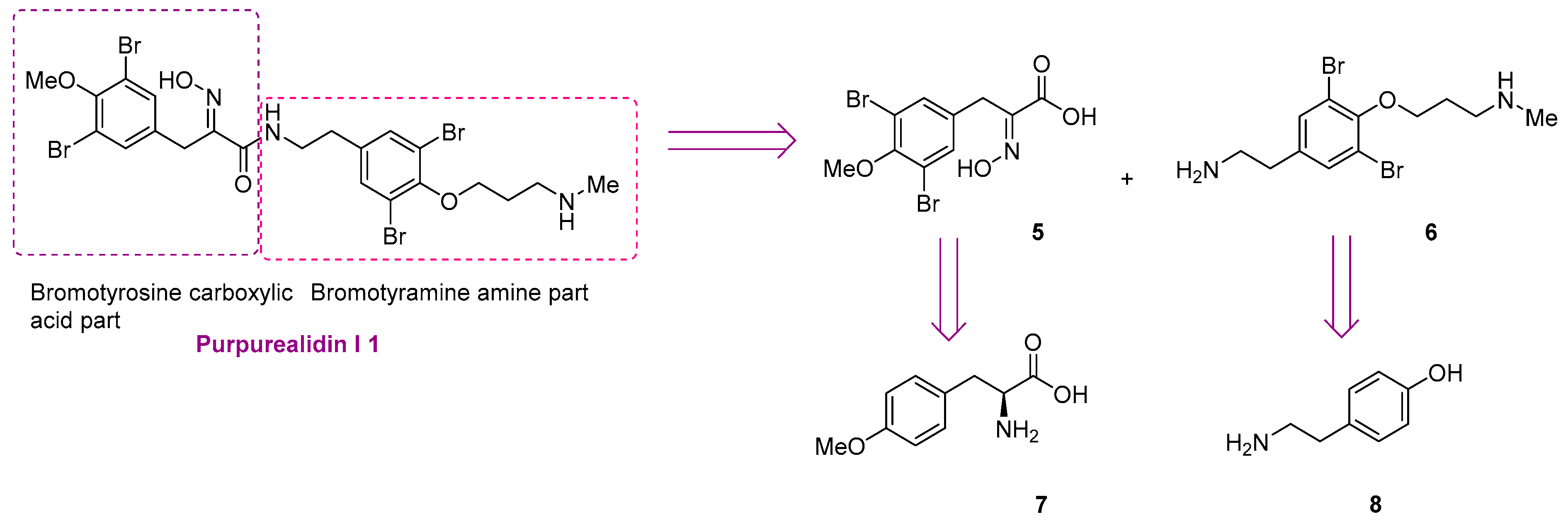
4.1.2. Experimental Procedures
General Procedure for the Formation of Azlactones
Aldehyde 19a–d, acetylglycine (1.5 equiv.) and anhyd. NaOAc (1.5 equiv.) were dissolved in Ac2O (10–15 mL) and the reaction mixture was stirred at 80 °C overnight. Afterwards, the reaction mixture was cooled to room temperature and poured into water (50 mL). The formed precipitate was filtered, washed with water (4 × 20 mL) and dried in vacuo. The obtained crude product 20a–d was used in the subsequent step without further purification.
4-(3,5-Dibromo-4-methoxybenzylidene)-2-methyloxazol-5(4H)-one (20a).
3,5-Dibromo-4-methoxybenzaldehyde
19a (1.57 g, 5.33 mmol) gave
20a as a grey solid (1.94 g, 97%).
1H NMR (400 MHz, CDCl
3)
δ 8.26 (s, 2H), 6.92 (s, 1H), 3.93 (s, 3H), 2.43 (s, 3H).
1H NMR is in accordance with the literature [
26].
4-(3-Bromo-4-methoxybenzylidene)-2-methyloxazol-5(4H)-one (20b).
3-Bromo-4-methoxybenzaldehyde
19b (2.00 g, 9.30 mmol) gave
20b as a yellow solid (2.70 g, 98%)
1H NMR (400 MHz, CDCl
3)
δ 8.43 (d,
J = 2.1 Hz, 1H), 7.98–7.94 (m, 1H), 7.02 (s, 1H), 6.95 (d,
J = 8.6 Hz, 1H), 3.96 (s, 3H), 2.41 (d,
J = 0.7 Hz, 3H).
1H NMR is in accordance with the literature [
25].
4-(3-Chloro-4-methoxybenzylidene)-2-methyloxazol-5(4H)-one (20c).
3-Chloro-4-methoxybenzaldehyde 19c (2.43 g, 14.3 mmol) gave 20c as a yellow solid (2.66 g, 74%). 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 2.1 Hz, 1H), 7.89 (dd, J = 8.6, 2.1 Hz, 1H), 7.02 (s, 1H), 6.98 (d, J = 8.6 Hz, 1H), 3.97 (s, 3H), 2.41 (s, 3H).
4-(4-Methoxybenzylidene)-2-methyloxazol-5(4H)-one (20d).
4-Methoxybenzaldehyde 19d (3.03 g, 22.3 mmol) gave 20d as a yellow solid (2.21 g, 46%). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.5 Hz, 2H), 7.11 (s, 1H), 6.96 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H), 2.39 (s, 3H).
General Method for the Hydrolysis of Azlactones
Azlactone 20a–d was dissolved in a 10% solution of HCl in H2O (30 mL). A capillary tube was introduced in the flask to allow the reflux despite a solid layer forming while heating. The reaction mixture was refluxed overnight. The reaction mixture was cooled to room temperature and poured into cold water (2 × 10 mL). The resulting precipitate was filtered, washed with water (4 × 20 mL) and dried in vacuo. The obtained crude product 21a–d was used in the subsequent step without further purification.
3-(3,5-Dibromo-4-methoxyphenyl)-2-oxopropanoic acid (21a)
Azlactone
20a (1.74 g, 4.63 mmol) gave acid
21a as a yellowish solid (1.46 g, 89%).
1H NMR (400 MHz,
d6-DMSO)
δ 8.06 (s, 2H), 3.80 (s, 3H), 3.32 (s, 2H);
13C NMR (101 MHz,
d6-DMSO)
δ 165.7, 151.8, 143.3, 134.5, 132.9, 117.3, 106.1, 60.5. NMR showed the enol tautomer.
1H NMR is in accordance with the literature [
25].
3-(3-Bromo-4-methoxyphenyl)-2-oxopropanoic acid (21b)
Azlactone
20b (3.33 g, 9.11 mmol) gave
21b as a dark red solid (1.89 g, 79%).
1H NMR (400 MHz,
d6-DMSO)
δ 9.26 (bs, 1H), 8.10 (d,
J = 2.1 Hz, 1H), 7.69–7.64 (m, 1H), 7.10 (d,
J = 8.7 Hz, 1H), 6.35 (s, 1H), 3.85 (s, 3H). NMR showed the enol tautomer.
