Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508


Abstract
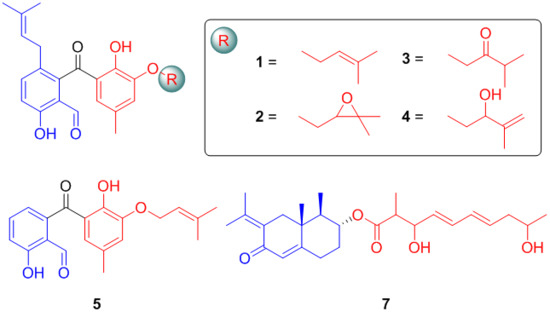
:
1. Introduction
2. Results and Discussion
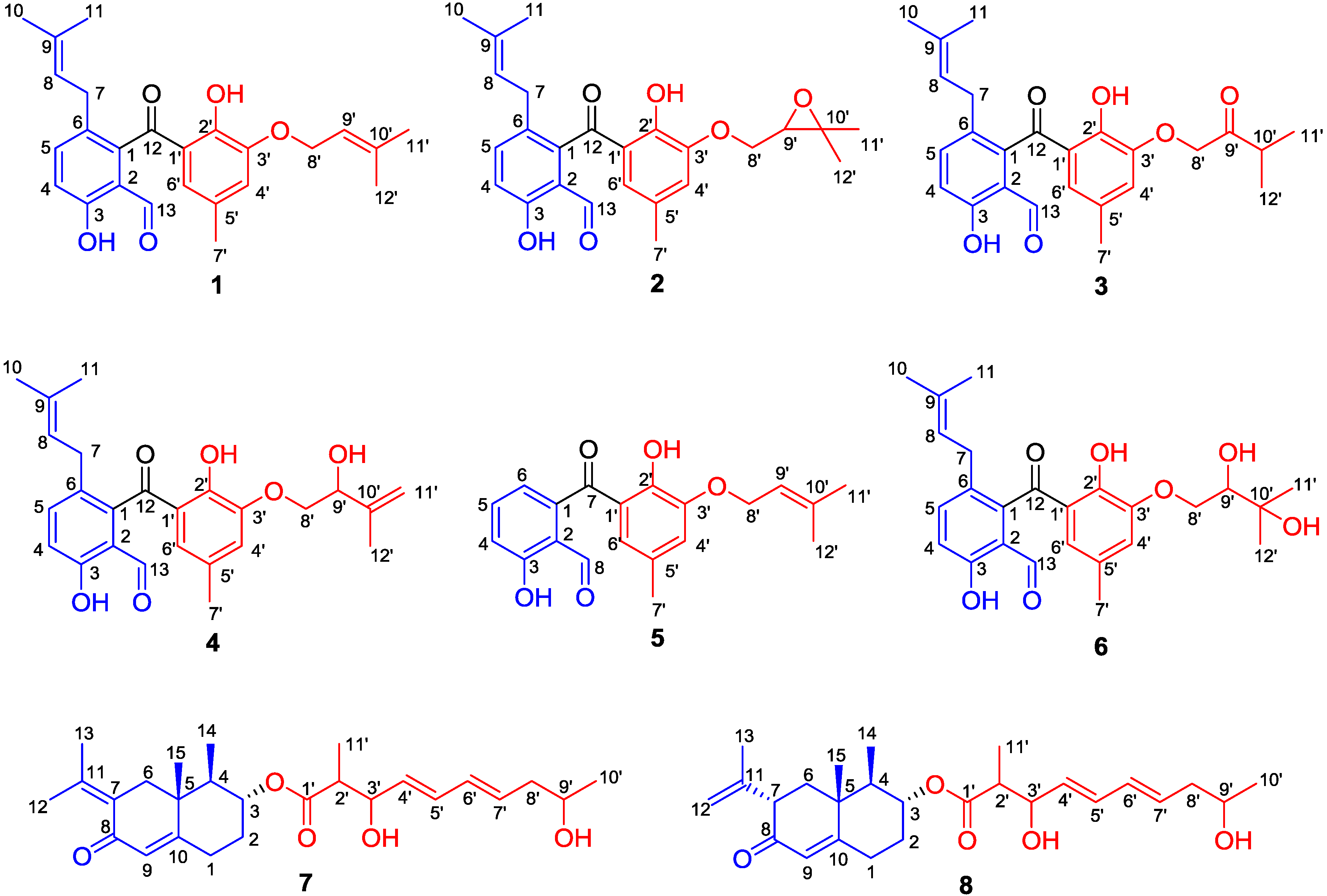
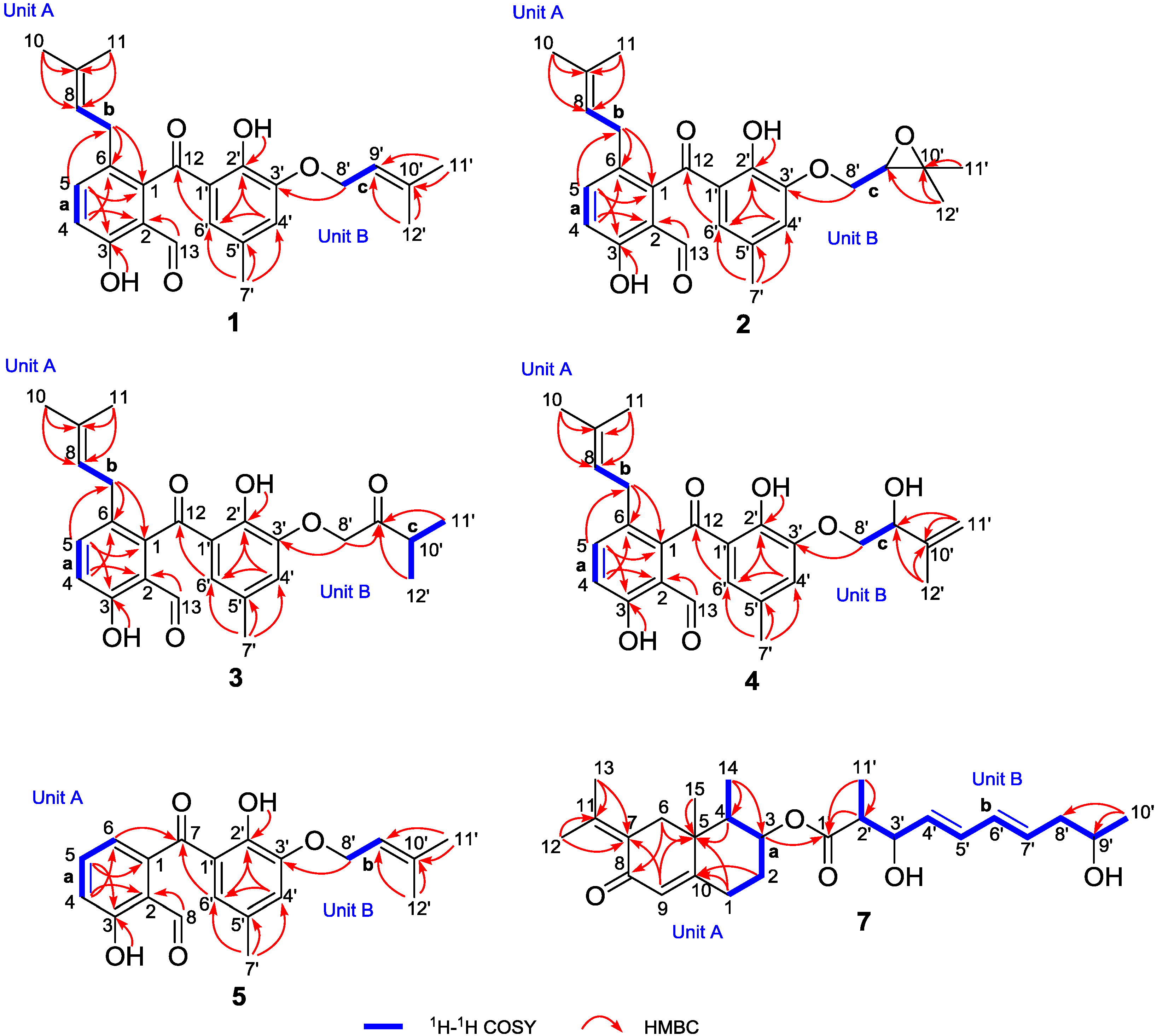
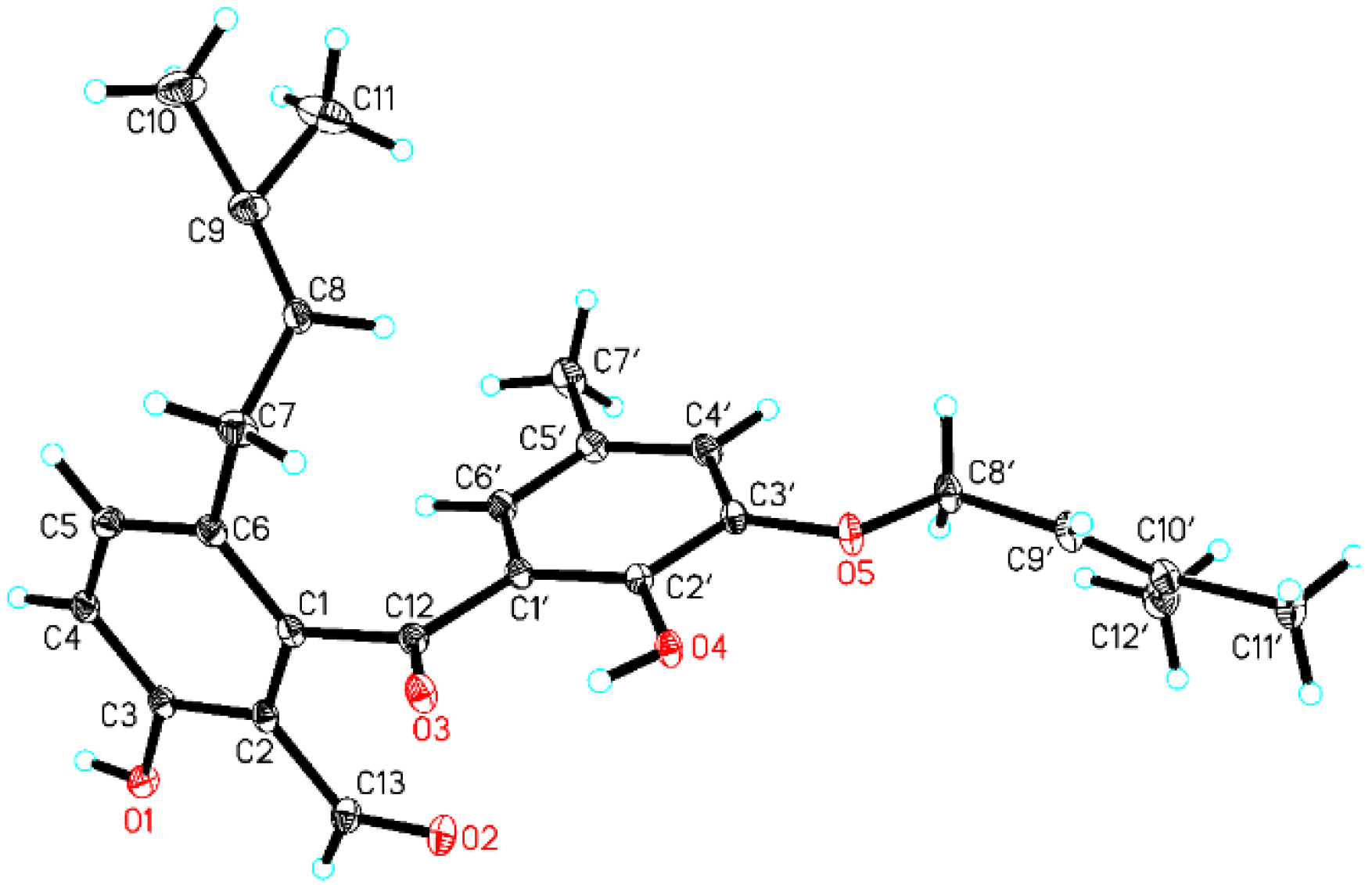
2.1. Structure Elucidation
2.2. Cytotoxicity Assay
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction and Isolation
3.4. X-Ray Analysis of Tenellone D (1)
3.5. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, X.D.; Li, X.M.; Li, X.; Xu, G.M.; Liu, Y.; Wang, B.G. Aspewentins D–H, 20-nor-isopimarane derivatives from the deep sea sediment-derived fungus Aspergillus wentii SD-310. J. Nat. Prod. 2016, 79, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guo, L.; Hao, J.J.; Wang, L.P.; Zhu, W.M. α-glucosidase inhibitors from the marine-derived fungus Aspergillus flavipes HN4-13. J. Nat. Prod. 2016, 79, 2977–2981. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Prakash, V.; Ranjan, N. Marine fungi: A source of potential anticancer compounds. Front. Microbiol. 2018, 8, 2536. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.J.; Bae, S.Y.; Won, T.H.; You, M.J.; Kim, S.H.; Oh, D.C.; Lee, S.K.; Oh, K.B.; Shin, J. Asperphenins A and B, lipopeptidyl benzophenones from a marine-derived Aspergillus sp. fungus. Org. Lett. 2017, 19, 2066–2069. [Google Scholar] [CrossRef] [PubMed]
- Soldatou, S.; Baker, B.J. Cold-water marine natural products, 2006 to 2016. Nat. Prod. Rep. 2017, 34, 585–626. