Nile Tilapia Derived TP4 Shows Broad Cytotoxicity toward to Non-Small-Cell Lung Cancer Cells

Abstract
:1. Introduction
2. Results
2.1. Cationic TP4 is Toxic to NSCLC Cells
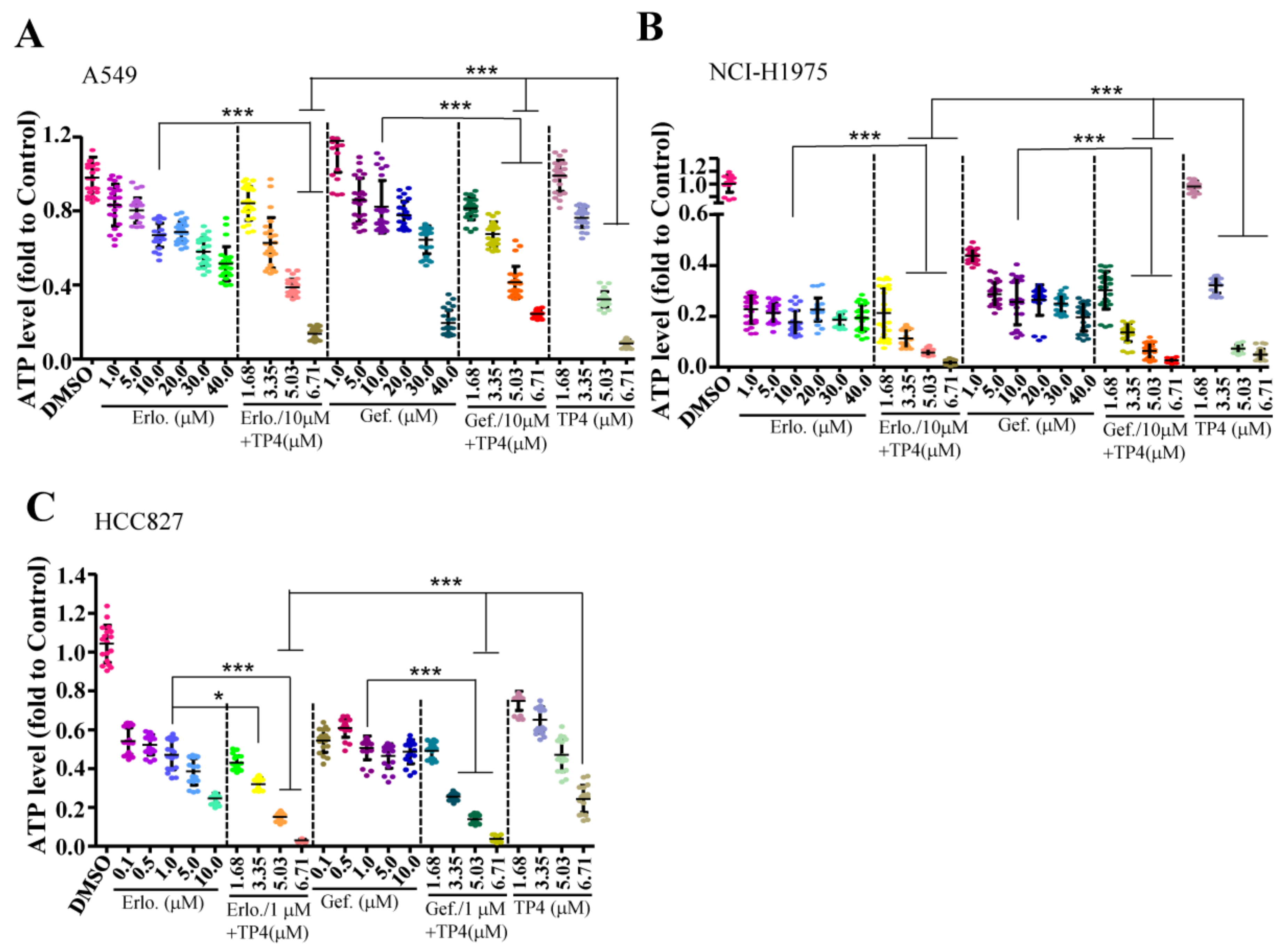
2.2. Combining TP4 with Potent EGFR Tyrosine Kinase Inhibitors (TKIs) Enhances Toxicity
2.3. TP4 Induces Necrotic Death in NSCLC Cells
3. Discussion
4. Material and Methods
4.1. Reagents and Peptide Sequence Analysis
4.2. Cell Culture and Cell Viability Assay
4.3. Western Blotting
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975-2015, National Cancer Institute. Bethesda, MD. Available online: https://seer.cancer.gov/csr/1975_2015/ (accessed on 10 September 2018).