1H NMR is in accordance with the literature [
25].
3-(3-Chloro-4-methoxyphenyl)-2-oxopropanoic acid (21c)
Azlactone 20c (2.66 g, 10.6 mmol) gave 21c as a brown solid (1.92 g, 79%). 1H NMR (400 MHz, CD3OD) δ 7.91 (d, J = 2.1 Hz, 1H), 7.59 (ddd, J = 8.6, 2.1, 0.5 Hz, 1H), 7.03 (d, J = 8.7 Hz, 1H), 6.40 (s, 1H), 3.89 (s, 3H). NMR showed the enol tautomer.
3-(4-Methoxyphenyl)-2-oxopropanoic acid (21d)
Azlactone
20d (2.21 g, 10.2 mmol) gave product
21d as a brown solid (1.78 g, 90%).
1H NMR (400 MHz, CDCl
3)
δ 7.75 (d,
J = 8.7 Hz, 2H), 6.92 (d,
J = 8.9 Hz, 2H), 6.64 (s, 1H), 3.84 (s, 3H). NMR showed the enol tautomer.
1H NMR is in accordance with the literature [
31].
General Procedure for THP-Protection
Carboxylic acid 21a–d and THPONH2 (2 equiv.) were dissolved in dry ethanol (15–20 mL). The reaction mixture was stirred at room temperature for 18–48 h under argon atmosphere. The reaction mixture was concentrated under reduced pressure and then EtOAc (20 mL) was added to the residue. The organic layer was washed with a 2 M solution of HCl in H2O (2 × 20 mL). The aqueous layer was extracted back with EtOAc (2 × 10 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The obtained crude product 22a–d was used in the subsequent step without further purification.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanoic acid (22a)
Carboxylic acid 21a (1.16 g, 3.30 mmol) and THPONH2 (0.97 g, 8.3 mmol, 2 equiv.) were used. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (DCM/MeOH, 0→10%) to give 22a as a yellow oil (1.44 g, 97%). 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 2H), 5.50–5.41 (m, 1H), 3.95–3.88 (m, 2H), 3.84 (s, 3H), 3.69–3.54 (m, 2H), 1.93–1.83 (m, 2H), 1.77–1.65 (m, 2H), 1.64–1.58 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 163.7, 163.3, 153.2, 149.9, 133.6, 118.2, 102.2, 62.4, 60.8, 29.6, 28.1, 24.9, 18.6.
(E)-3-(3-Bromo-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanoic acid (22b)
Carboxylic acid 21b (2.00 g, 7.32 mmol) and THPONH2 (1.71 g, 14.6 mmol, 2 equiv.) were used. 22b was obtained as an oil (2.74 g, quant.). 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 2.2 Hz, 1H), 7.23 (dd, J = 2.2, 8.5 Hz, 1H), 6.81 (d, J = 8.5 Hz, 1H), 5.46 (d, J = 3.1 Hz, 1H), 3.94–3.87 (m, 2H), 3.86 (s, 3H), 3.69–3.65 (m, 2H), 1.91–1.81 (m, 2H), 1.76–1.66 (m, 2H), 1.65–1.56 (m, 2H).
(E)-3-(3-Chloro-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanoic acid (22c)
Carboxylic acid 21c (1.86 g, 8.13 mmol) and THPONH2 (1.90 g, 16.3 mmol, 2 equiv.) were used. 22c was obtained as a yellow oil (3.05 g, quant). 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 2.2 Hz, 1H), 7.18 (dd, J = 8.4, 2.2 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 5.47 (d, J = 3.1 Hz, 1H), 3.87 (s, 3H), 3.86 (s, 2H), 3.63 (dd, J = 7.6, 3.4 Hz, 2H), 1.91–1.82 (m, 2H), 1.77–1.66 (m, 2H), 1.65–1.52 (m, 2H).
(E)-3-(4-Methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanoic acid (22d)
Carboxylic acid 21d (1.73 g, 7.98 mmol) and THPONH2 (1.87 g, 16.0 mmol, 2 equiv.) were used. 22d was obtained as a yellow oil (2.78 g, quant). 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 8.7 Hz, 2H), 5.47–5.43 (t, 1H), 3.87 (s, 2H), 3.76 (s, 3H), 3.67–3.59 (m, 2H), 1.93–1.80 (m, 2H), 1.72–1.65 (m, 2H), 1.60–1.51 (m, 2H).
(E)-N-[3,5-Dibromo-4-[3-(2,2,2-trifluoro-N-methylacetamido)propoxy]phenethyl]-3-(3,5-dibromo-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (26)
A 20 mL Biotage MW tube was charged with carboxylic acid
22a (0.24 g, 0.53 mmol), amine
17 [
15] (0.25 g, 0.53 mmol), EDC·HCl (0.15 g, 0.79 mmol, 1.5 equiv.), HOBt (0.11 g, 0.79 mmol, 1.5 equiv.), DIPEA (0.10 g, 0.79 mmol, 1.5 equiv.) and dry DCM (15 mL). The reaction was MW irradiated for 2 h at 60 °C (5 bar). The reaction mixture was diluted with DCM (25 mL), washed with water (2 × 15 mL) and a 1 M solution of HCl in H
2O (15 mL). The organic layer was dried over Na
2SO
4 and concentrated in vacuo. The crude product was purified by flash chromatography twice, 25 g, gradient elution: (heptane/EtOAc, 0→40%) to
23 as a pale yellow viscous liquid (0.16 g, 34%).
1H NMR (400 MHz, CDCl
3)
δ 7.50 (s, 2H), 7.34 and 7.33 (2 s, 2H, rotamers ratio 2:1), 6.92 (t,
J = 6.2 Hz, 1H), 5.38 (t,
J = 2.8 Hz, 1H), 4.06–4.00 (m, 2H), 3.93 (d,
J = 13.1 Hz, 1H), 3.84 (s, 3H), 3.83 (d,
J = 13.1 Hz, 1H), 3.80–3.72 (m, 2H), 3.65–3.54 (m, 3H), 3.43 (ddt,
J = 12.9, 8.2, 6.6 Hz, 1H), 3.23 (m) and 3.11 (m) N-CH
3 rotamers 2:1, 2.77 (td,
J = 7.3, 1.7 Hz, 2H), 2.23–2.09 (m, 2H), 1.89–1.54 (m, 6H).
