Halogenated and Nonhalogenated Metabolites from the Marine-Alga-Endophytic Fungus Trichoderma asperellum cf44-2


Abstract
:1. Introduction
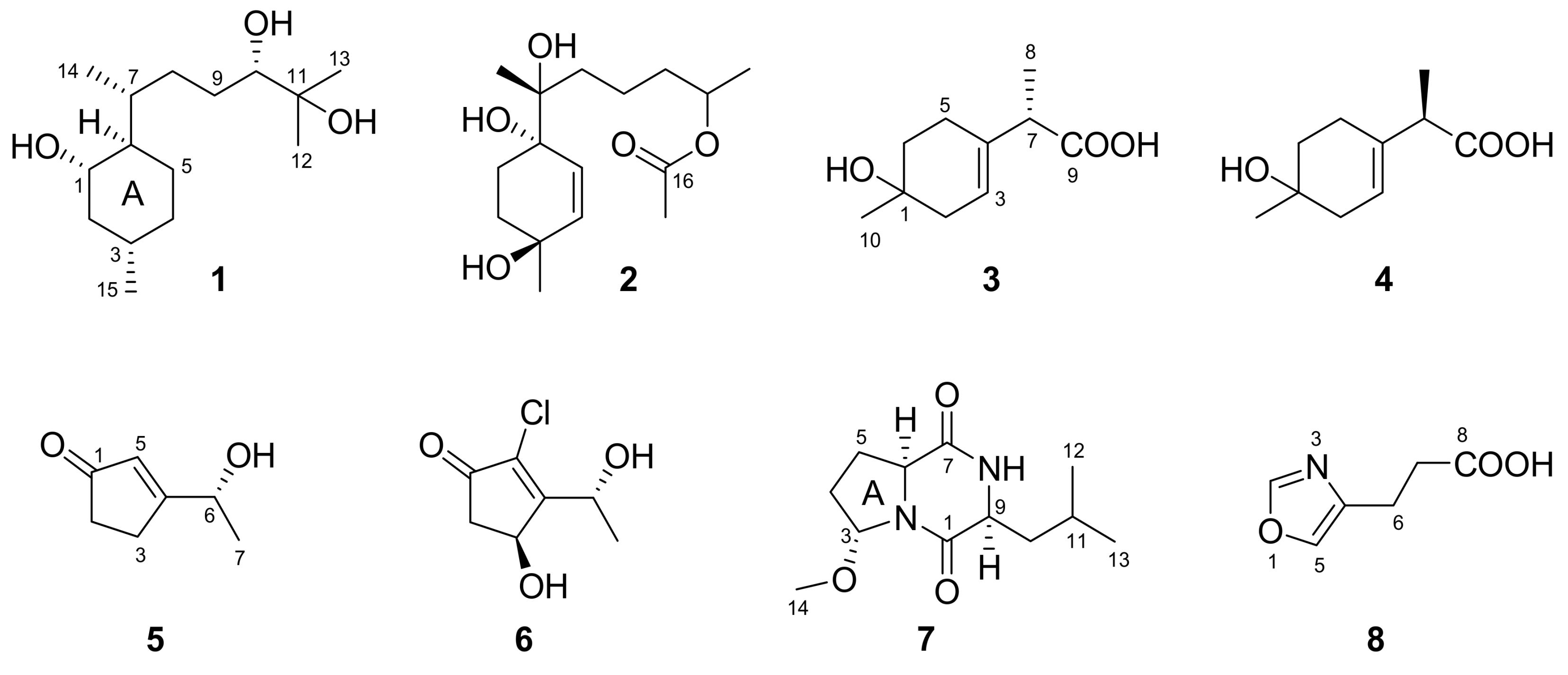
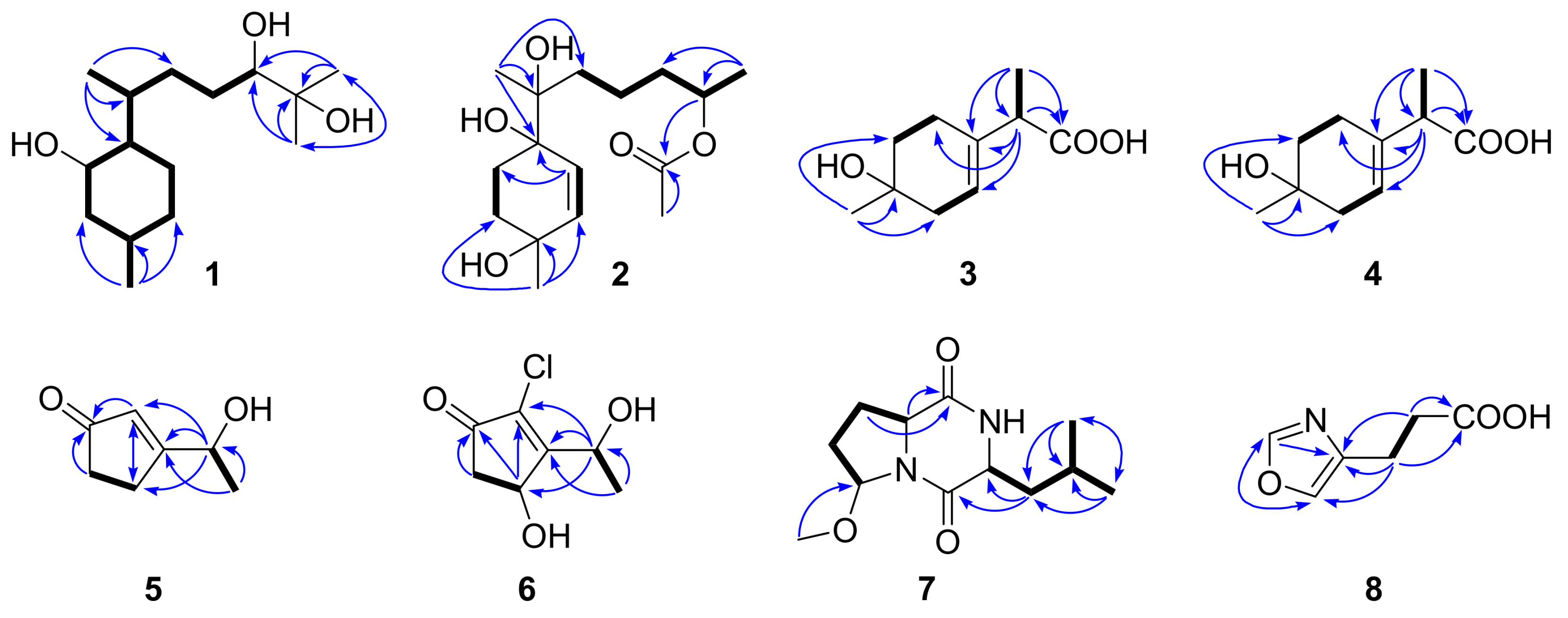
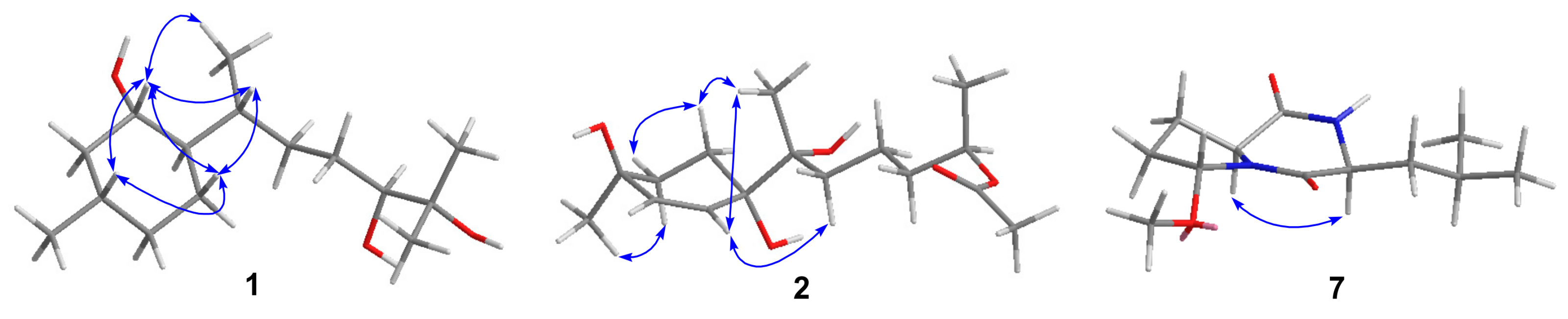
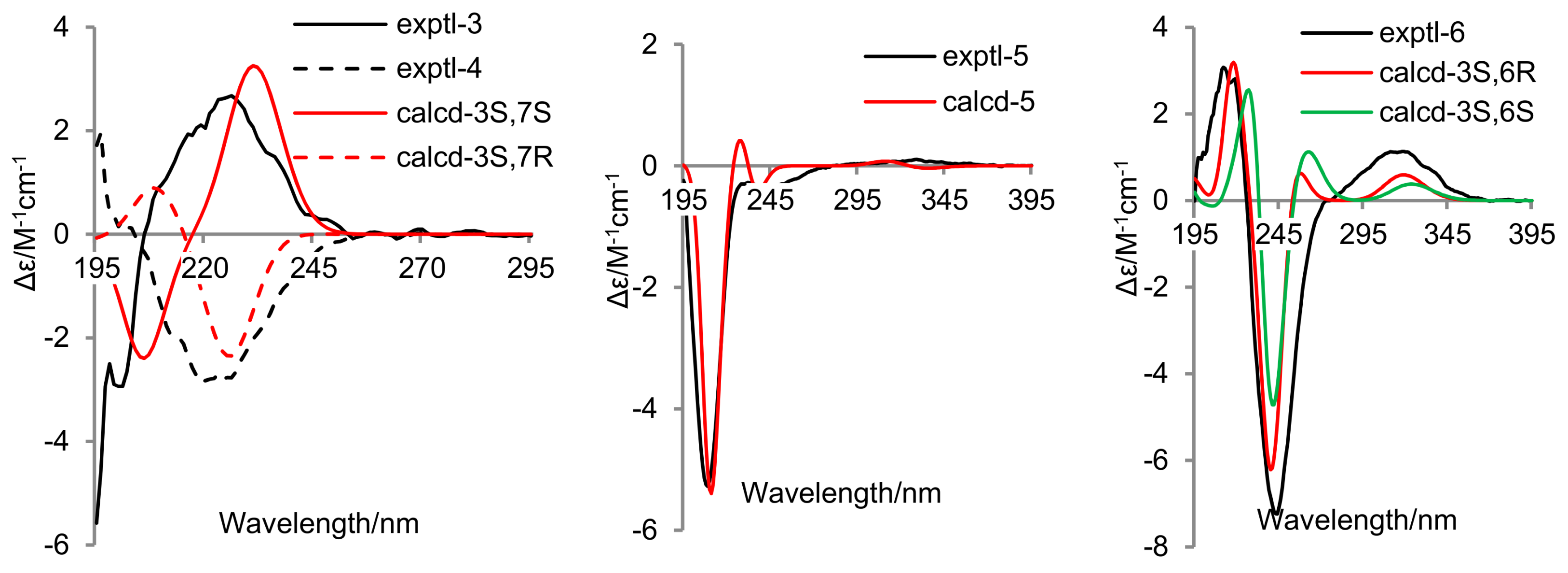
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material and Fermentation
3.3. Extraction and Isolation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrita, M.T.; Vale, C.; Rauter, A.P. Halogenated compounds from marine algae. Mar. Drugs 2010, 8, 2301–2317. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.G.; Gloer, J.B.; Ji, N.Y.; Zhao, J.C. Halogenated organic molecules of Rhodomelaceae origin: Chemistry and biology. Chem. Rev. 2013, 113, 3632–3685. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.Y.; Wang, B.G. Mycochemistry of marine algicolous fungi. Fungal Divers. 2016, 80, 301–342. [Google Scholar] [CrossRef] [Green Version]
- Garo, E.; Starks, C.M.; Jensen, R.R.; Fenical, W.; Lobkovsky, E.; Clardy, J. Trichodermamides A and B, cytotoxic modified dipeptides from the marine-derived fungus Trichoderma virens. J. Nat. Prod. 2003, 66, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Rotinsulu, H.; Narita, R.; Takahashi, R.; Namikoshi, M. Induced production of halogenated epidithiodiketopiperazines by a marine-derived Trichoderma cf. brevicompactum with sodium halides. J. Nat. Prod. 2015, 78, 2319–2321. [Google Scholar] [CrossRef] [PubMed]
