Kinase-Based Screening of Marine Natural Extracts Leads to the Identification of a Cytotoxic High Molecular Weight Metabolite from the Mediterranean Sponge Crambe tailliezi




Abstract
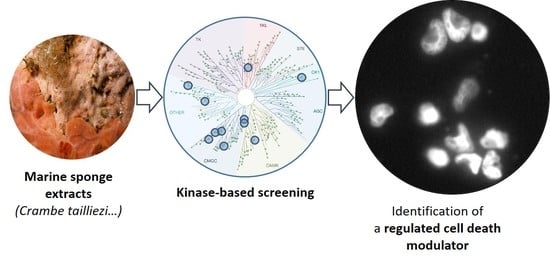
:
1. Introduction
2. Results
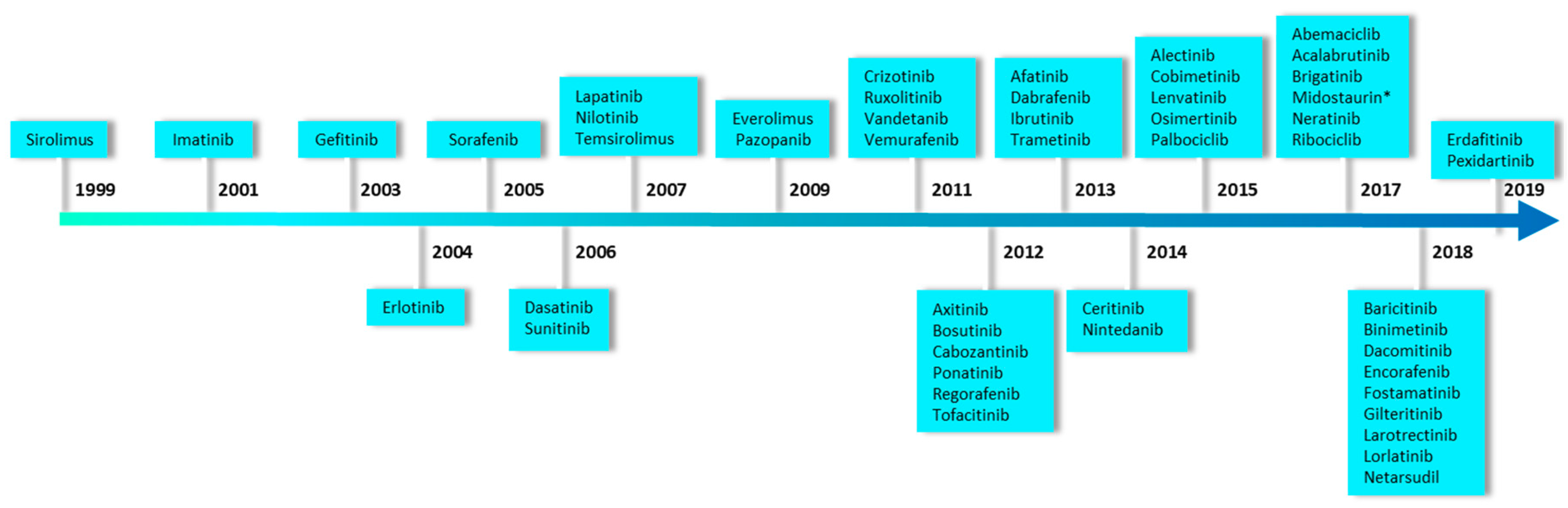