![Marinedrugs 16 00481 i014]()
(E)-N-[3,5-Dibromo-4-[3-(2,2,2-trifluoro-N-methylacetamido)propoxy]phenethyl]-3-(3,5-dibromo-4-methoxyphenyl)-2-(hydroxyimino)propanamide (28)
THP ether 26 (0.14 g, 0.16 mmol), a 2 M solution of HCl in Et2O (4 mL), dry DCM (4 mL) and dry MeOH (0.3 mL) were added to a 20-mL sealed tube and heated in an oil bath at 70 °C for 3 h. The reaction mixture was then concentrated in vacuo. The crude product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 10 g, gradient elution: (heptane/EtOAc, 0→30%) to give 28 as a pale yellow viscous liquid (0.068 g, 54%). 1H NMR (400 MHz, CD3OD) δ 7.48 (s, 2H), 7.43 and 7.42 (2 s, 2H, rotamers ratio 2:1), 4.00 (t, J = 6.0 Hz, 2H), 3.82 (m, 5H), 3.78–3.70 (m, 2H), 3.44 (t, J = 7.1 Hz, 2H), 3.24 (q) and 3.10 (m) N-CH3 rotamers 2:1, 2.76 (t, J = 7.1 Hz, 2H), 2.25–2.08 (m, 2H).
(E)-N-[3,5-Dibromo-4-[3-(methylamino)propoxy]phenethyl]-3-(3,5-dibromo-4-methoxyphenyl)-2-(hydroxyimino)propenamide, purpurealidin I, (1)
Compound 28 (0.050 g, 0.062 mmol) and K2CO3 (0.018 g, 0.12 mmol, 2.0 equiv.) in MeOH (5 mL) and H2O (0.5 mL) were refluxed for 2.5 h. The reaction mixture was concentrated in vacuo and partitioned between EtOAc (10 mL) and water (4 mL). The aqueous layer was back-extracted with EtOAc (10 mL). The combined organic layers ware dried over Na2SO4, filtered and the solvent was removed in vacuo to give 1, as a pale yellow viscous liquid (0.040 -g, 91%). 1H NMR (300 MHz, CDCl3) δ 7.49 (s, 2H), 7.26 (s, 2H), 4.16–4.11 (m, 2H), 3.84–3.83 (m, 5H), 3.42–3.35 (m, 2H), 2.93 (t, J = 7.2 Hz, 2H), 2.71 (t, J = 6.9 Hz, 2H), 2.53 (s, 3H), 2.09–2.00 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 163.6, 152.5, 151.3, 150.4, 138.2, 135.9, 133.5, 133.0, 118.3, 117.8, 71.8, 60.7, 49.0, 40.0, 35.5, 34.3, 29.0, 28.0. HRMS (ESI+): calcd. for C22H26N3O4Br4 [M + H]+, 711.8657; found, 711.8660.
![Marinedrugs 16 00481 i016]()
(E)-N-[3,5-Dibromo-4-[3-(dimethylamino)propoxy]phenethyl]-3-(3,5-dibromo-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (27)
A 20-mL Biotage MW tube was charged with purpurealidin E
25 [
15,
27] (0.17 g, 0.44 mmol), carboxylic acid
22a (0.20 g, 0.44 mmol), EDC·HCl (0.13 g, 0.66 mmol, 1.5 equiv.), HOBt (0.10 g, 0.66 mmol, 1.2 equiv.), DIPEA (0.12 mL, 0.66 mmol, 1.2 equiv.) and dry DCM (15 mL). The tube was sealed and microwave irradiated at 60 °C for 5 h. The reaction mixture was diluted with DCM (10 mL) and washed with water (2 × 15 mL), a 1 M solution of HCl in H
2O (15 mL) and brine (15 mL). The organic phase was dried over Na
2SO
4 (anhyd.), filtered and volatiles were removed in vacuo. The crude product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, isocratic elution: (DCM/MeOH, 7:3) to give
27 as a yellow oil (0.30 g, 83%).
1H NMR (400 MHz, CDCl
3)
δ 7.48 (s, 2H), 7.31 (s, 2H), 6.94 (t,
J = 6.2 Hz, 1H), 5.36 (t,
J = 2.8 Hz, 1H), 4.03 (t,
J = 5.7 Hz, 2H), 3.89 (s, 1H), 3.82 (s, 3H), 3.58 (dd,
J = 4.9, 8.1 Hz, 3H), 3.42 (m, 1H), 3.23–3.10 (m, 2H), 2.75 (t,
J = 7.3 Hz, 2H), 2.69 (s, 6H), 2.29 (m, 2H), 1.89–1.59 (m, 6H).
![Marinedrugs 16 00481 i017]()
(E)-N-[3,5-Dibromo-4-[3-(dimethylamino)propoxy]phenethyl]-3-(3,5-dibromo-4-methoxyphenyl)-2-(hydroxyimino)propanamide (29)
THP ether 27 (0.30 g, 0.36 mmol) was dissolved to dry DCM (7 mL), and TFA (3 mL) was added under argon atmosphere. The reaction mixture was stirred at room temperature for 17 h. It was then diluted by adding DCM (10 mL) and washed with a 2 M solution of NaOH in H2O (15 mL) and water (15 mL) until pH was neutral. The aqueous phase was back extracted with DCM (10 mL) and the combined organic phase was dried over Na2SO4, filtered and concentrated in vacuo to give crude product (0.21 g, 80%). The crude product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (DCM/MeOH, 2→20%) to give 29 as a yellow oil (0.074 g, 28%). 1H NMR (400 MHz, CD3OD) δ 7.48 (s, 2H), 7.43 (s, 2H), 4.02 (t, J = 5.9 Hz, 2H), 3.82 (s, 3H), 3.44 (dd, J = 6.7, 7.6 Hz, 2H), 3.35 (s, 2H), 3.15–2.98 (m, 2H), 2.85–2.69 (m, 2H), 2.64 (s, 6H), 2.15 (dq, J = 5.8, 7.7 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 165.4, 153.8, 152.5, 152.1, 140.0, 137.4, 134.5, 134.4, 118.8, 118.6, 71.8, 61.0, 57.3, 44.6, 41.4, 35.2, 28.8, 27.7. HRMS (ESI+): calcd. for C23H27Br4N3O4 [M+H]+ 725.8813, found: 725.8809.