- Pujar, B.G.; Bhattacharyya, P.K. Microbiological transformation of terpenes: Part XVII−fermentation of 1-p-menthene & 3-p-menthene by a soil pseudomonad (PL-Strain). Indian J. Biochem. Biophys. 1973, 10, 164–169. [Google Scholar] [PubMed]
- Usami, Y.; Ikura, T.; Amagata, T.; Numata, A. First total syntheses and configurational assignments of cytotoxic trichodenones A−C. Tetrahedron Asymmetry 2000, 11, 3711–3725. [Google Scholar] [CrossRef]
- Naresh, K.; Reddy, B.M.; Bahu, V.H. Economical and eco-friendly method for the reduction of heterocyclics possessing α,β-unsaturated acid systems using novel reduction method. Eur. J. Biomed. Pharm. Sci. 2014, 1, 446–451. [Google Scholar]
- Kreuzer, T.; Metz, P. Enantioselective synthesis of the hydroazulene core of 3α-Hydroxy-15-rippertene. Eur. J. Org. Chem. 2008, 572–579. [Google Scholar] [CrossRef]
- Liang, X.R.; Miao, F.P.; Song, Y.P.; Liu, X.H.; Ji, N.Y. Citrinovirin with a new norditerpene skeleton from the marine algicolous fungus Trichoderma citrinoviride. Bioorg. Med. Chem. Lett. 2016, 26, 5029–5031. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Greca, M.D.; Fiorentino, A.; Monaco, P.; Previtera, L. Cycloartane triterpenes from Juncus effusus. Phytochemistry 1994, 35, 1017–1022. [Google Scholar] [CrossRef]
- Inada, A.; Murayta, H.; Inatomi, Y.; Nakanishi, T. Cycloartane triterpenes from the leaves of Aglaia harmsiana. J. Nat. Prod. 1995, 58, 1143–1146. [Google Scholar] [CrossRef]
