2. Results and Discussion
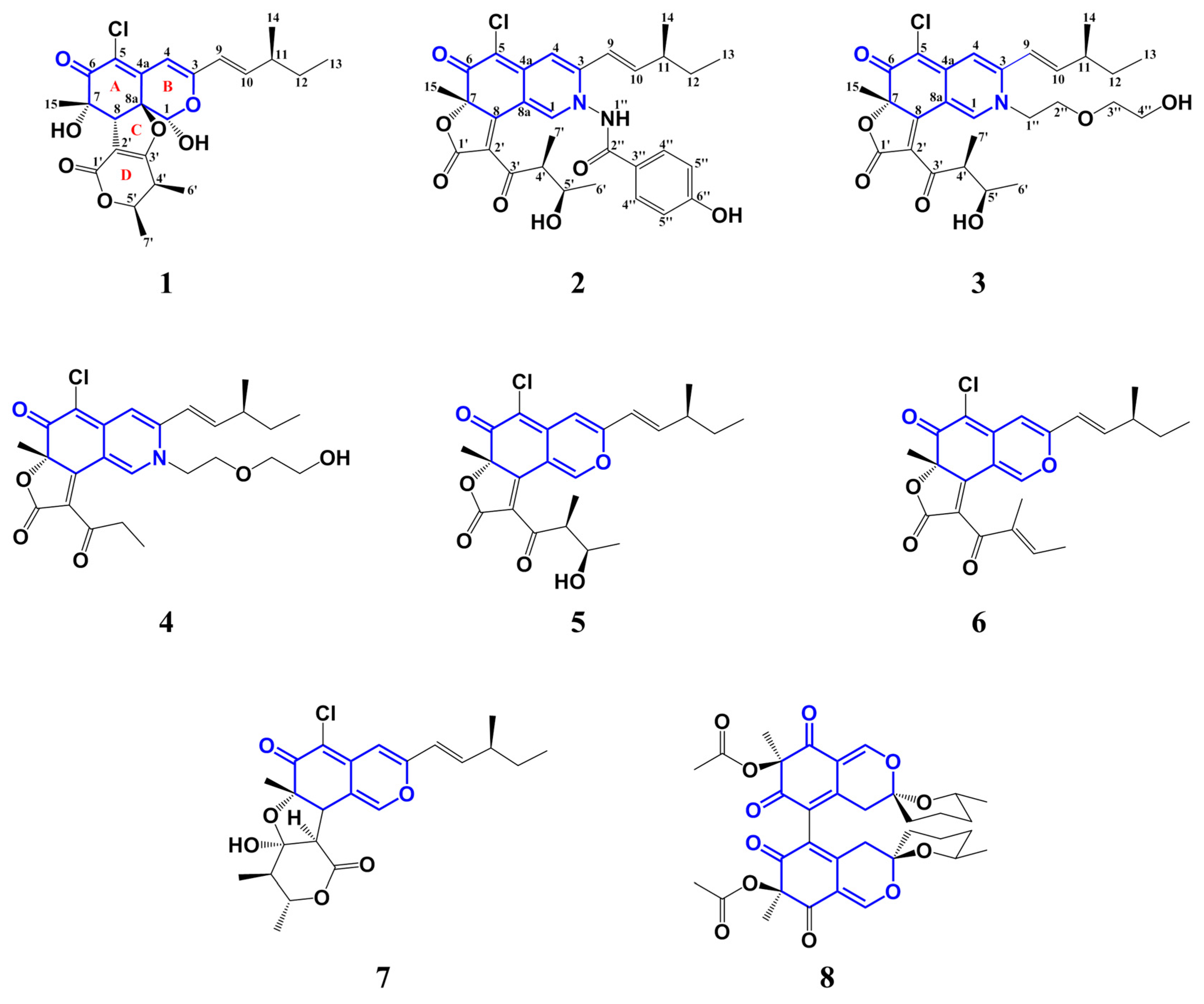
Chaephilone C (1), chaetoviridides A–C (2–4) were characterized as azaphilone derivatives with a chlorine atom at C-5, a methyl unit at C-7 and a branched pentenyl side chain at C-3. Chromium trioxide oxidation of compounds 1–5 gave 2-methylbutanoic acid showing (+)-rotation. Thus, the stereochemistry of C-11 for compounds 1–4 in the pentenyl side chain was established as (S).