General Method for the Amide Coupling
Carboxylic acid (0.20 g), aniline or amine (1–1.5 equiv.), EDC⋅HCl (1.5 equiv.), HOBt (1.5 equiv.), and DIPEA (1.5 equiv.) were dissolved in dry DCM (5 mL). The mixture was irradiated by microwaves for 2 h at 60 °C. The TLC indicated the completion of the reaction using vanillin as a visualization reagent. The reaction mixture was diluted with DCM (20 mL) and washed with water (2 × 15 mL) and a 2 M solution of HCl in H2O (2 × 15 mL). The organic layer was dried over anhyd. Na2SO4, filtered and concentrated in vacuo. The products were purified with flash column chromatography.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(3,4-dichlorophenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]-imino]propanamide (30-THP)
Carboxylic acid 22a (0.20 g, 0.44 mmol) and 3,4-dichloroaniline (0.11 g, 0.67 mmol, 1.5 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g and Ultra 25 g, gradient elution: (heptane/EtOAc, 12→100%) to give 30-THP as a crude product (0.19 g, 73%). 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 7.87 (d, J = 1.9 Hz, 1H), 7.52 (s, 2H), 7.41–7.38 (m, 2H), 5.49 (t, J = 2.6 Hz, 1H), 3.99 (d, J = 13.1 Hz, 1H), 3.89 (d, J = 13.1 Hz, 1H), 3.84 (s, 3H), 3.65–3.59 (m, 2H), 1.93–1.59 (m, 6H).
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(3,4-dichlorobenzyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]-imino]propanamide (31-THP)
Carboxylic acid 22a (0.20 g, 0.44 mmol) and 3,4-dichlorobenzylamine (0.090 mL, 0.67 mmol, 1.5 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (heptane/EtOAc, 7→60%) to give 31-THP as a light yellow solid (0.23 g, 84%). 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 2H), 7.39 (d, J = 8.2 Hz, 1H), 7.37 (d, J = 2.0 Hz, 1H), 7.19 (t, J = 5.9 Hz, 1H), 7.12 (dd, J = 2.1, 8.2 Hz, 1H), 5.38 (s, 1H), 4.54 (dd, J = 6.7, 15.2 Hz, 1H), 4.35 (dd, J = 5.8, 15.2 Hz, 1H), 3.95 (d, J = 13.1 Hz, 1H), 3.85 (d, 13.1 Hz, 1H), 3.84 (s, 1H), 3.61–3.55 (m, 2H), 1.87–1.56 (m, 6H).
(E)-N-[4-Chloro-3-(trifluoromethyl)phenyl]-3-(3,5-dibromo-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (32-THP)
Carboxylic acid 22a (0.19 g, 0.42 mmol) and 4-chloro-3-(trifluoromethyl)aniline (0.12 g, 0.63 mmol, 1.5 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (heptane/EtOAc, 7→60%). to give 32-THP as an oil (0.19 g, 72%). 1H NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 7.95 (d, J = 2.6 Hz, 1H), 7.79 (dd, J = 2.6, 8.7 Hz, 1H), 7.52 (s, 2H), 7.46 (d, J = 8.8 Hz, 1H), 5.50 (t, J = 2.7 Hz, 1H), 4.00 (d, J = 13.2 Hz, 1H), 3.90 (d, J = 13.2 Hz, 1H), 3.84 (s, 3H), 3.66–3.59 (m, 2H), 1.95–1.66 (m, 6H).
(E)-N-(3-Chloro-4-methoxyphenyl)-3-(3,5-dibromo-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (33-THP)
Carboxylic acid 22a (0.20 g, 0.44 mmol) and 3-chloro-4-methoxyaniline (0.11 g, 0.67 mmol, 1.5 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (hexane/EtOAc, 7→60%) to give 33-THP as a light yellow solid (0.20 g, 77%). 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 7.70 (d, J = 2.6 Hz, 1H), 7.53 (s, 2H), 7.44 (dd, J = 2.6, 8.9 Hz, 1H), 6.89 (d, J = 8.9 Hz, 1H), 5.50–5.46 (m, 1H), 3.99 (d, J = 13.2 Hz, 1H), 3.89 (d, J = 13.2 Hz, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.65–3.60 (m, 2H), 1.92–1.66 (m, 6H).
(E)-N-(3-Bromo-4-methoxyphenyl)-3-(3,5-dibromo-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (34-THP)
Carboxylic acid 22a (0.20 g, 0.44 mmol) and 3-bromo-4-methoxyaniline (0.13 g, 0.67 mmol, 1.5 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (heptane/EtOAc, 7→60%) to give 34-THP as a light yellow solid (0.20 g, 71%). 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 7.85 (d, J = 2.6 Hz, 1H), 7.53 (s, 2H), 7.51 (dd, J = 2.6, 8.9 Hz, 1H), 6.86 (d, J = 8.9 Hz, 1H), 5.47 (d, J = 2.9 Hz, 1H), 3.99 (d, J = 13.1 Hz, 1H), 3.92–3.87 (m, 4H), 3.84 (s, 3H), 3.65–3.60 (m, 2H), 1.91–1.66 (m, 6H).
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(3,5-dichlorophenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]-imino]propenamide (35-THP)
Carboxylic acid 22a (0.20 g, 0.44 mmol) and 3,5-dichloroaniline (0.11 g, 0.67 mmol, 1.5 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g and SNAP KP-NH 11g, gradient elution: (heptane/EtOAc, 7→60%). to give 35-THP as a yellow oil (0.18 g, 69%). 1H NMR (400 MHz, CDCl3) δ 8.64 (s, 1H), 7.58 (d, J = 1.8 Hz, 2H), 7.52 (s, 2H), 7.12 (t, J = 1.8 Hz, 1H), 5.48 (t, J = 2.7 Hz, 1H), 3.98 (d, J = 13.2 Hz, 1H), 3.88 (d, J = 13.2 Hz, 1H) 3.84 (s, 3H), 3.65–3.59 (m, 2H), 1.92–1.66 (m, 6H).
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(pyridin-2-yl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino] propanamide (36-THP)
Carboxylic acid 22a (0.20 g, 0.44 mmol) and 2-aminopyridine (0.063 g, 0.67 mmol, 1.5 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (hexane/EtOAc, 12→60%) to give 36-THP as a light yellow solid (0.11 g, 45%). 1H NMR (400 MHz, CDCl3) δ 9.26 (s, 1H), 8.32–8.28 (m, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.76–7.70 (m, 1H), 7.53 (s, 2H), 7.06 (ddd, J = 0.8, 4.9, 7.3 Hz, 1H), 5.47 (t, J = 2.9 Hz, 1H), 3.99 (d, J = 13.2 Hz, 1H), 3.89 (d, J = 13.2 Hz, 1H), 3.83 (s, 3H), 3.64–3,61 (m, 2H), 1.91–1.64 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 160.5, 153.0, 151.8, 150.7, 148.1, 138.6, 134.6, 133.7, 120.2, 118.0, 114.2, 102.3, 62.5, 60.7, 28.8, 28.5, 25.1, 19.0. HRMS (ESI+): calculated 525.9977 (C20H22Br2N3O4), found 525.9972.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(pyridin-3-yl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino] propanamide (37-THP)
Carboxylic acid 22a (0.42 g, 0.94 mmol), 3-aminopyridine (0.11 g, 1.12 mmol, 1.2 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography Biotage SNAP Cartridge KP-Sil 25 g eluent (DCM/MeOH (0→10%) gradient to give the product 37-THP as an oil (0.34 g, 68%). 1H NMR (400 MHz, CDCl3) δ 8.71 (d, J = 10.1 Hz, 1H), 8.38 (s, 1H), 8.20 (ddd, J = 1.4, 2.7, 8.4 Hz, 1H), 7.53 (s, 2H), 7.30 (dd, J = 4.6, 8.4 Hz, 1H), 5.51 (t, J = 2.9 Hz, 1H), 4.06–3.87 (m, 2H), 3.84 (s, 3H), 3.67–3.59 (m, 2H), 1.90–1.59 (m, 6H).
