For a Correct Application of the CD Exciton Chirality Method: The Case of Laucysteinamide A


Abstract
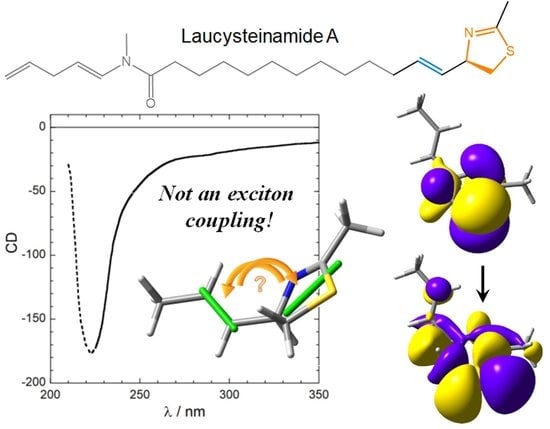
:
1. Introduction
2. Results
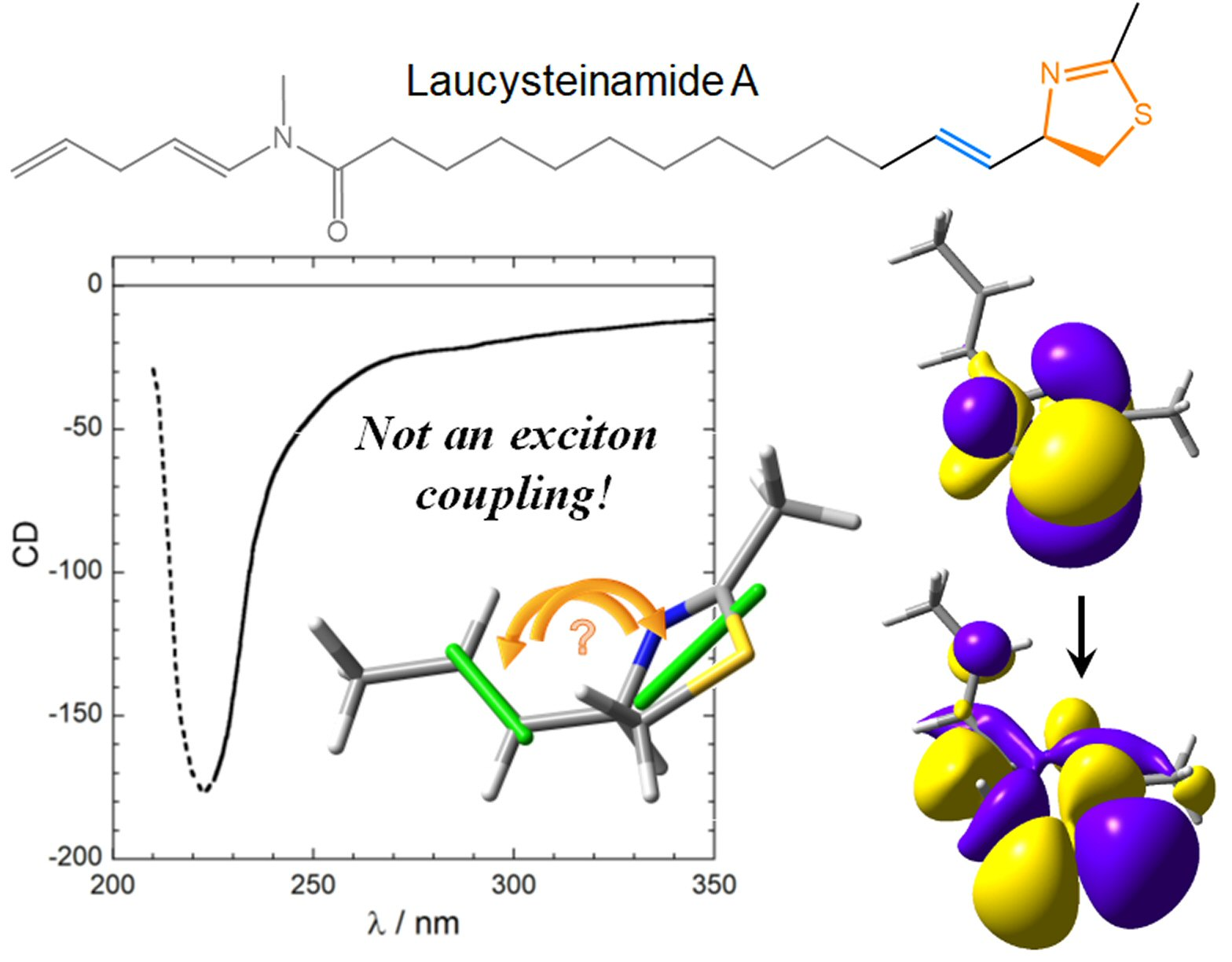
- The nature and polarization of the transition of the thiazoline chromophore responsible for the CD band above 220 nm were not discussed; it was not clear whether this is a π–π* transition, namely, the kind of transition which is expected to be involved in exciton coupling;