Chaephilone C (
1) was obtained as a yellow amorphous solid. Its molecular formula was established as C
23H
27O
7Cl by high-resolution electrospray ionization mass spectroscopy (HR-ESI-MS) (
m/
z 449.1373 [M − H]
−; calcd. for C
23H
26O
7Cl, 449.1367; ∆ + 1.3 ppm) and the ratio of isotope peaks ([M − H]
−/[M − H + 2]
−), implying ten degrees of unsaturation. The
13C/DEPT and HSQC spectrum revealed the presence of one primary methyl group (C-13), three secondary methyl groups (C-6′, C-7′ and C-14), one tertiary methyl group (C-15), one methylene group (C-12), five sp
3-hybridized methine groups (C-1, C-4′, C-5′, C-8 and C-11) including two oxygen-bearing carbons (C-1 and C-5′), three sp
2-hybridized methine groups (C-4, C-9 and C-10), two sp
3-hybridized quaternary oxygen-bearing carbons (C-7 and C-8a), five sp
2-hybridized quaternary carbons (C-3, C-4a, C-5, C-2′ and C-3′) including one oxygen-bearing carbon (C-3′) and two carbonyl carbons (C-6 and C-1′) (
Figures S3 and S6). The
1H-
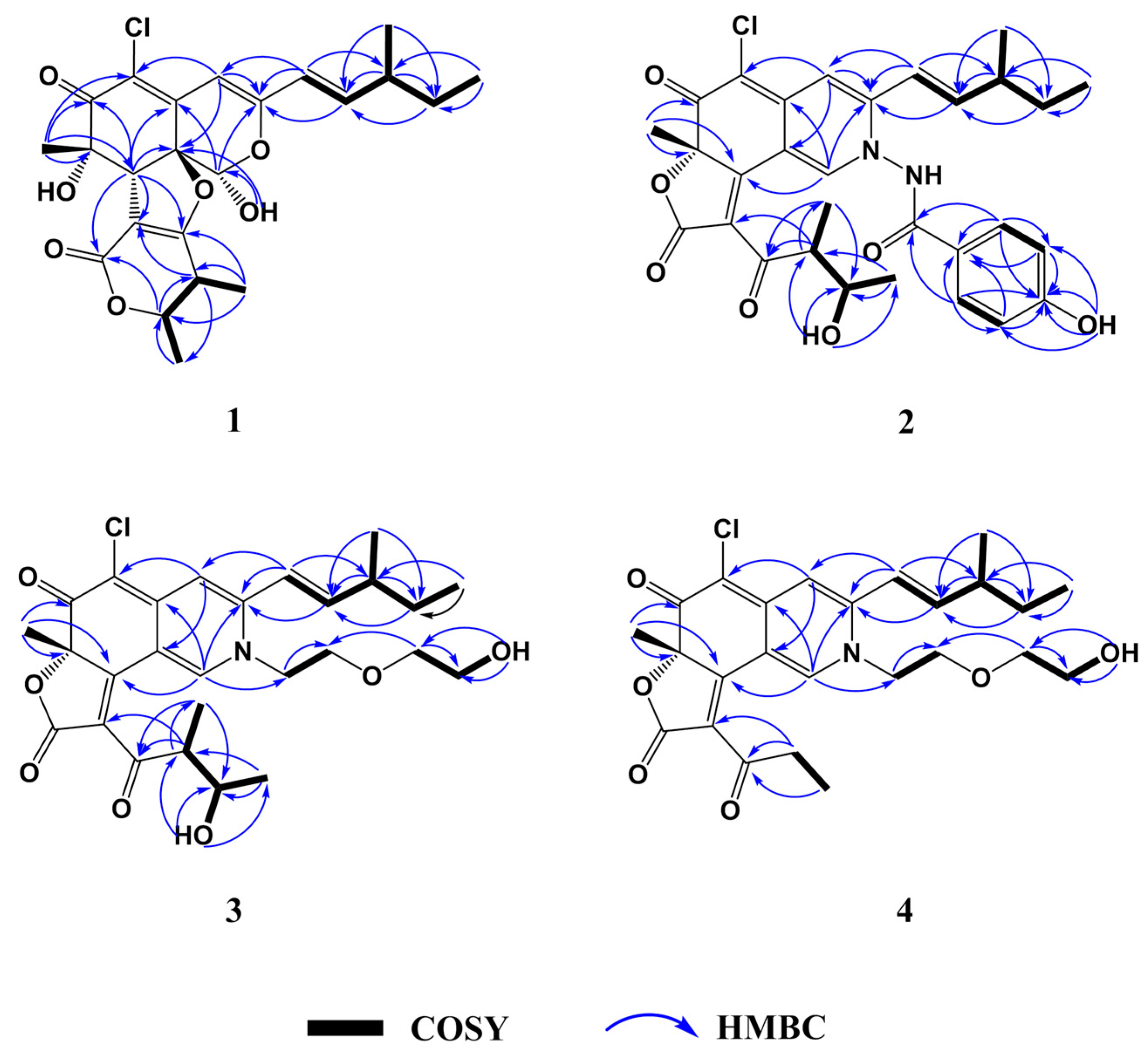
1H COSY spectrum allowed the elucidation of two partial units as shown by bold-faced lines in
Figure 2. The geometrical configuration of the double bond (C-9–C-10) was deduced as
trans from the coupling constant of the olefinic protons
3J9,10 (15.95) (
Table 1). The connection of these units and the remaining groups was established based on the HMBC correlations as shown in
Figure 2. The 3-methyl-1-pentenyl chain was connected to C-3 by HMBC correlations of H-9 with C-3 and C-4 and of H-10 with C-3. The connection of a chlorine atom to C-5 was reasonable from its chemical shift (
δC 124.2). Therefore, the planar structure of
1 was assigned and named chaephilone C (
1).
