131.6, CH | 131.7, CH | ||
| 6 | 126.7, CH | 7.91, dd (7.6, 1.8) | 127.0, CH | 7.91, d (7.6, 1.7) |
| 7 | 128.9, CH 127.8, CH | 7.63, m | 129.2, CH 128.0, CH | 7.65, m |
| 8 | 7.66, m | 7.67, m | ||
| 9 | 129.0, CH | 8.05, d (8.3) | 129.4, CH | 8.05, d (8.0) |
| 10 | 140.8, C | 141.2, C | ||
| 3-OH | 12.01, s | 12.05, s | ||
| 1′ | 171.1, C | 172.7, C | ||
| 2′ | 54.6, CH | 4.61, ddd (8.1, 4.2, 3.3) | 52.3, CH | 4.53, ddd (8.6, 8.0, 3.7) |
| 3′ | 60.8, CH2 | 3.98, dd (11.2, 4.2) 3.89, dd (11.2, 3.3) | 26.5, CH2 | 2.22, m 2.11, m |
| 4′ | - | - | 31.6, CH2 | 2.22, m, 2H |
| 5′ | 173.8, C | |||
| NH | 9.19, d (8.1) | 9.53, d (8.8) | ||
| COOH | 13.11, br s | - | ||
| NH2 | - | 7.32, br s/6.80, br s | ||
| Position | 3 | 6 | ||
|---|---|---|---|---|
| δC, Type * | δH (J in Hz) | δC, Type * | δH (J in Hz) | |
| 1 | 159.1, C | 159.1, C | ||
| 2 | 134.5, C | 134.1, C | ||
| 3 | 150.2, C | 150.2, C | ||
| 4 | 119.8, CH | 7.96, br s | 119.7, CH | 7.92, br s |
| 5 | 130.4, C | 130.4, C | ||
| 6 | 127.0, CH | 7.89, dd (7.4, 1.8) | 127.0, CH | 7.89, dd (7.1, 1.6) |
| 7 | 128.9, CH 128.0, CH | 7.59, m | 128.9, CH 128.0, CH | 7.59, ddd (7.1, 6.8, 1.4) |
| 8 | 7.63, m | 7.62, ddd (8.1, 6.8, 1.6) | ||
| 9 | 129.4, CH | 8.03, br d (7.8) | 129.4, CH | 8.00, br d (8.1) |
| 10 | 142.5, C | 142.4, C | ||
| 3-OH | 10.61, s | 10.45, s | ||
| 1′ | 159.2, C | 159.2, C | ||
| 2′ | 136.2, C | 136.0, C | ||
| 3′ | 143.6, CH | 8.96, s | 143.8, CH | 8.90, s |
| 1″ | 172.0, C | 172.0, C | ||
| 2″ | 55.3, CH | 4.58, m | 51.6, CH | 4.79, ddd (8.5, 4.5, 4.3) |
| 3″ | 25.6, CH2 | 3.08, m 2.96, m | 39.3, CH2 | 3.38, m 3.19, dd (12.6, 4.5) |
| NH | 9.20, d (8.5) | |||
| COOH | 13.09, br s | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-López, F.J.; Alcalde, E.; Sarmiento-Vizcaíno, A.; Díaz, C.; Cautain, B.; García, L.A.; Blanco, G.; Reyes, F. New 3-Hydroxyquinaldic Acid Derivatives from Cultures of the Marine Derived Actinomycete Streptomyces cyaneofuscatus M-157. Mar. Drugs 2018, 16, 371. https://doi.org/10.3390/md16100371
Ortiz-López FJ, Alcalde E, Sarmiento-Vizcaíno A, Díaz C, Cautain B, García LA, Blanco G, Reyes F. New 3-Hydroxyquinaldic Acid Derivatives from Cultures of the Marine Derived Actinomycete Streptomyces cyaneofuscatus M-157. Marine Drugs. 2018; 16(10):371. https://doi.org/10.3390/md16100371
Chicago/Turabian StyleOrtiz-López, Francisco Javier, Elsa Alcalde, Aida Sarmiento-Vizcaíno, Caridad Díaz, Bastien Cautain, Luis A. García, Gloria Blanco, and Fernando Reyes. 2018. "New 3-Hydroxyquinaldic Acid Derivatives from Cultures of the Marine Derived Actinomycete Streptomyces cyaneofuscatus M-157" Marine Drugs 16, no. 10: 371. https://doi.org/10.3390/md16100371
APA StyleOrtiz-López, F. J., Alcalde, E., Sarmiento-Vizcaíno, A., Díaz, C., Cautain, B., García, L. A., Blanco, G., & Reyes, F. (2018). New 3-Hydroxyquinaldic Acid Derivatives from Cultures of the Marine Derived Actinomycete Streptomyces cyaneofuscatus M-157. Marine Drugs, 16(10), 371. https://doi.org/10.3390/md16100371