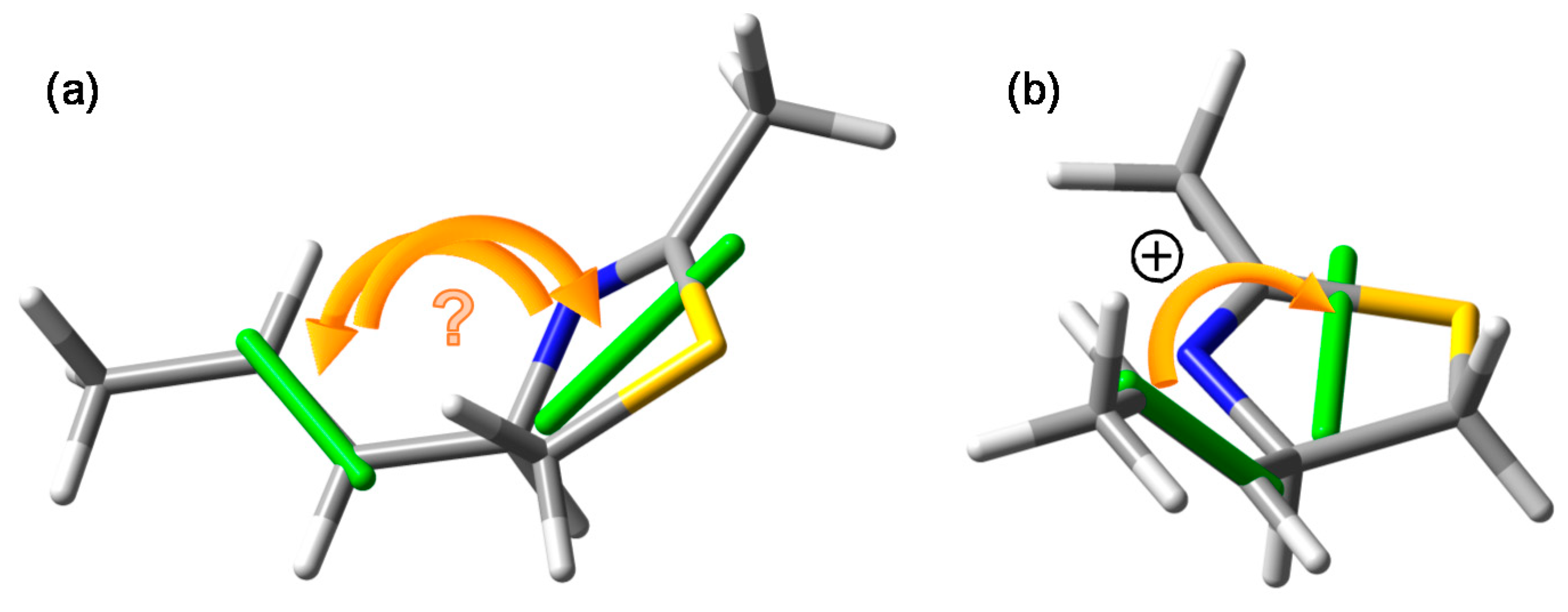
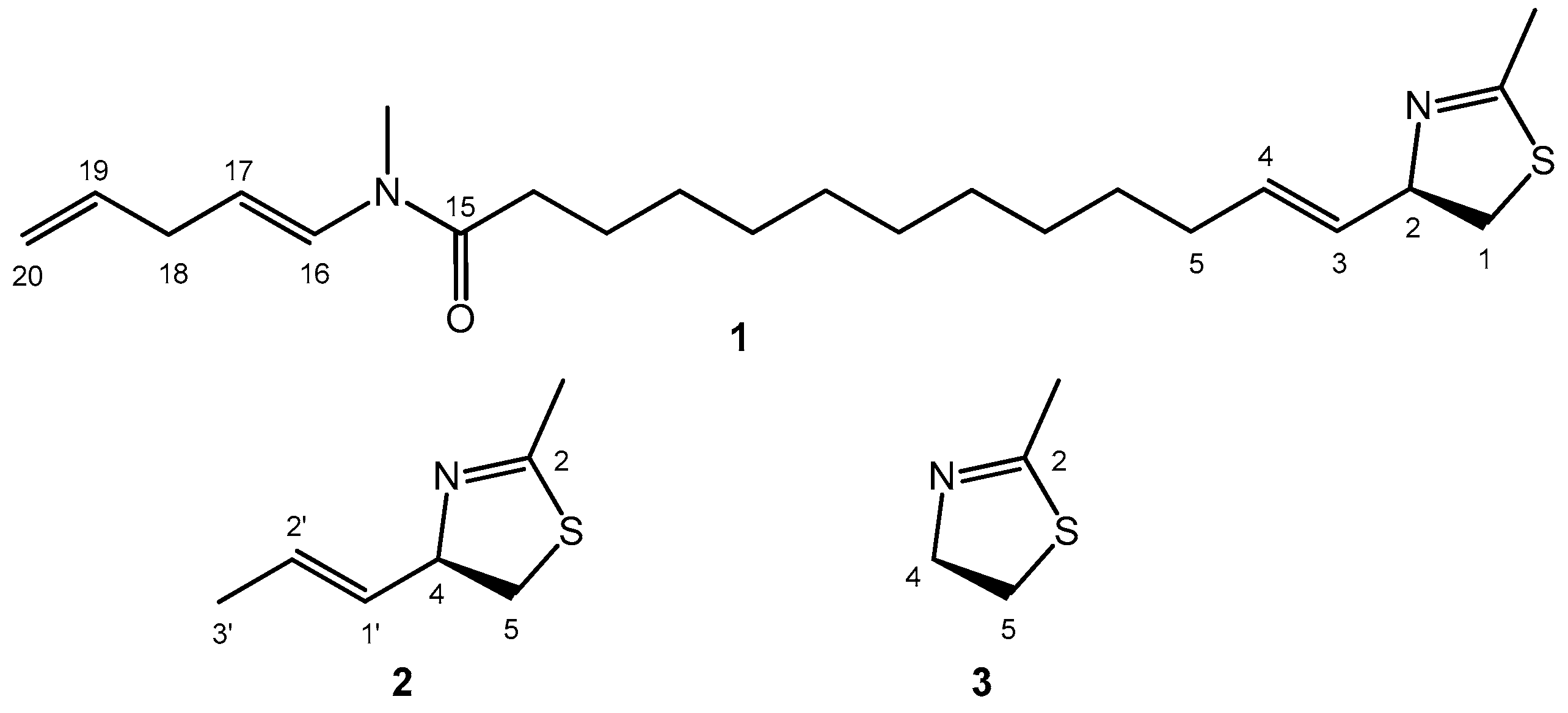
- In the drawing of 1 used to establish the chirality associated with the NDEC, the direction of the transition moment allied with the thiazoline chromophore was not shown, and an ambiguous viewpoint was assumed;
- No conformational analysis of 1 was run, or at least it is not reported; ECM was applied to a single conformer whose relative population is unclear.