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Honecker, F. Marine compounds and cancer: 2017 updates. Mar. Drugs 2018, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Xue, Y.R.; Liu, C.H. A brief review of bioactive metabolites derived from deep-sea fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, J.; Zhai, H.J.; Zhu, K.K.; Yu, J.H.; Zhang, Y.Y.; Wang, Y.Y.; Jiang, C.S.; Zhang, X.Y.; Zhang, Y.; Zhang, H. Bioactive pyridone alkaloids from a deep-sea-derived fungus Arthrinium sp. UJNMF0008. Mar. Drugs 2018, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.J.; Cheng, W.; Heydari, H.; Wang, B.; Zhu, K.; Konuklugil, B.; Lin, W.H. Sorbicillinoid-based metabolites from a sponge-derived fungus Trichoderma saturnisporum. Mar. Drugs 2018, 16, 226. [Google Scholar] [CrossRef] [PubMed]
- Daletos, G.; Ebrahim, W.; Ancheeva, E.; El-Neketi, M.; Song, W.G.; Lin, W.H.; Proksch, P. Natural products from deep-sea-derived fungi a new source of novel bioactive compounds? Curr. Med. Chem. 2018, 25, 186–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhang, H.; Gigant, B.; Yu, Y.M.; Wu, Y.P.; Chen, X.Z.; Lai, Q.H.; Yang, Z.Y.; Chen, Q.; Yang, J.L. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.X.; Wu, Y.; Xie, S.S.; Sun, W.G.; Guo, Y.; Li, X.N.; Liu, J.J.; Li, H.; Wang, J.P.; Luo, Z.W.; et al. Phomopsterones A and B, two functionalized ergostane-type steroids from the endophytic fungus Phomopsis sp. TJ507A. Org. Lett. 2017, 19, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Raju, R.; Salim, A.A.; Khalil, Z.G.; Capon, R.J. Cytochalasins from an Australian marine sediment-derived Phomopsis sp. (CMB-M0042F): Acid-mediated intramolecular cycloadditions enhance chemical diversity. J. Org. Chem. 2017, 82, 9704–9709. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Krohn, K.; Ahmed, I.; Draeger, S.; Schulz, B.; Di Pietro, S.; Pescitelli, G. Phomopsinones A–D: Four new pyrenocines from endophytic fungus Phomopsis sp. Eur. J. Org. Chem. 2012, 2012, 1783–1789. [Google Scholar] [CrossRef]