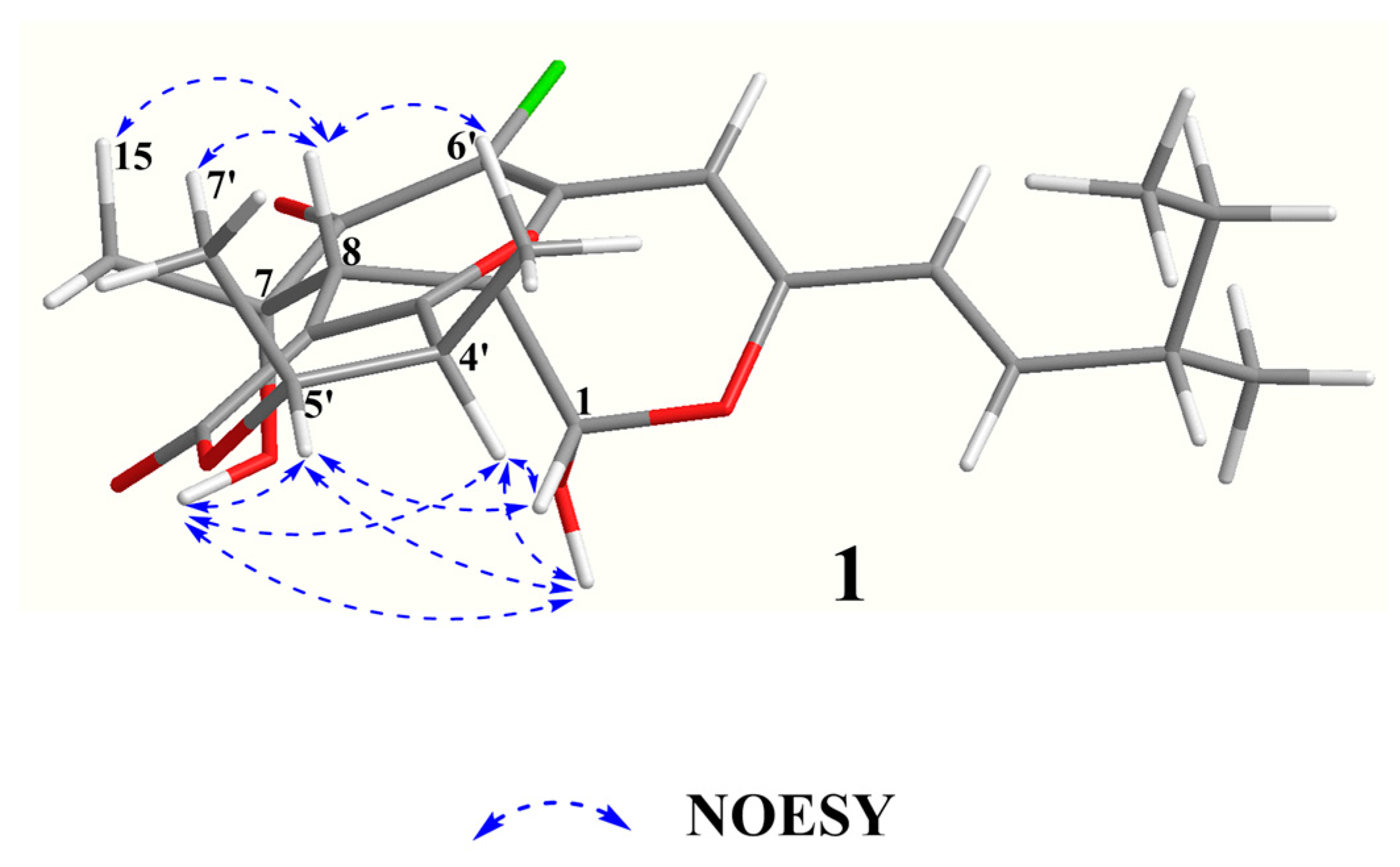
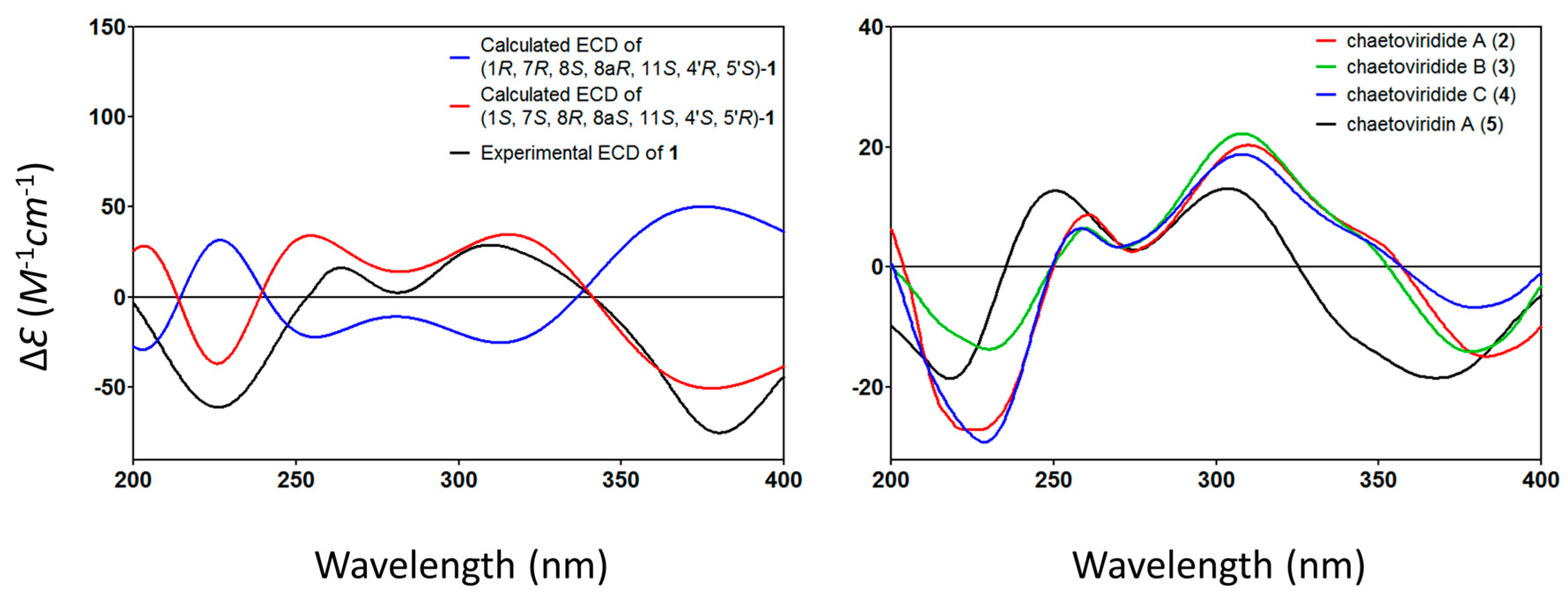
The relative configuration of
1 was elucidated based on NOESY spectra (
Figure 3). The strong NOESY correlations of OH-1 with OH-7, H-4′ and H-5′, of H-1 with H-4′ and H-5′, of H-8 with H-15, H-6′ and H-7′ and no NOE correlations of H-8 with H-4′ and H-5′, indicated that the unit of C-1-OH, OH-7, H-4′ and H-5′ faced to the same side of the fused A/C/D rings and H-8, H-15, H-6′ and H-7′ oriented to the opposite side. Therefore, two possible isomers of (1
S, 7
S, 8
R, 8a
S, 11
S, 4′
S, 5′
R)-
1 and (1
R, 7
R, 8
S, 8a
R, 11
S, 4′
R, 5′
S)-
1 were proposed and their ECD spectra were calculated by time-dependent density functional theory (TD-DFT). The experimental ECD spectrum of
1 was in good agreement with the calculated ECD spectrum of (1
S, 7
S, 8
R, 8a
S, 11
S, 4′
S, 5′
R)-
1 (
Figure 4). Thus, the absolute configuration at C-1, C-7, C-8, C-8a, C-4′ and C-5′ of
1 was established as
S,
S,
R,
S,
S,
S and
R, respectively and the structure of
1 was shown in
Figure 1.
Chaetoviridide A (
2) was obtained as a red amorphous solid. Its molecular formula was determined as C
30H
31N
2O
7Cl by HR-ESI-MS (
m/
z 565.1735 [M − H]
−; calcd. for C
30H
30N
2O
7Cl, 565.1742; ∆ − 1.2 ppm) and the ratio of isotope peaks ([M − H]
−/[M − H + 2]
−), implying sixteen degrees of unsaturation. Analysis of
1H-
1H COSY spectrum allowed the assignment of the 3-methyl-1-pentenyl and 2-butanol-3-yl moieties. 3-methyl-1-pentenyl was attached to C-3 by HMBC correlations to C-3 from H-9 and H-10. 2-butanol-3-yl was connected to the conjugated carbonyl C-3′ by HMBC correlations of H-4′ to C-2′ and of H-7′ to C-3′ (