2.1. Electronic Transitions of the Model Thiazoline Chromophore 3
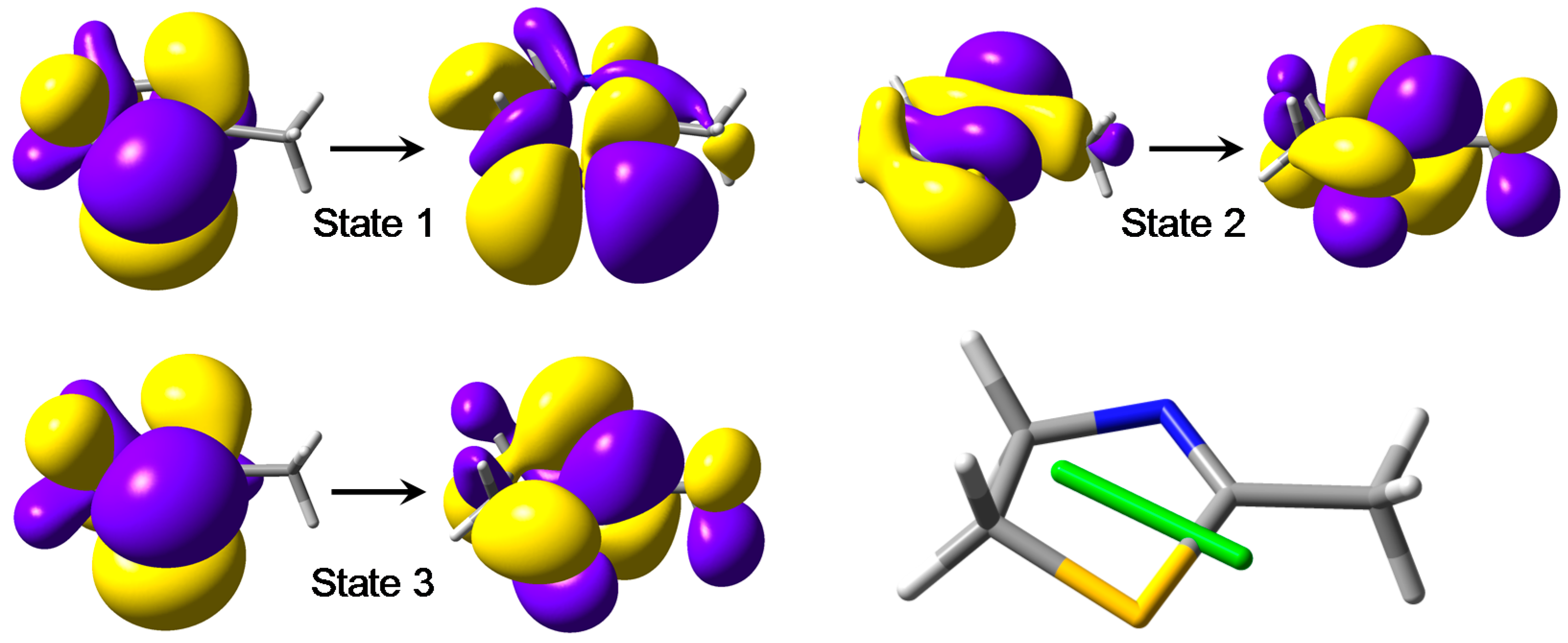
- the first calculated transition is a magnetic-dipole-allowed sulfur-centered n–σ*;
- the second calculated transition is a magnetic-dipole-allowed nitrogen-centered n–π*;
- the third calculated transition is a π–π* transition; this is an electric-dipole-allowed transition associated with a rather weak electric transition dipole (oscillator strength f ~ 0.05).