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(pyridin-4-yl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino] propanamide (38-THP)
Carboxylic acid 22a (0.20 g, 0.45 mmol) and 4-aminopyridine (0.040 g, 0.45 mmol) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography Biotage SNAP Cartridge KP-Sil 10 g, eluent (DCM/MeOH, 0→10%) gradient to give the product 38-THP as an oil (0.049 g, 21%). 1H NMR (400 MHz, CDCl3) δ 8.55 (d, J = 5.6 Hz, 2H), 7.64 (d, J = 5.8 Hz, 2H), 7.52 (s, 2H), 5.51 (t, 1H), 4.04–3.86 (m, 2H), 3.84 (s, 3H), 3.66–3.59 (m, 2H), 1.85–1.60 (m, 6H).
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(pyridin-3-ylmethyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino) propanamide (39-THP)
Carboxylic acid 22a (0.20 g, 0.44 mmol) and 3-picolylamine (0.068 mL, 0.67 mmol, 1.5 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (DCM/MeOH, 0→10%) to give 39-THP as an oil (0.20 g, 82%). 1H NMR (400 MHz, CDCl3) δ 8.55 (br, 2H), 7.63 (d, J = 7.8 Hz, 1H), 7.51 (s, 2H), 7.30–7.25 (m, 1H), 7.22 (t, J = 6.1 Hz, 1H), 5.36 (t, J = 2.4 Hz, 1H), 4.61 (dd, J = 6.6, 15.1 Hz, 1H), 4.44 (dd, J = 5.8, 15.1 Hz, 1H), 3.94 (d, J = 13.2 Hz, 1H), 3.88–3.81 (m, 4H), 3.60–3.55 (m, 2H), 1.88–1.62 (m, 6H).
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(4-(dimethylamino)phenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (40-THP)
Carboxylic acid 22a (0.22 g, 0.49 mmol and 4-(dimethylamino)aniline (0.060 g, 0.49 mmol) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (EtOAc/MeOH, 0→20%) to give 40-THP as an oil (0.16 g, 56%). 1H NMR (400 MHz, CDCl3) δ 7.55 (s, 2H), 7.45 (d, J = 9.0 Hz, 2H), 6.72 (d, J = 8.5 Hz, 2H), 5.47 (t, 1H), 4.03-3.89 (m, 2H), 3.83 (s, 3H), 3.65–3.60 (m, 2H), 2.93 (s, 6H), 1.91–1.57 (m, 6H)
(E)-N,3-Bis(3-bromo-4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propenamide (41-THP)
Carboxylic acid 22b (0.40 g, 1.07 mmol) and 3-bromo-4-methoxyaniline (0.12 g, 1.29 mmol, 1.2 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography Biotage SNAP Cartridge KP-Sil 25 g eluent (DCM/MeOH (0→10%) gradient to give 41-THP as an oil (0.43 g, 72%). 1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 7.86 (d, J = 2.6 Hz, 1H), 7.58 (d, J = 2.2 Hz, 1H), 7.49 (dd, J = 2.6, 8.9 Hz, 1H), 7.29 (dd, J = 2.2, 8.4 Hz, 1H), 6.85 (d, J = 8.9 Hz, 1H), 6.80 (d, J = 8.5 Hz, 1H), 5.46 (t, J = 2.8 Hz, 1H), 3.99 (d, J = 13.0 Hz, 1H), 3.91 (d, J = 13.0 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.71–3.58 (m, 2H), 1.92–1.60 (m, 6H).
(E)-3-(3-Bromo-4-methoxyphenyl)-N-(pyridin-2-yl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (42-THP)
Carboxylic acid 22b (0.40 g, 1.07 mmol) and 2-aminopyridine (0.12 g, 1.29 mmol, 1.2 equiv.) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography Biotage SNAP Cartridge KP-Sil 25 g eluent (DCM/MeOH (0→10%) gradient to give 42-THP as an oil (0.31 g, 64%). 1H NMR (400 MHz, CDCl3) δ 9.27 (s, 1H), 8.29 (ddd, J = 0.9, 2.0, 4.9 Hz, 1H), 8.23 (dt, J = 1.0, 8.4 Hz, 1H), 7.70 (ddd, J = 1.9, 7.4, 8.5 Hz, 1H), 7.57 (d, J = 2.2 Hz, 1H), 7.07–7.01 (m, 1H), 6.80 (d, J = 8.4 Hz, 1H), 5.47–5.44 (m, 1H), 3.99 (d, J = 13.1 Hz, 1H), 3.91 (d, J = 13.1 Hz, 1H), 3.84 (s, 3H), 3.68–3.60 (m, 2H), 1.90–-1.61 (m, 6H).
(E)-N-(3,4-Dichlorobenzyl)-3-(4-methoxyphenyl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (43-THP)
Carboxylic acid 22d (0.40 g, 1.36 mmol) and 3,4-dichlorobenzylamine (0.11 g, 1.12 mmol, 1.2 equiv.) were reacted according to the general procedure for amide coupling. 43-THP was obtained as light brown solid (0.54 g, 87%). 1H NMR (400 MHz CDCl3) δ 7.37 (d, J = 8.2 Hz, 1H), 7.34 (d, J = 2.1 Hz, 1H), 7.24 (s, 1H), 7.11–7.06 (m, 1H), 6.83–6.79 (m, 2H), 5.38–5.35 (m, 1H), 4.52 (dd, J = 6.7, 15.3 Hz, 1H), 4.34 (dd, J = 5.8, 15.2 Hz, 1H), 3.95 (d, J = 13.1 Hz, 1H), 3.85 (d, J = 13.1 Hz, 1H), 3.77 (s, 3H), 3.69–3.55 (m, 2H), 1.88–1.54 (m, 6H).