Figure 2). Partial spectra data of
2 were similar to chaetoviridin A, except that C-1, C-3 and C-8a were shifted to upfield and C-4a to downfield (
Table 1). Chemical shift differences and the detailed 2D NMR (
1H-
1H COSY, HSQC and HMBC) correlations of the unassigned carbons suggested the nitrogen at position of
2 bearing a
p-hydroxybenzamide moiety which was confirmed by the NOE correlations from H-1′′ to H-1, H-9, H-10 and H-4′′ (
Figure S17). Therefore, the planar structure of
2 was deduced and named chaetoviridide A (
2).
Chaetoviridide B (
3) was obtained as a red amorphous solid. Its molecular formula was determined as C
27H
34NO
7Cl by HR-ESI-MS (
m/
z 520.2106 [M + H]
+; calcd. for C
27H
35NO
7Cl, 520.2102; ∆ + 0.8 ppm) and the ratio of isotope peaks ([M + H]
+/[M + H + 2]
+), implying eleven degrees of unsaturation. The
1H-
1H COSY spectrum allowed the elucidation of four partial units as shown by bold-faced lines in
Figure 2. The connection of these units and the remaining groups was established based on the HMBC correlations as shown in
Figure 2. By comparison of NMR data with chaetoviridin A,
3 was characterized as a nitrogenated chaetoviridin A derivative with a 2-hydroxyethoxy-ethyl group attached to N-2, which was confirmed by HMBC correlations from H-1 to C-1′′. The planar structure of
3 was established as N-2-(hydroxyethoxy)ethyl chaetoviridin A and named chaetoviridide B (
3).