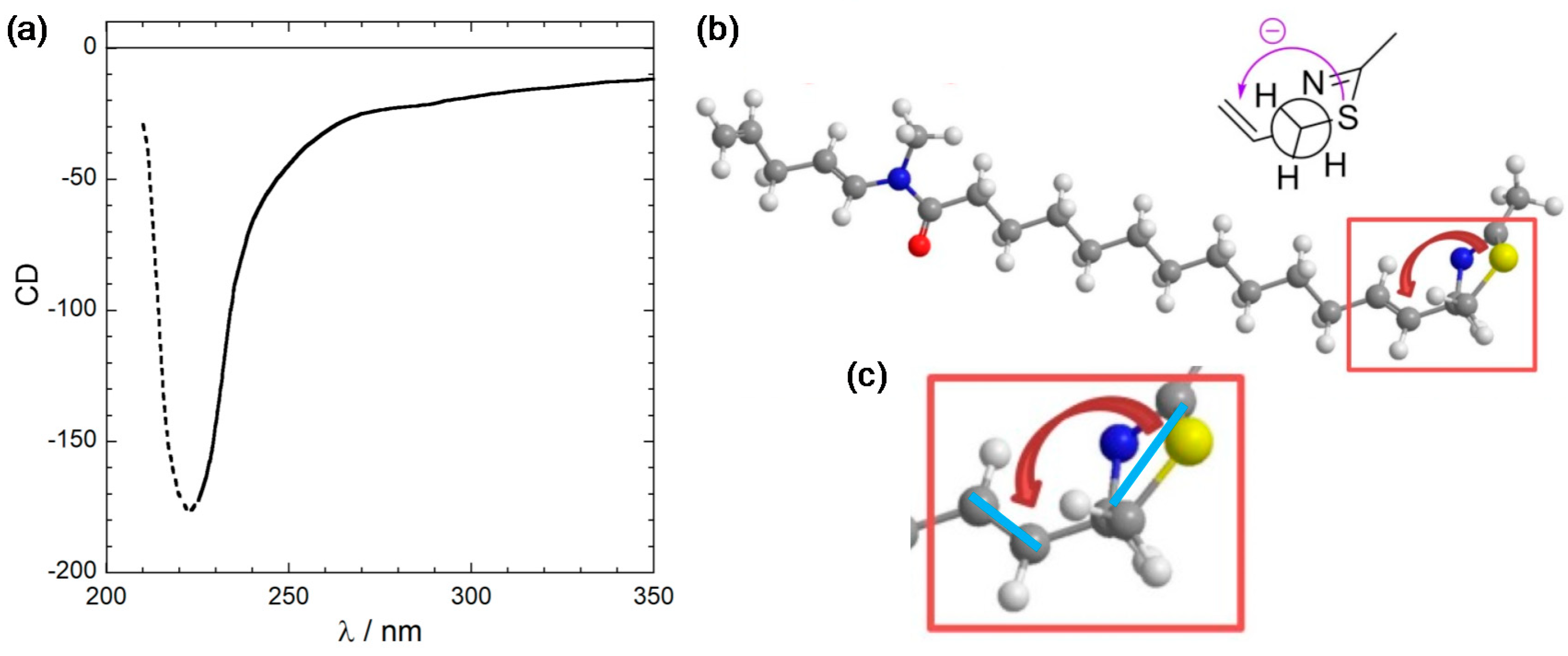
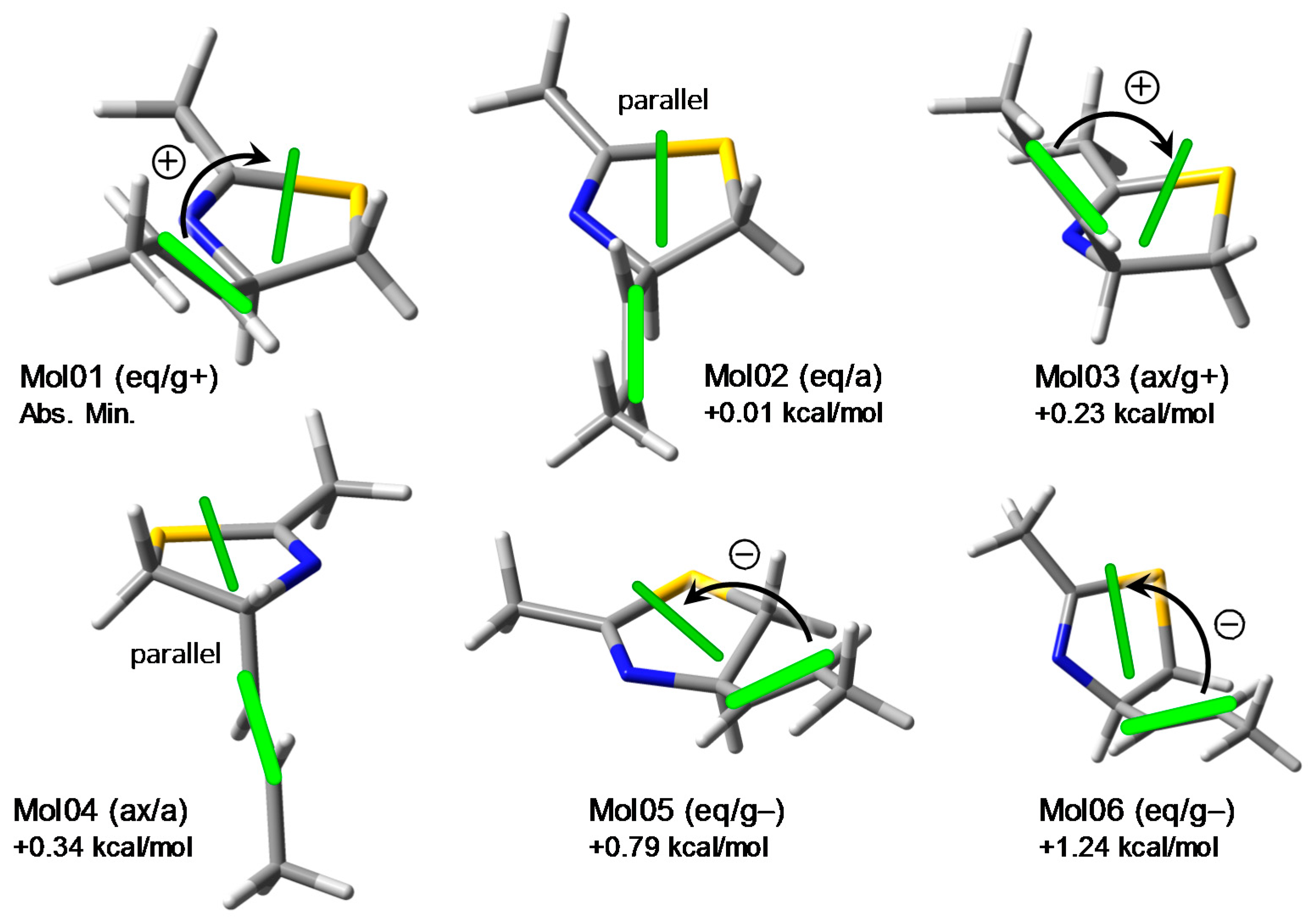
2.2. Conformational Analysis and Geometry Optimizations of Truncated Model 2
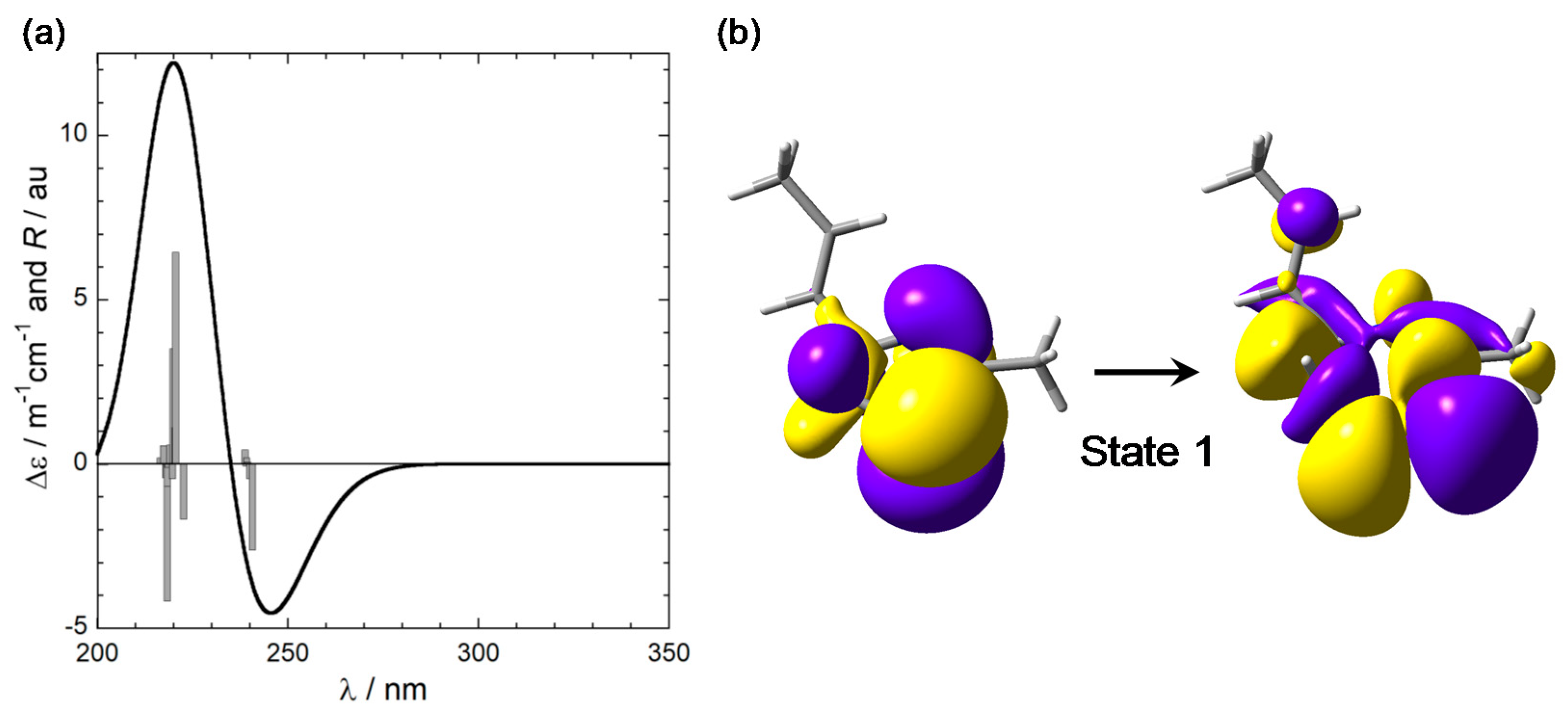
2.3. CD Calculations on Truncated Model 2 and Transition Analysis
3. Discussion
3.1. Missing Conformational Analysis
3.2. Nature of the Transition Involved
3.3. Viewpoint Used to Establish the Transition Moment Chirality
4. Materials and Methods
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Krastel, P.; Petersen, F.; Roggo, S.; Schmitt, E.; Schuffenhauer, A. Aspects of Chirality in Natural Products Drug Discovery. In Chirality in Drug Research; Francotte, E., Lindner, W., Eds.; Wiley-VCH: Weinheim, Germany, 2006; pp. 67–94. [Google Scholar]
- Lin, G.-Q.; You, Q.-D.; Cheng, J.-F. (Eds.) Chiral Drugs: Chemistry and Biological Action; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albright, A.L.; White, J.M. Determination of Absolute Configuration Using Single Crystal X-Ray Diffraction. In Metabolomics Tools for Natural Product Discovery: Methods and Protocols; Roessner, U., Dias, A.D., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 149–162. [Google Scholar]
- Berova, N.; Polavarapu, P.L.; Nakanishi, K.; Woody, R.W. (Eds.) Comprehensive Chiroptical Spectroscopy; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Polavarapu, P.L. Chiroptical Spectroscopy: Fundamentals and Applications; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Superchi, S.; Scafato, P.; Górecki, M.; Pescitelli, G. Absolute Configuration Determination by Quantum Mechanical Calculation of Chiroptical Spectra: Basics and Applications to Fungal Metabolites. Curr. Med. Chem. 2018, 25, 287–320. [Google Scholar] [CrossRef] [PubMed]
- Hopmann, K.H.; Šebestík, J.; Novotná, J.; Stensen, W.; Urbanová, M.; Svenson, J.; Svendsen, J.S.; Bouř, P.; Ruud, K. Determining the Absolute Configuration of Two Marine Compounds Using Vibrational Chiroptical Spectroscopy. J. Org. Chem. 2012, 77, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Joseph-Nathan, P.; Gordillo-Román, B. Vibrational Circular Dichroism Absolute Configuration Determination of Natural Products. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Falk, H., Kobayashi, J., Eds.; Springer: Cham, Switzerland, 2015; Volume 100, pp. 311–452. [Google Scholar]