- Amagata, T.; Usami, Y.; Minoura, K.; Ito, T.; Numata, A. Cytotoxic substances produced by a fungal strain from a sponge: Physico-chemical properties and structures. J. Antibiot. 1998, 51, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.L.; Kuo, P.C.; Hwang, T.L.; Wu, T.S. Anti-inflammatory principles from Cordyceps sinensis. J. Nat. Prod. 2011, 74, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Thaning, M.; Wistrand, L.G. Synthetic application of electrochemically produced α-methoxyamides, part 2, oxidation of hydroxyproline derivatives. Helv. Chim. Acta 1986, 69, 1711–1717. [Google Scholar] [CrossRef]
- Takahashi, S.; Hashimoto, R.; Hamano, K.; Suzuki, T.; Nakagawa, A. Melanoxazal, new melanin biosynthesis inhibitor discovered by using the larval haemolymph of the silkworm, Bombyx mori. Production, isolation, structural elucidation, and biological properities. J. Antibiot. 1996, 49, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Bruhn, T.; Hemberger, Y.; Schaumlöffel, A.; Bringmann, G. SpecDis, Version 1.51; University of Wuerzburg: Wuerzburg, Germany, 2011. [Google Scholar]
- Miao, F.P.; Liang, X.R.; Yin, X.L.; Wang, G.; Ji, N.Y. Absolute configurations of unique harziane diterpenes from Trichoderma species. Org. Lett. 2012, 14, 3815–3817. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Imamura, N.; Nishijima, M.; Adachi, K.; Sakai, M.; Sano, H. Halymecins, new antimicroalgal substances produced by fungi isolated from marine algae. J. Antibiot. 1996, 49, 998–1005. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pos. | 1 (in CDCl3) | 2 (in CD3OD) | ||
|---|---|---|---|---|
| δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | |
| 1a | 3.44, td (10.5, 4.3) | 71.5, CH | 1.97, td (13.4, 2.9) | 27.8, CH2 |
| 1b | 1.57, m | |||
| 2a | 1.97, dddd (12.1, 4.2, 3.6, 2.0) | 45.3, CH2 | 1.88, td (13.6, 3.0) | 34.3, CH2 |
| 2b | 0.97, ddd (12.1, 12.1, 10.7) | 1.68, m | ||
| 3 | 1.42, m | 31.8, CH | 67.3, C | |
| 4a | 1.66, dm (12.9) | 34.7, CH2 | 5.73, dd (10.2, 1.6) | 136.8, CH |
| 4b | 0.84, m | |||
| 5a | 1.59, dq (13.2, 3.3) | 23.5, CH2 | 5.90, dd (10.2, 1.6) | 131.3, CH |
| 5b | 1.02, qd (13.1, 3.5) | |||
| 6 | 1.20, m | 49.0, CH | 74.9, C | |
| 7 | 2.03, m | 30.8, CH | 77.1, C | |
| 8a | 1.53, m | 32.7, CH2 | 1.73, m | 36.3, CH2 |
| 8b | 1.31, m | 1.40, m | ||
| 9a | 1.46, m | 29.8, CH2 | 1.51, m | 20.5, CH2 |
| 9b | 1.36, m | 1.41, m | ||
| 10a | 3.36, dd (9.9, 2.0) | 78.9, CH | 1.60, m | 37.7, CH2 |
| 10b | 1.52, m | |||
| 11 | 73.3, C | 4.89, m | 72.6, CH | |
| 12 | 1.16, s | 23.3, CH3 | ||
| 13 | 1.21, s | 26.7, CH3 | 1.22, d (6.3) | 20.2, CH3 |
| 14 | 0.81, d (6.9) | 14.0, CH3 | 1.10, s | 21.2, CH3 |
| 15 | 0.91, d (6.6) | 22.3, CH3 | 1.27, s | 30.3, CH3 |
| 16 | 172.8, C | |||
| 17 | 2.01, s | 21.3, CH3 | ||
| Pos. | 3 (in CDCl3) | 4 (in CDCl3) | 5 (in CDCl3) | 6 (in CD3OD) | 7 (in CD3OD) | 8 (in CD3OD) |
|---|---|---|---|---|---|---|
| 2a | 2.19, d (18.0) | 2.16, brs | 2.45, m | 2.88, ddd (18.6, 6.3, 0.9) | 8.11, s | |
| 2b | 2.12, d (17.6) | 2.30, dd (18.6, 1.7) | ||||
| 3 | 5.54, brs | 5.54, brs | 2.64, m | 5.14, dd (6.3, 1.5) | 5.24, d (5.0) | |
| 4a | 2.05, dt (13.2, 3.3) | |||||
| 4b | 1.91, m | |||||