(E)-3-(4-Methoxyphenyl)-N-(pyridin-2-yl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (44-THP)
Carboxylic acid 22d (0.40 g, 1.37 mmol), 2-aminopyridine (0.13 g, 1.37 mmol) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography Biotage SNAP Cartridge KP-Sil 25 g and 10 g, eluent (EtOAc/MeOH, 0→10%) gradient to give 44-THP as an oil (0.095 g, 19%). 1H NMR (400 MHz, CD3OD) δ 8.28 (ddd, J = 5.0, 1.9, 0.9 Hz, 1H), 8.18 (dt, J = 8.4, 1.0 Hz, 1H), 7.81 (ddd, J = 8.4, 7.4, 1.9 Hz, 1H), 7.25 (d, J = 8.8 Hz, 2H), 7.13 (ddd, J = 7.4, 5.0, 1.0 Hz, 1H), 6.83 (d, J = 8.8 Hz, 2H), 3.96-3.79 (m, 2H), 3.73 (s, 3H), 3.66–3.48 (m, 2H), 1.91–1.46 (m, 6H).
(E)-3-(3-Chloro-4-methoxyphenyl)-N-(pyridin-2-yl)-2-[[(tetrahydro-2H-pyran-2-yl)oxy]imino]propanamide (45-THP)
Carboxylic acid 22c (0.32 g, 0.98 mmol) and 2-aminopyridine (0.093 g, 0.98 mmol) were reacted according to the general procedure for amide coupling. The product was purified by column chromatography Biotage SNAP Cartridge KP-Sil 10 g, eluent (DCM/MeOH, 0→10%) gradient to give 45-THP as an oil (0.29 g, 74%). 1H NMR (400 MHz, CDCl3) δ 8.28 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 8.24 (dt, J = 8.4, 1.0 Hz, 1H), 7.71 (ddd, J = 8.4, 7.4, 1.9 Hz, 1H), 7.40 (d, J = 2.2 Hz, 1H), 7.22 (dd, J = 8.4, 2.2 Hz, 1H), 7.04 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 6.82 (d, J = 8.5 Hz, 1H), 5.46 (t, 1H), 3.99 (d, J = 13.1 Hz, 1H), 3.91 (d, J = 13.1 Hz, 1H), 3.85 (s, 3H), 3.68–3.56 (m, 2H), 1.90–1.57 (m, 6H).
General Method for THP Deprotection
THP ethers 30-THP–45-THP (0.16 g, 0.27 mmol) and TFA (3 mL) were dissolved in dry DCM (7 mL). The reaction mixture was stirred under argon atmosphere for 3 d. Subsequently, the reaction mixture was quenched with a 2 M solution of NaOH in H2O (15 mL) and extracted with DCM (2 × 15 mL). The combined organic layers were dried over anhyd. Na2SO4, filtered and concentrated in vacuo. The crude products were purified using flash column chromatography.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(3,4-dichlorophenyl)-2-(hydroxyimino)propanamide (30)
The general procedure for THP deprotection was used, starting from ether 30 (0.16 g, 0.27 mmol). The worked-up reaction mixture was attempted to be purified using column chromatography Biotage SNAP Cartridge KP-Sil 25 g and SNAP Ultra 10 g without success. A successful purification was achieved by using Biotage SNAP Cartridge KP-NH 11 g, gradient elution: (heptane/EtOAc, 12→100%). The obtained oil was recrystallized from CHCl3 to give 30 as a white solid (0.052 g, 36%). Mp: 198 °C (decomposed).1H NMR (400 MHz, d6-DMSO) δ 12.38 (s, 1H), 10.30 (s, 1H), 8.08 (d, J = 2.4 Hz, 1H), 7.71 (dd, J = 2.5, 8.9 Hz, 1H), 7.57 (d, J = 8.8 Hz, 1H), 7.50 (s, 2H), 3.86 (s, 2H), 3.76 (s, 3H). 13C NMR (101 MHz, d6-DMSO) δ 162.2, 151.9, 151.3, 138.4, 136.0, 132.9, 130.8, 130.5, 125.3, 121.4, 120.3, 117.2, 60.4, 27.9. HRMS (ESI+): calculated 508.8670 (C16H13Br2Cl2N2O3), found 508.8670.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-(3,4-dichlorobenzyl)-2-(hydroxyimino)propanamide (31)
Unlike in the general procedure, compound 31-THP (0.20 g, 0.33 mmol) was deprotected using a 2 M solution of HCl in Et2O (5 mL) in dry DCM (5 mL) under various conditions (sealed tube, 2 h, 60 °C, MW; sealed tube, 2 h, 70 °C, oil bath; sealed tube, 12 h, 30 °C, oil bath; reflux under argon, 60 h). The worked-up reaction mixture was purified twice by column chromatography, using Biotage SNAP KP-Sil 25 g, gradient elution: (heptane/EtOAc, 12→100%) to give 31 as a light yellow solid (0.038 g, 22%). 1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H), 7.49 (s, 2H), 7.39 (d, J = 8.2 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.10 (dd, J = 2.0, 8.2 Hz, 1H), 7.02 (t, J = 5.5 Hz, 1H), 4.45 (d, J = 6.2 Hz, 2H), 3.90 (s, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 162.5, 153.0, 152.6, 138.1, 134.6, 133.6, 132.9, 131.9, 130.9, 129.7, 127.1, 118.1, 60.7, 42.6, 28.1. HRMS (ESI+): calculated 522.8825 (C17H15Br2Cl2N2O3), found 522.8827.
![Marinedrugs 16 00481 i036]()
(E)-N-[4-Chloro-3-(trifluoromethyl)phenyl]-3-(3,5-dibromo-4-methoxyphenyl)-2-(hydroxyimino) propanamide (32)
The general procedure for THP deprotection was employed to deprotect 32-THP (0.19 g, 0.30 mmol). The worked-up reaction mixture was purified by column chromatography, Biotage SNAP Cartridge KP-NH 11 g, gradient elution: (heptane/EtOAc, 12→100%) to give 32 as a light yellow solid (0.055 g, 33%). 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 1H), 8.13 (s, 1H), 7.90 (d, J = 2.6 Hz, 1H), 7.77 (dd, J = 2.6, 8.7 Hz, 1H), 7.51 (s, 2H), 7.46 (d, J = 8.7 Hz, 1H), 3.95 (s, 2H), 3.84 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.2, 153.0, 152.4, 135.9, 134.1, 133.5, 132.1, 129.1, 128.8, 127.2 (m), 123.6, 118.7 (q, JCF = 5.7), 118.0, 60.6, 27.7. HRMS (ESI+): calculated 542.8934 (C17H13Br2ClF3N2O3), found 542.8937.