- Srebro-Hooper, M.; Autschbach, J. Calculating Natural Optical Activity of Molecules from First Principles. Annu. Rev. Phys. Chem. 2017, 68, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Nakanishi, K. Circular Dichroic Spectroscopy—Exciton Coupling in Organic Stereochemistry; University Science Books: Mill Valley, CA, USA, 1983. [Google Scholar]
- Harada, N.; Nakanishi, K. Exciton chirality method and its application to configurational and conformational studies of natural products. Acc. Chem. Res. 1972, 5, 257–263. [Google Scholar] [CrossRef]
- Berova, N.; Ellestad, G.A.; Harada, N. Characterization by Circular Dichroism Spectroscopy. In Comprehensive Natural Products II; Mander, L., Liu, H.-W., Eds.; Elsevier: Oxford, UK, 2010; Volume 9, pp. 91–146. [Google Scholar]
- Harada, N.; Nakanishi, K.; Berova, N. Electronic CD Exciton Chirality Method: Principles and Applications. In Comprehensive Chiroptical Spectroscopy; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 115–166. [Google Scholar]
- Meng, J.; Cheng, W.; Heydari, H.; Wang, B.; Zhu, K.; Konuklugil, B.; Lin, W. Sorbicillinoid-Based Metabolites from a Sponge-Derived Fungus Trichoderma saturnisporum. Mar. Drugs 2018, 16, 226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Naman, C.; Engene, N.; Gerwick, W. Laucysteinamide A, a Hybrid PKS/NRPS Metabolite from a Saipan Cyanobacterium, cf. Caldora penicillata. Mar. Drugs 2017, 15, 121. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zou, Y.; Wang, R.-P.; Hamann, M.; Zhang, H.-J.; Jiao, W.-H.; Han, B.-N.; Song, S.-J.; Lin, H.-W. Relative and Absolute Stereochemistry of Diacarperoxides: Antimalarial Norditerpene Endoperoxides from Marine Sponge Diacarnus megaspinorhabdosa. Mar. Drugs 2014, 12, 4399–4416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, L.; Zhuang, Y.; Kong, F.; Zhang, C.; Zhu, W. Phenolic Polyketides from the Co-Cultivation of Marine-Derived Penicillium sp. WC-29-5 and Streptomyces fradiae 007. Mar. Drugs 