| 5a | 2.28, m | 2.18, m | 6.13, q (1.7) | 2.15, m | 7.69, s | |
| 5b | 2.04, brd (17.4) | 2.12, m | 2.15, m | |||
| 6a | 1.72, dt (13.0, 5.0) | 1.73, dt (13.1, 5.2) | 4.66, q (6.7) | 4.97, q (6.9) | 4.26, t (8.8) | 2.83, t (7.4) |
| 6b | 1.60, ddd (12.9, 8.5, 6.4) | 1.60, dt (13.1, 6.7) | ||||
| 7 | 3.13, q (6.8) | 3.13, q (6.9) | 1.45, d (6.7) | 1.51, d (6.9) | 2.64, t (7.4) | |
| 8 | 1.29, d (7.1) | 1.29, d (7.1) | ||||
| 9 | 4.09, dd (8.1, 4.0) | |||||
| 10a | 1.26, s | 1.25, s | 1.93, m | |||
| 10b | 1.52, m | |||||
| 11 | 1.89, m | |||||
| 12 | 0.99, d (6.4) | |||||
| 13 | 0.97, d (6.3) | |||||
| 14 | 3.37, s |
| Pos. | 3 (in CDCl3) | 4 (in CDCl3) | 5 (in CDCl3) | 6 (in CD3OD) | 7 (in CD3OD) | 8 (in CD3OD) |
|---|---|---|---|---|---|---|
| 1 | 68.6, C | 68.7, C | 209.7, C | 200.1, C | 171.1, C | |
| 2 | 39.8, CH2 | 39.8, CH2 | 35.4, CH2 | 44.0, CH2 | 153.4, CH | |
| 3 | 121.8, CH | 122.0, CH | 27.9, CH2 | 68.4, CH | 88.5, CH | |
| 4 | 135.6, C | 135.6, C | 184.4, C | 175.4, C | 31.8, CH2 | 140.1, C |
| 5 | 24.3, CH2 | 24.1, CH2 | 128.1, CH | 131.5, C | 25.3, CH2 | 136.5, CH |
| 6 | 35.4, CH2 | 35.4, CH2 | 68.0, CH | 66.3, CH | 60.5, CH | 22.4, CH2 |
| 7 | 46.3, CH | 46.3, CH | 22.2, CH3 | 21.8, CH3 | 173.6, C | 33.7, CH2 |
| 8 | 15.6, CH3 | 15.7, CH3 | 176.2, C | |||
| 9 | 179.5, C | 179.7, C | 55.0, CH | |||
| 10 | 28.6, CH3 | 28.4, CH3 | 38.8, CH2 | |||
| 11 | 25.9, CH | |||||
| 12 | 23.4, CH3 | |||||
| 13 | 22.0, CH3 | |||||
| 14 | 57.4, CH3 |
| IC50 (μg/mL) | Inhibitory Zone Diameter (mm) at 20 μg/disk | |||||||