(E)-N-(3-Chloro-4-methoxyphenyl)-3-(3,5-dibromo-4-methoxyphenyl)-2-(hydroxyimino)propanamide (33)
The general procedure for THP deprotection was used, starting from ether 33-THP (0.20 g, 0.34 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-NH 11 g, gradient elution: (cyclohexane/EtOAc, 12→100%) to give 33 as a light orange solid (0.086 g, 50%). 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 8.07 (s, 1H), 7.64 (d, J = 2.6 Hz, 1H), 7.52 (s, 2H), 7.42 (dd, J = 2.6, 8.9 Hz, 1H), 6.88 (d, J = 8.9 Hz, 1H), 3.94 (s, 2H), 3.88 (s, 3H), 3.84 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.0, 153.0, 152.7, 152.2, 134.5, 133.6, 130.8, 122.8, 122.4, 119.6, 118.1, 112.4, 60.7, 56.6, 27.9. HRMS (ESI+): calculated 504.9165 (C17H16Br2ClN2O4), found 504.9164.
(E)-N-(3-Bromo-4-methoxyphenyl)-3-(3,5-dibromo-4-methoxyphenyl)-2-(hydroxyimino)propanamide (34)
The general procedure for THP deprotection was used, starting from ether 34-THP (0.20 g, 0.31 mmol). The worked-up reaction mixture was attempted to be purified using Biotage SNAP Cartridge KP-Sil 25 g and SNAP Ultra 10 g. A successful purification was achieved by column chromatography using Biotage SNAP Cartridge KP-NH 11 g, gradient elution: (heptane/EtOAc, 12→100%) to give 34 as a yellow oily solid (0.087 g, 50%). 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.42 (s, 1H), 7.78 (d, J = 2.6 Hz, 1H), 7.52 (s, 2H), 7.48 (dd, J = 2.6, 8.9 Hz, 1H), 6.85 (d, J = 8.9 Hz, 1H), 3.94 (s, 2H), 3.86 (s, 3H), 3.83 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.1, 153.2, 152.9, 152.6, 134.6, 133.6, 131.1, 125.4, 120.4, 118.1, 112.2, 111.8, 60.7, 56.6, 27.9. HRMS (ESI+): calculated 548.8660 (C17H16Br3N2O4), found 548.8660.
(E)-3-(3,5-Dibromo-4-methxyphenyl)-N-(3,5-dichlorophenyl)-2-(hydroxyimino)propanamide (35)
The general procedure for THP deprotection was used, starting from ether 35-THP (0.18 g, 0.30 mmol). The worked-up reaction mixture was purified by column chromatography, Biotage SNAP Cartridge KP-NH 11 g, gradient elution: (heptane/EtOAc, 12→100%) to give 35 as a white solid (0.021 mg, 14%). 1H NMR (400 MHz, CD3OD) δ 7.71 (d, J = 1.8 Hz, 2H), 7.53 (s, 2H), 7.14 (t, J = 1.9 Hz, 1H), 3.90 (s, 2H), 3.81 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 162.2, 152.6, 150.9, 140.2, 135.7, 134.7, 133.2, 123.2, 118.0, 117.3, 59.6, 27.3. HRMS (ESI+): calculated 508.8670 (C16H13Br2Cl2N2O3), found 508.8659.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-2-(hydroxyimino)-N-(pyridin-2-yl)propanamide (36)
The general procedure for THP deprotection was used, starting from ether 36-THP (0.087 g, 0.17 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-NH 11 g, gradient elution: (hexane/EtOAc, 12→100%). The obtained 36 as a white powder was further re-crystallized from CHCl3 for the X-ray to give transparent crystals (0.037 g, 51%). M.p.: 192–196 °C (decomposed). 1H NMR (400 MHz, CDCl3) δ 13.85 (s, 1H), 9.41 (s, 1H), 8.39 (d, J = 8.5 Hz, 1H), 8.32–8.27 (m, 1H), 7.89–7.84 (m, 1H), 7.57 (s, 2H), 7.19 (ddd, J = 0.8, 5.2, 7.3 Hz, 1H), 4.02 (s, 2H), 3.84 (s, 3H. 13C NMR (101 MHz, CDCl3) δ 161.7, 152.8, 151.7, 150.1, 146.4, 140.2, 135.2, 133.7, 120.4, 118.0, 115.7, 60.7, 28.2. HRMS (ESI+): calculated 441.9402 (C15H14Br2N3O3), found 441.9401. LC-MS: [M + H]+ m/z 442 (tR = 5.39 min), >99%.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-2-(hydroxyimino)-N-(pyridin-3-yl)propanamide (37)
The general procedure for THP deprotection was used, starting from ether 37-THP (0.34 g, 0.64 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (hexane/acetone, 0→100%) to give 37 as a pale yellow solid (0.052 g, 18%). 1H NMR (400 MHz, CD3OD) δ 8.84 (dd, J = 2.5, 19.4 Hz, 1H), 8.30–8.16 (m, 2H), 7.53 (d, J = 8.4 Hz, 2H), 7.44–7.35 (m, 1H), 3.92 (s, 2H), 3.80 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 163.8, 154.4, 153.9, 152.3, 145.3, 142.3, 137.1, 134.44, 129.41, 125.2, 118.7, 61.0, 28.7. HRMS (ESI+): calculated 441.9402 (C15H14N3O3Br2), found 441.9402.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)2-(hydroxyimino)-N-(pyridin-4-yl)propanamide (38)
The general procedure for THP deprotection was used, starting from ether 38-THP (0.49 g, 0.093 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 10 g, gradient elution: (DCM/MeOH, 0→10%) to give 38 as a brownish solid (0.0099 g, 24%). 1H NMR (400 MHz, d6-acetone) δ 8.47 (s, 2H), 7.73 (d, J = 5.5 Hz, 2H), 7.59 (s, 2H), 3.98 (s, 2H), 3.82 (s, 3H). 13C NMR (101 MHz, d6-acetone) δ 162.9, 153.5, 152.2, 151.3, 145.8, 136.7, 134.3, 118.3, 114.5, 60.9, 28.4. HRMS (ESI+): calculated 441.9402 (C15H14N3O3Br2), found 441.9400.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-2-(hydroxyimino)-N-(pyridin-3-ylmethyl)propanamide (39)
The general procedure for THP deprotection was used, starting from ether 39-THP (0.17 g, 0.32 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-NH 11 g, gradient elution: (DCM/MeOH, 0→10%) to give 39 as a yellowish solid (0.039 g, 26%). 1H NMR (400 MHz, CDCl3) δ 8.52 (br s, 2H), 7.75 (d, J = 7.8 Hz, 1H), 7.52 (s, 2H), 7.39–7.33 (m, 1H), 7.16 (t, J = 6.1 Hz, 1H), 4.54 (d, J = 6.1 Hz, 2H), 3.91 (s, 2H), 3.84 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 163.3, 152.7, 151.1, 148.0, 147.8, 137.5, 135.4, 134.7, 133.6, 124.4, 118.0, 60.7, 41.0, 28.2. HRMS (ESI+): calculated 455.9558 (C16H16Br2N3O3), found 455.9563.