2014, 12, 2079–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Kong, F.; Wang, Y.; Fu, P.; Zhu, W. Cladodionen, a Cytotoxic Hybrid Polyketide from the Marine-Derived Cladosporium sp. OUCMDZ-1635. Mar. Drugs 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xi, L.; Liu, P.; Wang, Y.; Wang, W.; Huang, Y.; Zhu, W. Diketopiperazine Derivatives from the Marine-Derived Actinomycete Streptomyces sp. FXJ7.328. Mar. Drugs 2013, 11, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berova, N.; Pescitelli, G.; Petrovic, A.G.; Proni, G. Probing molecular chirality by CD-sensitive dimeric metalloporphyrin hosts. Chem. Commun. 2009, 5958–5980. [Google Scholar] [CrossRef] [PubMed]
- Pescitelli, G.; Di Bari, L. Revision of the Absolute Configuration of Preussilides A-F Established by the Exciton Chirality Method. J. Nat. Prod. 2017, 80, 2855–2859. [Google Scholar] [CrossRef] [PubMed]
- Pescitelli, G.; Di Bari, L. Exciton coupling between enones: Quassinoids revisited. Chirality 2017, 29, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.-W.; Lu, Q.-Q.; Pescitelli, G.; Ivšić, T.; Zhou, J.-H.; Gao, J.-M. Three Sesquiterpenoid Dimers from Chloranthus japonicus: Absolute Configuration of Chlorahololide A and Related Compounds. Chirality 2016, 28, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Pescitelli, G.; Bari, L.D. Comment on “Breakdown of Exciton Splitting through Electron Donor–Acceptor Interaction: A Caveat for the Application of Exciton Chirality Method in Macromolecules”. J. Phys. Chem. C 2014, 118, 24197–24198. [Google Scholar] [CrossRef]
- Harada, N.; Iwabuchi, J.; Yokota, Y.; Uda, H.; Nakanishi, K. A chiroptical method for determining the absolute configuration of allylic alcohols. J. Am. Chem. Soc. 1981, 103, 5590–5591. [Google Scholar] [CrossRef]
- Iwahana, S.; Iida, H.; Yashima, E.; Pescitelli, G.; Di Bari, L.; Petrovic, A.G.; Berova, N. Absolute Stereochemistry of a 4a-Hydroxyriboflavin Analogue of the Key Intermediate of the FAD-Monooxygenase Cycle. Chem. Eur. J. 2014, 20, 4386–4395. [Google Scholar] [CrossRef] [PubMed]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Roschester, J.; Berg, U.; Pierrot, M.; Sandström, J. Conformational analysis of N-(1-phenylethyl)-Δ4-thiazoline-2-thiones and analogs. A proton NMR, circular dichroism, x-ray crystallographic, and molecular mechanics study. J. Am. Chem. Soc. 1987, 109, 492–507. [Google Scholar] [CrossRef]