- Xie, S.S.; Wu, Y.; Qiao, Y.B.; Guo, Y.; Wang, J.P.; Hu, Z.X.; Zhang, Q.; Li, X.N.; Huang, J.F.; Zhou, Q.; et al. Protoilludane, illudalane, and botryane sesquiterpenoids from the endophytic fungus Phomopsis sp. TJ507A. J. Nat. Prod. 2018, 81, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Gao, J.; Shen, Y.; Chu, Y.L.; Xu, Q.; Tan, R.X. Immunosuppressive diterpenes from Phomopsis sp. S12. Eur. J. Org. Chem. 2014, 2014, 5728–5734. [Google Scholar] [CrossRef]
- Li, L.Y.; Sattler, I.; Deng, Z.W.; Groth, I.; Walther, G.; Menzel, K.D.; Peschel, G.; Grabley, S.; Lin, W.H. A-seco-oleane-type triterpenes from Phomopsis sp. (strain HKI0458) isolated from the mangrove plant Hibiscus tiliaceus. Phytochemistry 2008, 69, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Bunyapaiboonsri, T.; Yoiprommarat, S.; Srikitikulchai, P.; Srichomthong, K.; Lumyong, S. Oblongolides from the endophytic fungus Phomopsis sp. BCC9789. J. Nat. Prod. 2010, 73, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.W.; Liu, H.X.; Sun, Z.H.; Chen, Y.C.; Tan, Y.Z.; Zhang, W.M. Secondary metabolites from the deep-sea derived fungus Acaromyces ingoldii FS121. Molecules 2016, 21, 371. [Google Scholar] [CrossRef]
- Fan, Z.; Sun, Z.H.; Liu, Z.; Chen, Y.C.; Liu, H.X.; Li, H.H.; Zhang, W.M. Dichotocejpins A–C: New diketopiperazines from a deep-sea-derived fungus Dichotomomyces cejpii FS110. Mar. Drugs 2016, 14, 164. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Zhang, L.; Chen, Y.C.; Li, S.N.; Tan, G.H.; Sun, Z.H.; Pan, Q.L.; Ye, W.; Li, H.H.; Zhang, W.M. Cytotoxic pimarane-type diterpenes from the marine sediment-derived fungus Eutypella sp. FS46. Nat. Prod. Res. 2017, 31, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Zhang, L.; Chen, Y.C.; Sun, Z.H.; Pan, Q.L.; Li, H.H.; Zhang, W.M. Monoterpenes and sesquiterpenes from the marine sediment-derived fungus Eutypella scoparia FS46. J. Asian Nat. Prod. Res. 2017, 19, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.L.; Tan, H.B.; Chen, Y.C.; Li, S.N.; Huang, Z.L.; Guo, H.; Li, H.H.; Gao, X.X.; Liu, H.X.; Zhang, W.M. Lithocarpins A–D: Four tenellone-macrolide conjugated [4 + 2] hetero-adducts from the deep-sea derived fungus Phomopsis lithocarpus FS508. Org. Chem. Front. 2018, 5, 1792–1797. [Google Scholar] [CrossRef]
- Zhang, C.W.; Ondeyka, J.G.; Herath, K.B.; Guan, Z.Q.; Collado, J.; Platas, G.; Pelaez, F.; Leavitt, P.S.; Gurnett, A.; Nare, B.; et al. Tenellones A and B from a Diaporthe sp. Two highly substituted benzophenone inhibitors of parasite cGMP-dependent protein kinase activity. J. Nat. Prod. 2005, 68, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.D.; Jiang, Z.D.; Gallagher, R.T. Pine root (Deuteromycetes) fungal metabolites and analogs and derivatives thereof for anticancer agents. USA 5932613 A, 1999. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 141.0, C | 140.6, C | ||
| 2 | 117.4, C | 117.3, C | ||
| 3 | 160.8, C | 160.8, C | ||
| 4 | 7.06, d, (8.8) | 119.5, CH | 7.07, d, (8.7) | 119.6, CH |
| 5 | 7.45, d, (8.8) | 138.5, CH | 7.45, d, (8.7) | 138.7, CH |
| 6 | 129.9, C | 129.9, C | ||
| 7 | 3.12, s | 31.0, CH2 | 3.12, s | 31.0, CH2 |
| 8 | 5.05, m | 121.9, CH | 5.05, m | 121.7, CH |
| 9 | 134.1, C | 134.2, C | ||
| 10 | 1.47, s | 17.8, CH3 | 1.47, s | 17.8, CH3 |
| 11 | 1.58, s | 25.7, CH3 | 1.59, s | 25.7, CH3 |
| 12 | 203.1, C=O | 203.1, C=O | ||
| 13 | 9.71, s | 194.5, C=O | 9.71, s | 194.4, C=O |