|---|---|---|---|---|---|---|---|---|
| Chattonella marina | Heterosigma akashiwo | Karlodinium veneficum | Prorocentrum donghaiense | Vibrio parahaemolyticus | Vibrio anguillarum | Vibrio harveyi | Vibrio Splendidus | |
| 1 | – a | 30 | 27 | – b | 6.7 | 6.5 | 6.5 | 6.3 |
| 2 | 15 | 8.4 | 10 | 14 | 6.4 | 7.5 | 7.0 | 6.3 |
| 5 | 4.2 | 7.2 | 8.5 | 6.9 | 6.2 | 7.0 | 6.5 | 6.2 |
| 6 | 30 | 35 | 39 | 37 | 6.7 | 8.5 | 8.0 | 6.5 |
| K2Cr2O7 | 0.46 | 0.98 | 0.89 | 1.92 | ||||
| chloramphenicol | 20 | 18 | 18 | 19 | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.-P.; Miao, F.-P.; Fang, S.-T.; Yin, X.-L.; Ji, N.-Y. Halogenated and Nonhalogenated Metabolites from the Marine-Alga-Endophytic Fungus Trichoderma asperellum cf44-2. Mar. Drugs 2018, 16, 266. https://doi.org/10.3390/md16080266
Song Y-P, Miao F-P, Fang S-T, Yin X-L, Ji N-Y. Halogenated and Nonhalogenated Metabolites from the Marine-Alga-Endophytic Fungus Trichoderma asperellum cf44-2. Marine Drugs. 2018; 16(8):266. https://doi.org/10.3390/md16080266
Chicago/Turabian StyleSong, Yin-Ping, Feng-Ping Miao, Sheng-Tao Fang, Xiu-Li Yin, and Nai-Yun Ji. 2018. "Halogenated and Nonhalogenated Metabolites from the Marine-Alga-Endophytic Fungus Trichoderma asperellum cf44-2" Marine Drugs 16, no. 8: 266. https://doi.org/10.3390/md16080266
APA StyleSong, Y. -P., Miao, F. -P., Fang, S. -T., Yin, X. -L., & Ji, N. -Y. (2018). Halogenated and Nonhalogenated Metabolites from the Marine-Alga-Endophytic Fungus Trichoderma asperellum cf44-2. Marine Drugs, 16(8), 266. https://doi.org/10.3390/md16080266