Chaetoviridide C (
4) was obtained as a red amorphous solid. Its molecular formula was determined as C
25H
30NO
6Cl by HRESI-MS (
m/
z 476.1828 [M + H]
+; calcd. for C
25H
31NO
6Cl, 476.1840; ∆ − 2.5 ppm) and the ratio of isotope peaks ([M + H]
+/[M + H + 2]
+), indicating eleven degrees of unsaturation attributed to 3 rings and 8 double bonds. By comparison of 1D NMR data with chaetoviridide B (
3) and analysis of 2D NMR data (
Figures S25–S30),
4 were characterized as a chaetoviridide B analog with an ethyl group attached to C-3′ instead of the 2-butanol-3-yl moiety. Therefore, the planar structure of
4 was deduced and named chaetoviridide C (
4).
The optical rotation values of
2–
4 exhibited the same sign compared to that of chaetoviridin A (
5) isolated here,
. In addition, in CD spectra, the negative cotton effects at wavelengths of 225 and 378 nm and positive of 256 and 305 nm for
2–
4, which were also similar to those of chaetoviridin A (
5). Furthermore, the NMR data of C-7, C-4′ and C-5′ for
2–
3 and of C-7 for
4 were almost identical to those of chaetoviridin A (
5). The above evidence sufficiently allowed the assignment of the remaining asymmetric centers (7
S, 4′
S, 5′
R). Therefore, the absolute configuration of
2–
4 was established and shown in
Figure 1.
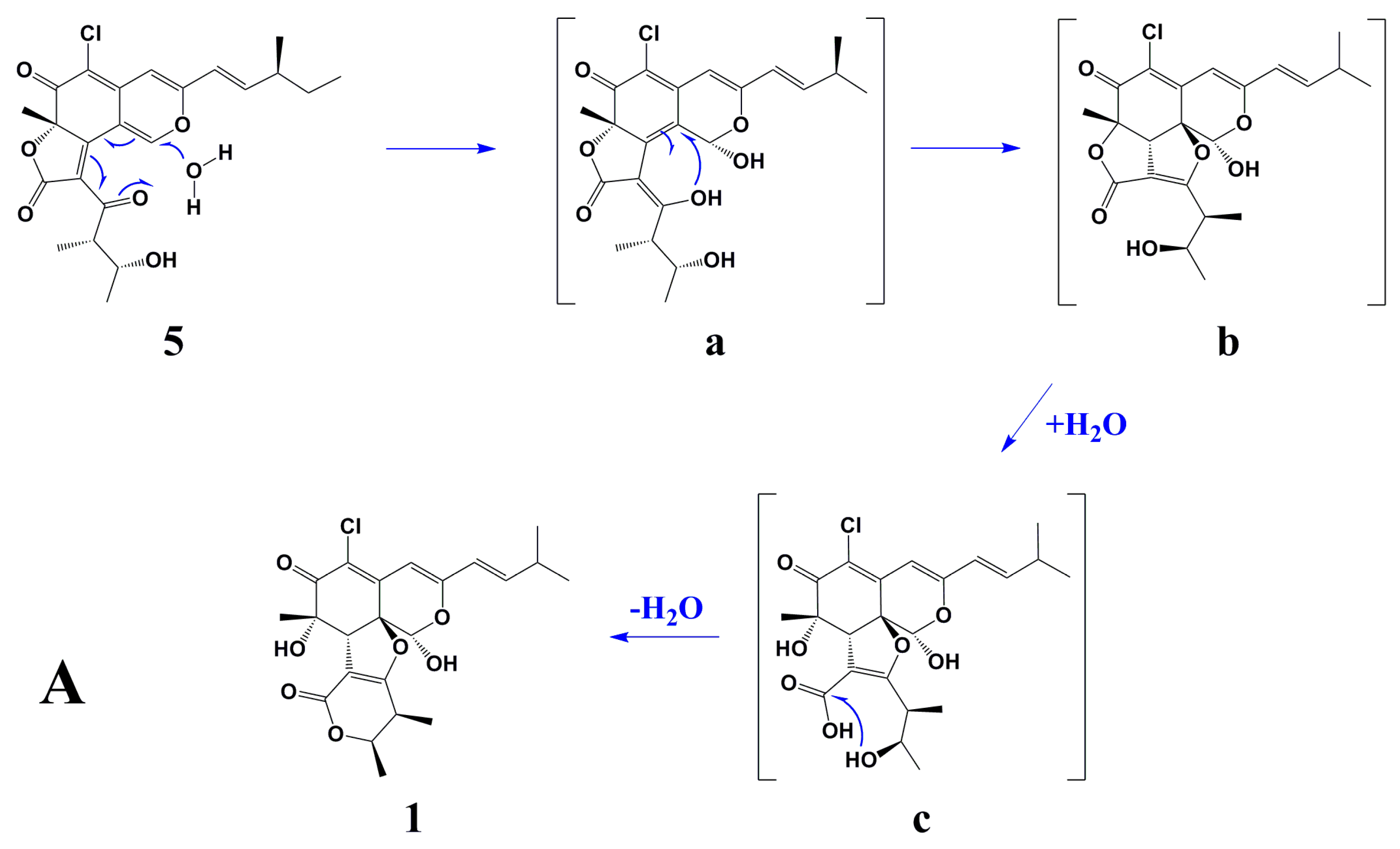
Scheme 1A showed the postulated biogenetic pathway for
1, which started with hydration of chaetoviridin A (
5) to form intermediate
a, followed by attack of 3′-OH on C-8a, hydrolytic opening of the γ-lactone [
2] and post dehydration to obtain chaephilone C (
1).