(E)-3-(3,5-Dibromo-4-methoxyphenyl)-N-[4-(dimethylamino)phenyl]-2-(hydroxyimino)propanamide (40)
The general procedure for THP deprotection was used, starting from ether 40-THP (0.16 g, 0.27 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (hexane/EtOAc, 7→60%). The obtained product was further re-crystallized from hexane and acetone to give 40 as a yellow solid (0.07 g, 50%). 1H NMR (400 MHz, d6-acetone) δ 7.61 (s, 2H), 7.56 (d, J = 9.1 Hz, 2H), 6.72 (d, J = 9.2 Hz, 2H), 3.96 (s, 2H), 3.82 (s, 3H), 2.90 (s, 6H). 13C NMR (101 MHz, d6-acetone) δ 161.2, 153.4, 153.0, 148.8, 137.2, 134.4, 129.1, 122.0, 118.2, 113.5, 60.9, 40.9, 28.5. HRMS (ESI+): calculated 483.9871 (C18H20N3O3Br2), found 483.9876.
(E)-N,3-Bis(3-bromo-4-methoxyphenyl)-2-(hydroxyimino)propanamide (41)
The general procedure for THP deprotection was used, starting from ether 41-THP (0.43 g, 0.78 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (hexane/EtOAc, 0→60%) to give 41 as a yellow solid (0.087 g, 24%). 1H NMR (400 MHz, CD3OD) δ 7.85 (d, J = 2.5 Hz, 1H), 7.51–7.45 (m, 2H), 7.24 (dd, J = 2.2, 8.5 Hz, 1H), 6.92 (d, J = 8.9 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 3.88 (s, 2H), 3.81 (s, 3H), 3.78 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 163.4, 155.9, 154.1, 153.3, 134.8, 133.0, 131.6, 130.5, 126.6, 121.9, 113.11, 113.09, 112.2, 112.1, 56.8, 56.6, 28.7. HRMS (ESI+): calculated 470.9555 (C17H17N2O4Br2), found 470.9554.
(E)-3-(3-Bromo-4-methoxyphenyl)-2-(hydroxyimino)-N-(pyridin-2-yl)propanamide (42)
The general procedure for THP deprotection was used, starting from ether 42-THP (0.31 g, 0.68 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (DCM/MeOH, 0→10%). The obtained solid was further re-crystallized from hexane to give 42 as a white solid (0.062 g, 25%). M.p.: 214–7 °C (decomposed). 1H NMR (400 MHz, d6-DMSO) δ 12.47 (s, 1H), 9.55 (s, 1H), 8.33 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 8.07 (dt, J = 8.3, 1.0 Hz, 1H), 7.83 (ddd, J = 8.5, 7.3, 1.9 Hz, 1H), 7.45 (d, J = 2.1 Hz, 1H), 7.23 (dd, J = 8.5, 2.2 Hz, 1H), 7.16 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H), 7.03 (d, J = 8.5 Hz, 1H), 3.84 (s, 2H), 3.80 (s, 3H). 13C NMR (101 MHz, d6-DMSO) δ 161.6, 153.9, 151.1, 150.6, 148.3, 138.5, 133.0, 130.0, 129.3, 120.0, 113.4, 112.7, 110.3, 56.2, 27.4. HRMS (ESI+): calculated 364.0297 (C15H15N3O3Br), found 364.0299.
(E)-N-(3,4-Dichlorobenzyl)-2-(hydroxyimino)-3-(4-methoxyphenyl)propanamide (43)
The general procedure for THP deprotection was used, starting from ether 43-THP (0.53 g, 1.18 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 25 g, gradient elution: (heptane/EtOAc, 0→100%) to give 43 as a yellow solid (0.057 g, 13%). 1H NMR (400 MHz, CD3OD) δ 7.39 (d, J = 8.3 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.22–7.15 (m, 2H), 7.13–7.07 (m, 1H), 6.82–6.72 (m, 2H), 4.36 (s, 2H), 3.87 (s, 2H), 3.74 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 166.3, 159.7, 153.9, 141.1, 133.2, 131.7, 131.5, 131.1, 130.3, 129.9, 128.1, 114.8, 55.6, 42.7, 29.2. HRMS (ESI+): calculated 367.0616 (C17H17N2O3Cl2), found 367.0615.
(E)-2-(Hydroxyimino)-3-(4-methoxyphenyl)-N-(pyridin-2-yl)propanamide (44)
The general procedure for THP deprotection was used, starting from ether 44-THP (0.095 g, 0.26 mmol). The product was purified by column chromatography, Biotage SNAP Cartridge KP-Sil 10 g, gradient elution: (DCM/MeOH, 2→10%). The obtained product was further re-crystallized from hexane and acetone to give 44 as a white solid (0.02 g, 28%). 1H NMR (400 MHz, CD3OD) δ 8.26 (ddd, J = 5.0, 1.9, 0.9 Hz, 1H), 8.19 (dt, J = 8.4, 1.0 Hz, 1H), 7.81 (ddd, J = 8.4, 7.4, 1.9 Hz, 1H), 7.30–7.21 (m, 2H), 7.12 (ddd, J = 7.4, 5.0, 1.1 Hz, 1H), 6.85–6.76 (m, 2H), 3.92 (s, 2H), 3.74 (s, 3H). 13C NMR (101 MHz, CD3OD) δ 163.4, 159.7, 153.2, 152.3, 149.0, 140.0, 131.2, 129.9, 121.1, 115.1, 114.8, 55.6, 28.6. HRMS (ESI+): calculated 286.1192 (C15H16N3O3), found 286.1195.
(E)-3-(3-Chloro-4-methoxyphenyl)-2-(hydroxyimino)-N-(pyridin-2-yl)propanamide (45)
The general procedure for THP deprotection was used, starting from ether 45-THP (0.29 g, 0.72 mmol). The obtained product was further re-crystallized from acetone to give 45 as a white solid (0.05 g, 20%). M.p.: 218–219 °C (decomposed) 1H NMR (400 MHz, d6-acetone) δ 8.29 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 8.20 (d, J = 8.3 Hz, 1H), 7.84–7.78 (m, 1H), 7.42–7.40 (m, 1H), 7.29 (ddt, J = 8.4, 2.2, 0.6 Hz, 1H), 7.12 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 7.02 (d, J = 8.5 Hz, 1H), 3.96 (s, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, d6-acetone) δ 162.0, 154.7, 152.8, 151.9, 149.2, 139.1, 131.5, 130.6, 129.7, 122.4, 120.7, 114.0, 113.3, 56.4, 28.2. HRMS (ESI+): calculated 320.0802 (C15H15N3O3Cl), found 320.0802.

 and
and 

























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















