- Langer, C.J.; Besse, B.; Gualberto, A.; Brambilla, E.; Soria, J.C. The evolving role of histology in the management of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 5311–5320. [Google Scholar] [CrossRef]
- Davidson, M.R.; Gazdar, A.F.; Clarke, B.E. The pivotal role of pathology in the management of lung cancer. J. Thorac. Dis. 2013, 5 (Suppl. 5), S463–S478. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef]
- Shigematsu, H.; Takahashi, T.; Nomura, M.; Majmudar, K.; Suzuki, M.; Lee, H.; Wistuba, I.I.; Fong, K.M.; Toyooka, S.; Shimizu, N.; et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005, 65, 1642–1646. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; Haggstrom, D.; Felip, E.; Kim, J.H.; Frewer, P.; Cantarini, M.; Brown, K.H.; Dickinson, P.A.; Ghiorghiu, S.; Ranson, M. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Sequist, L.V.; Soria, J.C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1700–1709. [Google Scholar] [CrossRef]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Janne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef]
- Romanidou, O.; Landi, L.; Cappuzzo, F.; Califano, R. Overcoming resistance to first/second generation epidermal growth factor receptor tyrosine kinase inhibitors and ALK inhibitors in oncogene-addicted advanced non-small cell lung cancer. Ther. Adv. Med. Oncol. 2016, 8, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Shih, J.Y.; Gow, C.H.; Yang, P.C. EGFR mutation conferring primary resistance to gefitinib in non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 207–208. [Google Scholar] [CrossRef]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; Ladanyi, M.; Miller, V.A.; Pao, W. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Tang, Z.; Du, R.; Jiang, S.; Wu, C.; Barkauskas, D.S.; Richey, J.; Molter, J.; Lam, M.; Flask, C.; Gerson, S.; et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br. J. Cancer 2008, 99, 911–922. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Smit, E.F.; Groen, H.J.; Horn, L.; Gettinger, S.; Camidge, D.R.; Riely, G.J.; Wang, B.; Fu, Y.; Chand, V.K.; et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014, 4, 1036–1045. [Google Scholar] [CrossRef]
- Soria, J.C.; Wu, Y.L.; Nakagawa, K.; Kim, S.W.; Yang, J.J.; Ahn, M.J.; Wang, J.; Yang, J.C.; Lu, Y.; Atagi, S.; et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): A phase 3 randomised trial. Lancet Oncol. 2015, 16, 990–998. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Doucette, C.D.; Pinto, D.M.; Patrzykat, A.; Douglas, S.; Hoskin, D.W. Pleurocidin-family cationic antimicrobial peptides are cytolytic for breast carcinoma cells and prevent growth of tumor xenografts. Breast Cancer Res. 2011, 13, R102. [Google Scholar] [CrossRef]