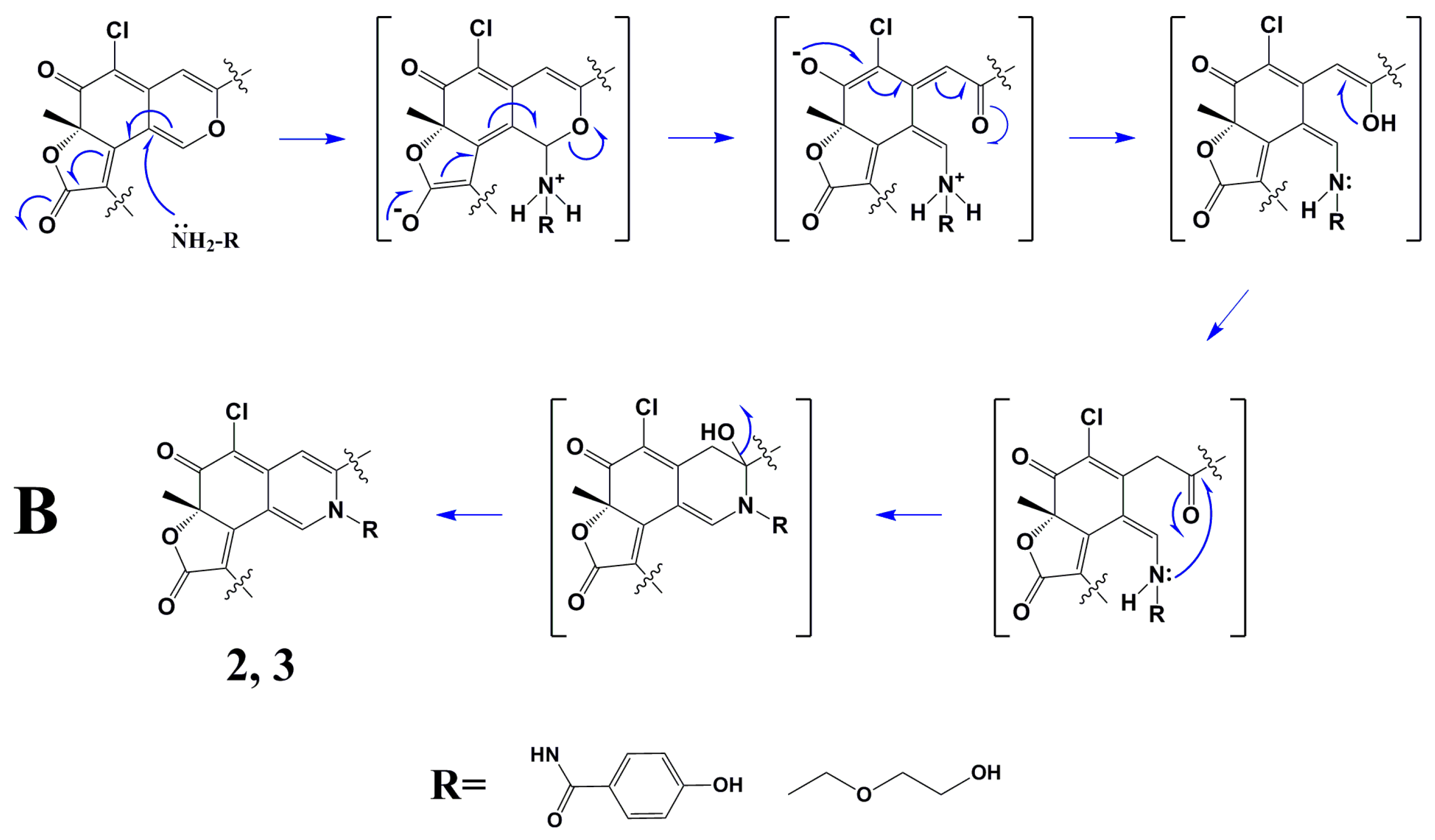
Scheme 1B showed the plausible mechanism for
2 and
3 via a Schiff base formation and dehydration reaction which has been reported previously [
3,
4,
5].
The known compounds (
5–
8) were identified as chaetoviridin A (
5) [
6], chaetoviridine E (
6) [
7], chaetomugilin D (
7) [
8] and cochliodone A (
8) [
7] respectively, by comparing their spectroscopic data with those reported in the literature.
The antimicrobial and cytotoxic activity of compounds
1–
8 was evaluated using cell lines of A549, HeLa and Hep G2 and strains of
Vibrio vulnificus MCCC E1758,
Vibrio rotiferianus MCCC E385 and
Vibrio campbellii MCCC E333, methicillin-resistant
Staphylococcus aureus (MRSA) (ATCC 43300, CGMCC 1.12409) (
Table 2). For strains of
Vibrio, compounds
1–
8 showed weaker activities than the positive control erythromycin. Compounds
2 and
3 exhibited relatively stronger activities than the other compounds against
V. rotiferianus and
V. vulnificus with MIC values ranging from 7 to 8 μg/mL, respectively. For strains of MRSA,
1,
3 and
4 showed similar activities in comparison to the positive control chloramphenicol with MIC values ranging from 7 to 8 μg/mL. For cytotoxicity, compounds
1–
8 displayed weaker activities than the positive control doxorubicin. Compound
2 exhibited the most potent cytotoxic activities towards Hep G2 cell with IC
50 value as 3.9 μM and compounds
1 and
3 demonstrated relatively stronger activities against HeLa cells with IC
50 values ranging from 5–8 μM.