| 1’ | 121.0, C | 121.3, C | ||
| 2’ | 151.7, C | 151.6, C | ||
| 3’ | 148.0, C | 147.7, C | ||
| 4’ | 6.93, d, (1.9) | 121.8, CH | 7.03, s | 123.7, CH |
| 5’ | 128.6, C | 129.1, C | ||
| 6’ | 6.47, d, (1.9) | 123.6, CH | 6.56, s | 125.1, CH |
| 7’ | 2.17, s | 21.1, CH3 | 2.19, s | 21.0, CH3 |
| 8α’ | 4.62, d, (5.7) | 66.3, CH2 | 4.12, dd, (9.5, 7.8) | 71.8, CH2 |
| 8β’ | 4.43, dd, (9.5, 2.8) | |||
| 9’ | 5.55, m | 119.4, CH | 4.07, dd, (7.8, 2.8) | 76.6, CH |
| 10’ | 138.7, C | 71.1, C | ||
| 11’ | 1.76, s | 18.4, CH3 | 1.69, s | 29.8, CH3 |
| 12’ | 1.80, s | 26.0, CH3 | 1.71, s | 28.4, CH3 |
| 3-OH | 11.51, s | 11.50, s | ||
| 2’-OH | 12.09, s | 12.13, s | ||
| No. | 3 | 4 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 140.6, C | 141.7, C | ||
| 2 | 117.3, C | 119.2, C | ||
| 3 | 160.8, C | 161.1, C | ||
| 4 | 7.08, d, (8.7) | 119.6, CH | 7.11, d, (8.6) | 119.6, CH |
| 5 | 7.46, d, (8.7) | 138.7, CH | 7.55, d, (8.6) | 139.2, CH |
| 6 | 129.9, C | 130.5, C | ||
| 7 | 3.13, s | 31.0, CH2 | 3.14, d, (7.2) | 31.4, CH2 |
| 8 | 5.06, m | 121.7, CH | 5.08, m | 122.3, CH |
| 9 | 134.2, C | 133.8, C | ||
| 10 | 1.48, s | 17.8, CH3 | 1.46, s | 17.6, CH3 |
| 11 | 1.59, s | 25.7, CH3 | 1.55, s | 25.7, CH3 |
| 12 | 203.2, C=O | 203.6, C=O | ||
| 13 | 9.72, s | 194.4, C=O | 9.98, s | 194.1, C=O |
| 1’ | 121.5, C | 123.2, C | ||
| 2’ | 151.5, C | 152.1, C | ||
| 3’ | 147.0, C | 148.8, C | ||
| 4’ | 6.90, d, (1.9) | 123.4, CH | 7.18, d, (2.0) | 123.0, CH |
| 5’ | 128.8, C | 129.2, C | ||
| 6’ | 6.57, s | 125.4, CH | 6.69, s | 124.6, CH |
| 7’ | 2.16, s | 21.0, CH3 | 2.16, s | 20.7, CH3 |
| 8α’ | 4.79, s | 72.9, CH2 | 4.01, dd, (9.8, 7.5) | 74.3, CH2 |
| 8β’ | 4.15, dd, (9.8, 3.9) | |||
| 9’ | 210.6, C=O | 4.47, dd, (7.5, 3.9) | 74.0, CH | |