- Ting, C.H.; Chen, Y.C.; Wu, C.J.; Chen, J.Y. Targeting FOSB with a cationic antimicrobial peptide, TP4, for treatment of triple-negative breast cancer. Oncotarget 2016, 7, 40329–40347. [Google Scholar] [CrossRef] [Green Version]
- Ting, C.H.; Huang, H.N.; Huang, T.C.; Wu, C.J.; Chen, J.Y. The mechanisms by which pardaxin, a,natural cationic antimicrobial peptide, targets the endoplasmic reticulum and induces c-FOS. Biomaterials 2014, 35, 3627–3640. [Google Scholar] [CrossRef]
- Peng, K.C.; Lee, S.H.; Hour, A.L.; Pan, C.Y.; Lee, L.H.; Chen, J.Y. Five Different Piscidins from Nile Tilapia, Oreochromis niloticus: Analysis of Their Expressions and Biological Functions. PLoS ONE 2012, 7, e50263. [Google Scholar] [CrossRef]
- Ayyub, A.; Saleem, M.; Fatima, I.; Tariq, A.; Hashmi, N.; Musharraf, S.G. Glycosylated Alpha-1-acid glycoprotein 1 as a potential lung cancer serum biomarker. Int. J. Biochem. Cell Biol. 2016, 70, 68–75. [Google Scholar] [CrossRef]
- Kucharska-Newton, A.M.; Rosamond, W.D.; Schroeder, J.C.; McNeill, A.M.; Coresh, J.; Folsom, A.R. HDL-cholesterol and the incidence of lung cancer in the Atherosclerosis Risk in Communities (ARIC) study. Lung Cancer 2008, 61, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Paredes-Gamero, E.J.; Martins, M.N.; Cappabianco, F.A.; Ide, J.S.; Miranda, A. Characterization of dual effects induced by antimicrobial peptides: Regulated cell death or membrane disruption. Biochim. Biophys. Acta 2012, 1820, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Aarbiou, J.; Tjabringa, G.S.; Verhoosel, R.M.; Ninaber, D.K.; White, S.R.; Peltenburg, L.T.; Rabe, K.F.; Hiemstra, P.S. Mechanisms of cell death induced by the neutrophil antimicrobial peptides alpha-defensins and LL-37. Inflamm. Res. 2006, 55, 119–127. [Google Scholar] [CrossRef]
- Tokumaru, S.; Sayama, K.; Shirakata, Y.; Komatsuzawa, H.; Ouhara, K.; Hanakawa, Y.; Yahata, Y.; Dai, X.; Tohyama, M.; Nagai, H.; et al. Induction of keratinocyte migration via transactivation of the epidermal growth factor receptor by the antimicrobial peptide LL-37. J. Immunol. 2005, 175, 4662–4668. [Google Scholar] [CrossRef]
- Kuo, H.M.; Tseng, C.C.; Chen, N.F.; Tai, M.H.; Hung, H.C.; Feng, C.W.; Cheng, S.Y.; Huang, S.Y.; Jean, Y.H.; Wen, Z.H. MSP-4, an Antimicrobial Peptide, Induces Apoptosis via Activation of Extrinsic Fas/FasL- and Intrinsic Mitochondria-Mediated Pathways in One Osteosarcoma Cell Line. Mar. Drugs 2018, 16, 8. [Google Scholar] [CrossRef]
- Attoub, S.; Arafat, H.; Mechkarska, M.; Conlon, J.M. Anti-tumor activities of the host-defense peptide hymenochirin-1B. Regul. Pept. 2013, 187, 51–56. [Google Scholar] [CrossRef]
- Mechkarska, M.; Attoub, S.; Sulaiman, S.; Pantic, J.; Lukic, M.L.; Conlon, J.M. Anti-cancer, immunoregulatory, and antimicrobial activities of the frog skin host-defense peptides pseudhymenochirin-1Pb and pseudhymenochirin-2Pa. Regul. Pept. 2014, 194-195, 69–76. [Google Scholar] [CrossRef]