3. Materials and Methods
3.1. General Experimental Procedures
1D NMR and 2D NMR spectra were measured on a Bruker Avance 600 MHz spectrometer. HRESIMS was carried out on a Xevo G2 Q-TOF mass spectrometer with an electrospray ionization source (Waters, Milford, MA, USA). Optical rotations were measured with a P-1020 digital polarimeter (JASCO Corporation, Tokyo, Japan). CD spectra were measured on a J-715 spectropolarimeter (JASCO Corporation). Optical rotations were recorded on a Rudolph IV Autopol automatic polarimeter (Hackettstown, NJ, USA). The UV spectra were recorded on a UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan). The high-performance liquid chromatography (HPLC) analysis was performed on a 1200 system (Agilent, Santa Clara, CA, USA). Thin-layer chromatography (TLC) plates (5 × 10 cm) were performed on GF254 (Qingdao Marine Chemical Co. Ltd., Qingdao, China) plates. For column chromatography (CC), RP-C18 (ODS-A, 50 µm, YMC, Kyoto, Japan), silica gel (200–300 mesh, 300–400 mesh, Qingdao Marine Chemical Co. Ltd., Qingdao, China) and Sephadex LH-20 (GE Healthcare Bio-Science AB, Pittsburgh, PA, USA) were used. Semi-preparative HPLC was run with a P3000 pump (CXTH, Beijing, China) and a UV3000 ultraviolet-visible detector (CXTH, Beijing, China), using a preparative RP-C18 column (5 µm, 20 × 250 mm, YMC, Kyoto, Japan).
3.2. Fungal Material
Strain NA-S01-R1 of
C. sp. was identified by ITS sequence homology (99% similarity with
Chaetomium globosum strain GYY2(1) with Genbank Accession No. KM268652.1 (max score 974,
e value 0.0, query cover 97%)). The fungal strain was inoculated into a 15 mL centrifuge tube containing 5 mL of potato dextrose (PD) medium and cultured at 28 °C at 120 rpm for 3 days. Total genomic DNA was extracted as described by Lai et al. [
9]. The internal transcribed spacer (ITS) region of rDNA was amplified by PCR using primers ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′). The PCR mixture consisted of 12.5 μL Taq premix (TaKaRa, Beijing, China), 0.25 μL (10 μM) of each primer, 0.75 μL dimethyl sulfoxide (DMSO), 10.25 μL dd. H
2O and 1 μL DNA template. After denaturation at 95 °C for 4 min, amplification was performed with 32 cycles of 30 s at 95 °C, 30 s at 55 °C and 40 s at 72 °C and a final extension at 72 °C for 7 min. The ITS1-5.8S-ITS2 rDNA sequence of the fungus has been submitted to GenBank with the accession number MG786198. A voucher specimen was deposited at the Third Institute of Oceanography, SOA, China. The working strain was prepared on potato dextrose agar (PDA) slants and stored at 4 °C.
3.3. Fermentation, Extraction and Isolation
Strain NA-S01-R1 was cultured on PDA plates at 28 °C for 3 days. Then, six plugs (5 mm diameter) were transferred to 12 Erlenmeyer flasks (1 L), each containing 500 mL Czapek’s medium (NaNO3 3.0 g/L, KH2PO4 1.0 g/L, MgSO4·7H2O 0.5 g/L, sucrose 30 g/L, FeSO4 0.01 g/L and KCl 0.5 g/L) in sterile conditions. Erlenmeyer flasks were shaken on a rotary shaker at 28 °C and 120 rpm for 3 days to form seed cultures (1 × 108 spores/mL). Next, seed cultures (45 × 100 mL) were transferred to flasks (45 × 1 L) containing 105 g of rice and 45 g of millet per flask. After 25 days, the fermented broth was dried, smashed and extracted with ethyl acetate (EtOAc). The organic solvent was evaporated under reduced pressure to afford the EtOAc extract (150 g), which was partitioned by 90% methanol in water and then extracted with petroleum ether (PE) three times. The methanol layer was concentrated to provide a defatted extract (50 g), which was subject to Sephadex LH-20 column chromatography, eluting with methanol to yield eight fractions, A–H, of which fraction B (500 mg) was in red color and exhibited better antibacterial and cytotoxic than the other fractions. Therefore, fraction B was applied to silica gel column chromatography using PE-acetone (9:1, 8.5:1.5, 8:2, 7.5:2.5, 6.5:3.5, V:V) to yield five subfractions, B1–B5. Fraction B1 was further purified by semi-preparative HPLC (90% methanol in H2O, flow rate 8 mL/min) and Sephadex LH-20 (90% methanol in water) to give compounds 7 (5 mg) and 4 (3.2 mg). Fraction B2 was further separated by semi-preparative HPLC (85% acetonitrile in H2O, flow rate 8 mL/min) to obtain compounds 1 (5 mg) and 8 (3 mg). Fraction B3 was subjected to Sephadex LH-20 (85% methanol in water) to yield compounds 3 (4.8 mg) and 5 (50 mg). Fraction B4 was purified by preparative HPLC (80% methanol in water) and Sephadex LH-20 to obtain compounds 2 (3.5 mg) and 6 (10 mg).