| 10’ | 3.00, m | 37.2, CH | 146.0, C | |
| 11α’ | 1.20, s | 18.1, CH3 | 4.92, s | 112.2, CH2 |
| 11β’ | 5.12, m | |||
| 12’ | 1.21, s | 29.9, CH3 | 1.83, s | 19.0, CH3 |
| 3-OH | 11.51, s | |||
| 2’-OH | 12.10, s | |||
| No. | δH (J in Hz) | δC | No. | δH (J in Hz) | δC |
|---|---|---|---|---|---|
| 1 | 142.5, C | 4’ | 6.96, s | 121.7, CH | |
| 2 | 117.9, C | 5’ | 128.3, C | ||
| 3 | 162.9, C | 6’ | 6.69, m | 124.2, CH | |
| 4 | 7.16, d, (8.5) | 120.5, CH | 7’ | 2.12, s | 21.2, CH3 |
| 5 | 7.59, dd, (8.5, 7.3) | 136.1, CH | 8’ | 4.62, d, (7.0) | 66.3, CH2 |
| 6 | 6.98, d, (7.3) | 120.0, CH | 9’ | 5.55, m | 119.5, CH |
| 7 | 201.0, C=O | 10’ | 138.8, C | ||
| 8 | 9.91, s | 195.0, C=O | 11 | 1.76, s | 18.4, CH3 |
| 1’ | 119.9, C | 12’ | 1.80, s | 26.0, CH3 | |
| 2’ | 152.3, C | 3-OH | 11.74, s | ||
| 3’ | 148.1, C | 2’-OH | 11.93, s |
| No. | δH (J in Hz) | δC | No. | δH (J in Hz) | δC |
|---|---|---|---|---|---|
| 1α | 2.34, m | 30.2, CH2 | 13 | 1.85, s | 22.3, CH3 |
| 1β | 2.45, m | 14 | 0.98, d, (6.7) | 10.8, CH3 | |
| 2α | 1.46, m | 31.7, CH2 | 15 | 1.03, s | 17.3, CH3 |
| 2β | 2.16, overlapped | 1’ | 175.2, C=O | ||
| 3 | 4.87, td, (11.2, 4.4) | 74.2, CH | 2’ | 2.56, m | 46.0, CH |
| 4 | 1.67, dt, (11.2, 6.7) | 46.1, CH | 3’ | 4.24, m | 74.5, CH |
| 5 | 42.3, C | 4’ | 5.59, dd, (15.0, 7.0) | 131.5, CH | |
| 6α | 2.16, overlapped | 41.2, CH2 | 5’ | 6.27, dd, (15.0, 10.4) | 132.5, CH |
| 6β | 2.91, d, (13.7) | 6’ | 6.12, dd, (15.0, 10.4) | 132.5, CH | |
| 7 | 127.2, C | 7’ | 5.72, dd, (15.0, 7.0) | 131.5, CH | |
| 8 | 191.7, C=O | 8’ | 2.26, m | 42.7, CH2 | |
| 9 | 5.77, d, (1.8) | 126.9, CH | 9’ | 3.86, m | 67.5, CH |
| 10 | 164.9, C | 10’ | 1.20, d (6.2) | 23.1, CH3 | |