- Roschester, J.; Sandström, J. Conformational analysis of N-(1-methoxycarbonylethyl)-Δ4-thiazoline-2-thiones by temperature-dependent circular dichroism and NMR spectroscopy and by molecular mechanics calculations. Tetrahedron 1989, 45, 5081–5100. [Google Scholar] [CrossRef]
- Orlova, G.; Goddard, J.D.; Brovko, L.Y. Theoretical Study of the Amazing Firefly Bioluminescence: The Formation and Structures of the Light Emitters. J. Am. Chem. Soc. 2003, 125, 6962–6971. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Superchi, S.; Giorgio, E.; Rosini, C. Structural determinations by circular dichroism spectra analysis using coupled oscillator methods: An update of the applications of the DeVoe polarizability model. Chirality 2004, 16, 422–451. [Google Scholar] [CrossRef] [PubMed]
- Garbisch, E.W. Conformations. VI. Vinyl-Allylic Proton Spin Couplings. J. Am. Chem. Soc. 1964, 86, 5561–5564. [Google Scholar] [CrossRef]
- Chisholm, J.D.; Golik, J.; Krishnan, B.; Matson, J.A.; Van Vranken, D.L. A Caveat in the Application of the Exciton Chirality Method to N,N-Dialkyl Amides. Synthesis and Structural Revision of AT2433-B1. J. Am. Chem. Soc. 1999, 121, 3801–3802. [Google Scholar] [CrossRef]
- Matile, S.; Berova, N.; Nakanishi, K. Intramolecular Porphyrin π,π-Stacking: Absolute Configurational Assignment of Acyclic Compounds with Single Chiral Centers by Exciton Coupled Circular Dichroism. Enantiomer 1996, 1, 1–12. [Google Scholar] [PubMed]
- Gargiulo, D.; Derguini, F.; Berova, N.; Nakanishi, K.; Harada, N. Unique ultraviolet-visible and circular dichroism behavior due to exciton coupling in a biscyanine dye. J. Am. Chem. Soc. 1991, 113, 7046–7047. [Google Scholar] [CrossRef]
- Pescitelli, G.; Di Bari, L.; Berova, N. Conformational Aspects in the Studies of Organic Compounds by Electronic Circular Dichroism. Chem. Soc. Rev. 2011, 40, 4603–4625. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Casarini, D. Recent trends in conformational analysis. WIREs Comput. Mol. Sci. 2012, 2, 613–641. [Google Scholar] [CrossRef]