Chaephilone C (
1): yellow amorphous solid;
−125 (
c 0.04, MeOH); UV λ
max (methanol) nm (log
ε): 300 (4.37), 380 (4.32);
1H NMR and
13C NMR data are shown in
Table 1; HR-ESI-MS:
m/
z 449.1373 [M − H]
− (Calcd. for 449.1367, C
23H
26O
7Cl, ∆ + 1.3 ppm).
Chaetoviridide A (
2): red amorphous solid;
+105 (
c 0.03, MeOH); UV λ
max (methanol) nm (log
ε): 297 (4.56), 380 (4.43);
1H NMR and
13C NMR data are shown in
Table 1; HR-ESI-MS:
m/
z 565.1735 [M − H]
− (Calcd. for 565.1742, C
30H
30N
2O
7Cl, ∆ − 1.2 ppm).
Chaetoviridide B (
3): red amorphous solid;
+95 (
c 0.03, MeOH); UV λ
max (methanol) nm (log
ε): 267 (4.55), 299 (4.58), 384 (4.53);
1H NMR and
13C NMR data are shown in
Table 1; HR-ESI-MS:
m/
z 520.2106 [M + H]
+ (Calcd. for 520.2102, C
27H
35NO
7Cl, ∆ + 0.8 ppm).
Chaetoviridide C (
4): red amorphous solid;
+90 (
c 0.03, MeOH); UV λ
max (methanol) nm (log
ε): 299 (4.42), 380 (4.35);
1H NMR and
13C NMR data are shown in
Table 1; HR-ESI-MS:
m/
z 476.1828 [M + H]
+ (Calcd. for 476.1840, C
25H
31NO
6Cl, ∆ − 2.5 ppm).
3.4. ECD Calculation
Conformational analysis was initially performed using Confab [
10] with systematic search at MMFF94 force field for undetermined relative configurations of compound
1. Room-temperature equilibrium populations were calculated according to the Boltzmann distribution law Equation (1). Dominative conformers with Boltzmann-based population over 1% were delivered to subsequent Quantum Mechanics (QM) calculations. The energies and populations of conformers were provided in
Table S1.
Ni is the number of conformer
i with energy
Ei and degeneracy
gi at temperature
T and
kB is the Boltzmann constant.
The theoretical calculations were carried out using Gaussian 09. At first, conformers were optimized at PM6 using semi-empirical theory method. The conformers with Boltzmann-based population lower than 1% were again filtered out and the remaining chosen for further optimization at B3LYP/6-311G(d,p) in methanol using the integral equation formalism polarizable continuum model (IEFPCM) (
Table S2). Vibrational frequency analysis confirmed the stable structures. Under the same condition, the ECD calculation was conducted using TD-DFT. Rotatory strengths for a total of 30 excited states were calculated. The ECD spectrum was simulated in SpecDis [
11] by overlapping Gaussian functions for each transition.
3.5. Antibacterial Assay
Antibacterial activities against
V. rotiferianus (MCCC E385),
V. vulnificus (MCCC E1758),
V. campbellii (MCCC E333) and MRSA (ATCC 43300, CGMCC 1.12409), were tested by continuous dilution in 96-well plates using resazurin as a surrogate indicator. Blue resazurin was reduced by metabolically active bacteria to pink resorufin. A mid-logarithmic-phase tested strain was added at a starting inoculum of 5 × 10
5 CFU/mL to the plate containing tested compound (final concentration ranging from 125 to 0.98 μg/mL in two-fold dilution) plus 10% resazurin solution (6.75 mg/mL in sterile water). The foil covered plate was incubated for 24 h with shaking at 37 °C. The MIC value was determined to be the lowest concentration that did not induce the color change [
12,
13,
14] by observing the blue-to-pink color change.
3.6. Cytotoxicity Assay
A549 (adenocarcinomic human alveolar basal epithelial cell), Hela (cervical cancer cell) and Hep G2 (human liver cancer cell) cells were maintained in F-12K, DMEM and MEM medium respectively and supplied with 10% FBS, 100 U/mL of penicillin and 100 mg/mL of streptomycin [
15]. Cells were grown in a humidified chamber with 5% CO
2 at 37 °C. For cytotoxicity assays, cells were seeded at a density of 5000 cells per well in 96-well plates, grown at 37 °C for 12 h and then treated with tested compound at five different concentrations (100 μL medium/well). The cytotoxicity was measured by Cell Counting Kit-8 (CCK-8) (DOJINDO) at 48 h post-treatment, following the manufacturer’s instructions.
CCK-8 assay is based on the conversion of a tetrazolium salt, 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2
H-tetrazolium, monosodium salt (WST-8) and a water-soluble formazan dye, upon reduction by dehydrogenases in the presence of an electron mediator [
16]. WST-8 is reduced by dehydrogenases in cells to give an orange colored product (formazan). The amount of the formazan dye is directly proportional to the number of living cells.
In brief, 10 μL of CCK-8 solution was added to each well of the 96-well plates. After incubation at 37 °C for 2 h, the absorbance at 450 nm was measured using a SpectraMAX M5 microplate reader. Wells with only culture medium and CCK-8 solution were used to determine the background and cells treated with DMSO were included as the negative controls [
15].









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}