- Liu, S.; Yang, H.; Wan, L.; Cai, H.W.; Li, S.F.; Li, Y.P.; Cheng, J.Q.; Lu, X.F. Enhancement of cytotoxicity of antimicrobial peptide magainin II in tumor cells by bombesin-targeted delivery. Acta Pharmacol. Sin. 2011, 32, 79–88. [Google Scholar] [CrossRef]
- Baker, M.A.; Maloy, W.L.; Zasloff, M.; Jacob, L.S. Anticancer Efficacy of Magainin2 and Analog Peptides. Cancer Res. 1993, 53, 3052–3057. [Google Scholar]
- Jiang, Z.Q.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef]
- Ting, C.H.; Liu, Y.C.; Lyu, P.C.; Chen, J.Y. Nile Tilapia Derived Antimicrobial Peptide TP4 Exerts Antineoplastic Activity Through Microtubule Disruption. Mar. Drugs 2018, 16, 462. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | IC50 (μM) | 95% Confidence Interval |
|---|---|---|
| BEAS-2B | ||
| 3 h | 33.100 ± 1.032 | 31.11 to 35.22 |
| 6 h | 32.040 ± 1.048 | 29.21 to 35.15 |
| 12 h | 29.760 ± 1.074 | 25.88 to 34.22 |
| 24 h | 26.500 ± 1.086 | 22.53 to 31.17 |
| MRC-5 | ||
| 3 h | 46.440 ± 1.049 | 42.32 to 50.96 |
| 6 h | 28.000 ± 1.068 | 24.65 to 31.80 |
| 12 h | 18.220 ± 1.079 | 15.67 to 21.17 |
| 24 h | 15.480 ± 1.063 | 13.72 to 17.46 |
| Treatment | IC50 (μM) | 95% Confidence Interval |
|---|---|---|
| A549 | ||
| 3 h | 27.620 ± 1.402 | 14.25 to 53.55 |
| 6 h | 17.080 ± 1.236 | 11.27 to 25.89 |
| 12 h | 4.089 ± 1.130 | 3.216 to 5.200 |
| 24 h | 1.922 ± 1.112 | 1.560 to 2.367 |
| NCI-H661 | ||
| 3 h | 14.170 ± 1.169 | 10.44 to 19.24 |
| 6 h | 11.890 ± 1.102 | 9.824 to 14.38 |
| 12 h | 5.276 ± 1.111 | 4.289 to 6.489 |
| 24 h | 3.769 ± 1.113 | 3.054 to 4.652 |
| NCI-H1975 | ||
| 3 h | 5.472 ± 1.112 | 4.445 to 6.737 |
| 6 h | 3.262 ± 1.143 | 2.513 to 4.236 |
| 12 h | 2.755 ± 1.202 | 1.920 to 3.954 |
| 24 h | 1.241 ± 1.174 | 0.9065 to 1.698 |
| HCC827 | ||
| 3 h | 18.520 ± 1.304 | 11.00 to 31.16 |
| 6 h | 18.370 ± 1.153 | 13.89 to 24.29 |
| 12 h | 16.070 ± 1.154 | 12.12 to 21.29 |
| 24 h | 10.610 ± 1.146 | 8.118 to 13.86 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ting, C.-H.; Chen, J.-Y. Nile Tilapia Derived TP4 Shows Broad Cytotoxicity toward to Non-Small-Cell Lung Cancer Cells. Mar. Drugs 2018, 16, 506. https://doi.org/10.3390/md16120506
Ting C-H, Chen J-Y. Nile Tilapia Derived TP4 Shows Broad Cytotoxicity toward to Non-Small-Cell Lung Cancer Cells. Marine Drugs. 2018; 16(12):506. https://doi.org/10.3390/md16120506
Chicago/Turabian StyleTing, Chen-Hung, and Jyh-Yih Chen. 2018. "Nile Tilapia Derived TP4 Shows Broad Cytotoxicity toward to Non-Small-Cell Lung Cancer Cells" Marine Drugs 16, no. 12: 506. https://doi.org/10.3390/md16120506
APA StyleTing, C. -H., & Chen, J. -Y. (2018). Nile Tilapia Derived TP4 Shows Broad Cytotoxicity toward to Non-Small-Cell Lung Cancer Cells. Marine Drugs, 16(12), 506. https://doi.org/10.3390/md16120506