- Mason, S.F.; Seal, R.H.; Roberts, D.R. Optical activity in the biaryl series. Tetrahedron 1974, 30, 1671–1682. [Google Scholar] [CrossRef]
- Di Bari, L.; Pescitelli, G.; Salvadori, P. Conformational study of 2,2′-homosubstituted 1,1′-binaphthyls by means of UV and CD spectroscopy. J. Am. Chem. Soc. 1999, 121, 7998–8004. [Google Scholar] [CrossRef]
- Koreeda, M.; Harada, N.; Nakanishi, K. Exciton chirality methods as applied to conjugated enones, esters, and lactones. J. Am. Chem. Soc. 1974, 96, 266–268. [Google Scholar] [CrossRef]
- Rosenfield, J.S.; Moscowitz, A. Optical activity data as an aid in the assignment of electronic transitions: Application to dialkyl sulphides. In Fundamental Aspects and Recent Developments in ORD and CD; Ciardelli, F., Salvadori, P., Eds.; Heyden: London, UK, 1967; pp. 41–49. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03, Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Pescitelli, G. SpecDis Version 1.71, SpecDis: Berlin, Germany.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Energy (eV) | Wavelength (nm) | Oscillator Strength |
|---|---|---|---|
| 1 | 5.208 | 238.1 | 0.0018 |
| 2 | 5.697 | 217.6 | 0.0040 |
| 3 | 5.775 | 214.7 | 0.0495 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pescitelli, G. For a Correct Application of the CD Exciton Chirality Method: The Case of Laucysteinamide A. Mar. Drugs 2018, 16, 388. https://doi.org/10.3390/md16100388
Pescitelli G. For a Correct Application of the CD Exciton Chirality Method: The Case of Laucysteinamide A. Marine Drugs. 2018; 16(10):388. https://doi.org/10.3390/md16100388
Chicago/Turabian StylePescitelli, Gennaro. 2018. "For a Correct Application of the CD Exciton Chirality Method: The Case of Laucysteinamide A" Marine Drugs 16, no. 10: 388. https://doi.org/10.3390/md16100388
APA StylePescitelli, G. (2018). For a Correct Application of the CD Exciton Chirality Method: The Case of Laucysteinamide A. Marine Drugs, 16(10), 388. https://doi.org/10.3390/md16100388