| 11 | 143.7, C | 11’ | 1.18, d (7.2) | 14.3, CH3 | |
| 12 | 2.10, s | 22.8, CH3 |
| Compounds | IC50 (μM) a | |||
|---|---|---|---|---|
| HepG-2 | MCF-7 | SF-268 | A549 | |
| 1 | >100 | >100 | >100 | >100 |
| 2 | >100 | >100 | >100 | >100 |
| 3 | >100 | >100 | >100 | >100 |
| 4 | 88.6 ± 3.1 | 85.7 ± 7.4 | 67.7 ± 3.1 | >100 |
| 5 | 16.0 ± 0.1 | 25.1 ± 1.1 | 23.0 ± 0.9 | 17.6 ± 0.3 |
| 6 | >100 | >100 | >100 | >100 |
| 7 | 90.9 ± 2.0 | 81.1 ± 2.8 | 92.5 ± 4.3 | 59.2 ± 2.1 |
| 8 | 26.2 ± 0.8 | 29.6 ± 4.6 | 28.8 ± 0.2 | 25.5 ± 0.4 |
| cisplatin | 2.4 ± 0.1 | 3.2 ± 0.1 | 3.3 ± 0.3 | 1.6 ± 0.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.-L.; Liu, H.-X.; Chen, Y.-C.; Tan, H.-B.; Guo, H.; Xu, L.-Q.; Li, S.-N.; Huang, Z.-L.; Li, H.-H.; Gao, X.-X.; et al. Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508. Mar. Drugs 2018, 16, 329. https://doi.org/10.3390/md16090329
Xu J-L, Liu H-X, Chen Y-C, Tan H-B, Guo H, Xu L-Q, Li S-N, Huang Z-L, Li H-H, Gao X-X, et al. Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508. Marine Drugs. 2018; 16(9):329. https://doi.org/10.3390/md16090329
Chicago/Turabian StyleXu, Jian-Lin, Hong-Xin Liu, Yu-Chan Chen, Hai-Bo Tan, Heng Guo, Li-Qiong Xu, Sai-Ni Li, Zi-Lei Huang, Hao-Hua Li, Xiao-Xia Gao, and et al. 2018. "Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508" Marine Drugs 16, no. 9: 329. https://doi.org/10.3390/md16090329
APA StyleXu, J. -L., Liu, H. -X., Chen, Y. -C., Tan, H. -B., Guo, H., Xu, L. -Q., Li, S. -N., Huang, Z. -L., Li, H. -H., Gao, X. -X., & Zhang, W. -M. (2018). Highly Substituted Benzophenone Aldehydes and Eremophilane Derivatives from the Deep-Sea Derived Fungus Phomopsis lithocarpus FS508. Marine Drugs, 16(9), 329. https://doi.org/10.3390/md16090329