2.1. Primary Screening of a Selected Set of Purified Marine Extracts Against a Panel of Disease-Related Kinases
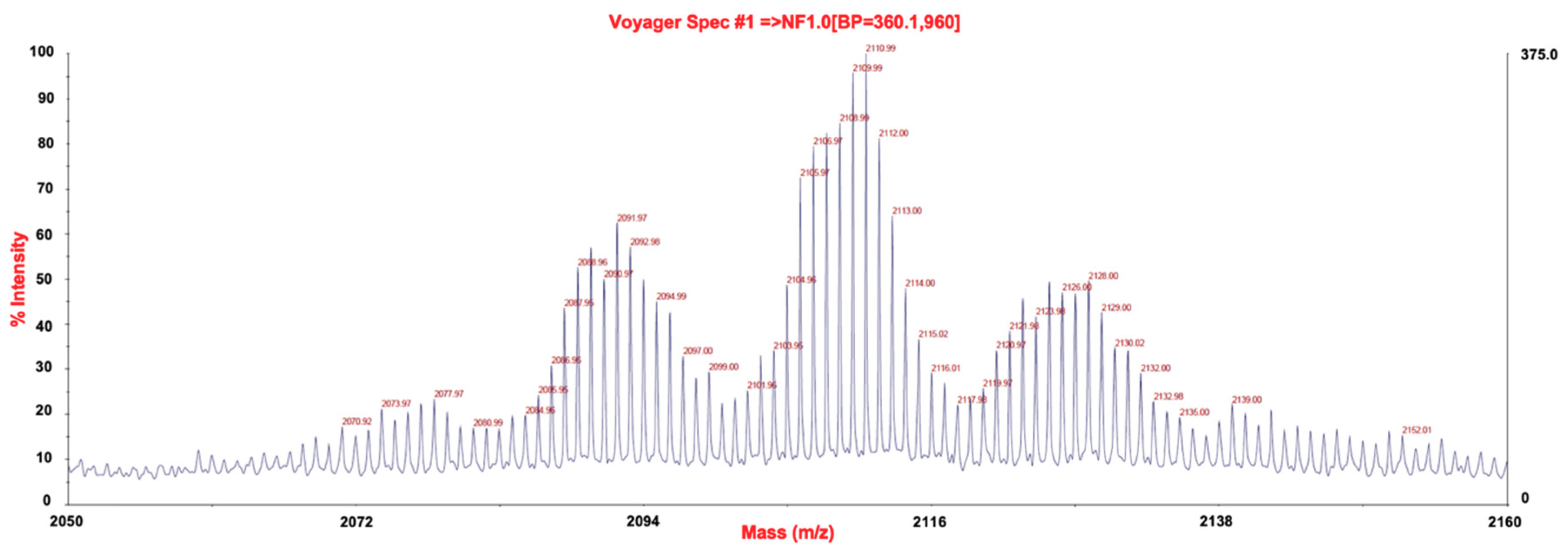
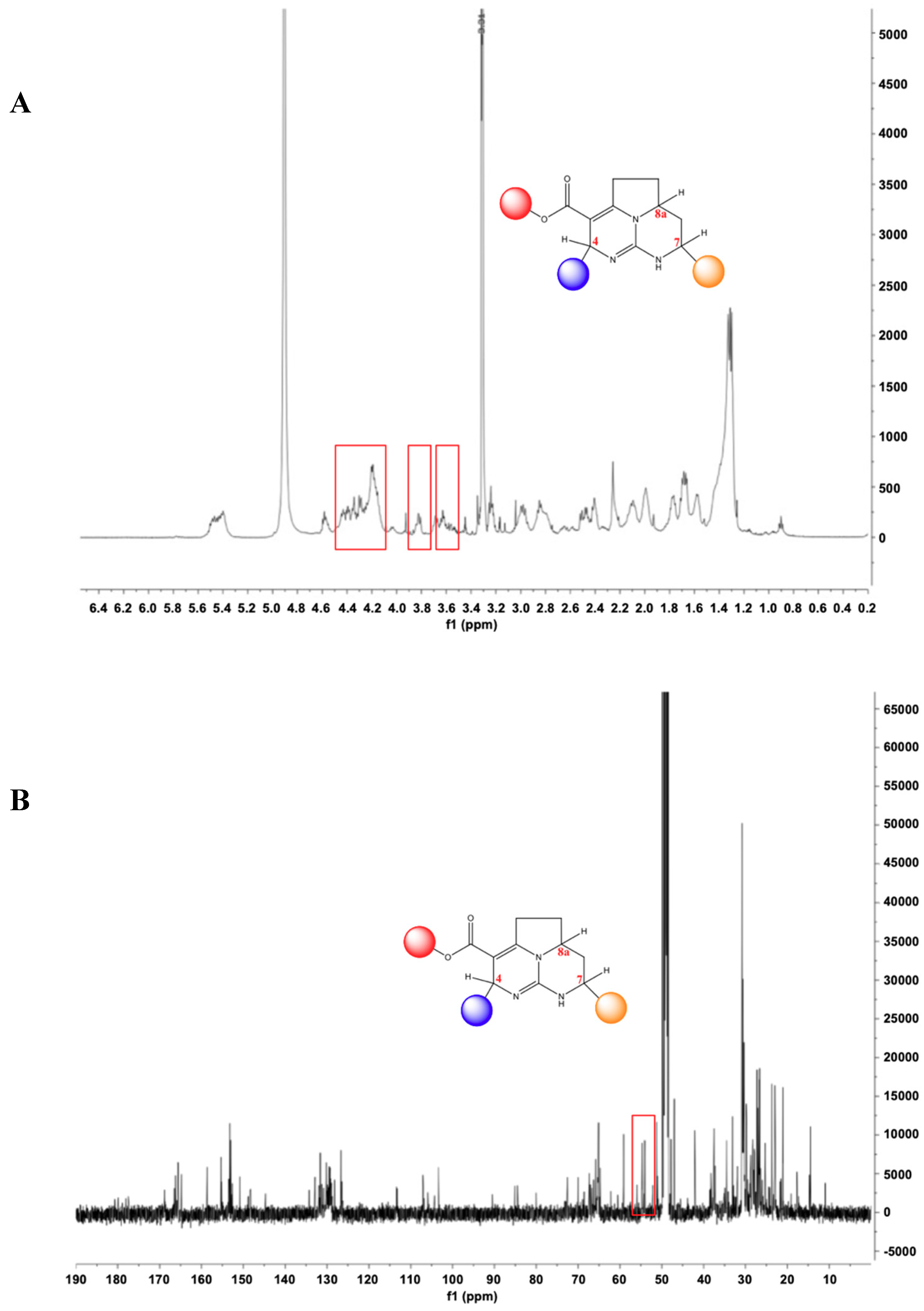
2.2. P3, a High Molecular Weight Natural Product Isolated from Crambe tailliezi Sponge, Selectively Inhibits Protein Kinases Aurora A and Aurora B
2.3. P3 Inhibits the Viability of Human Cancer Cell Lines
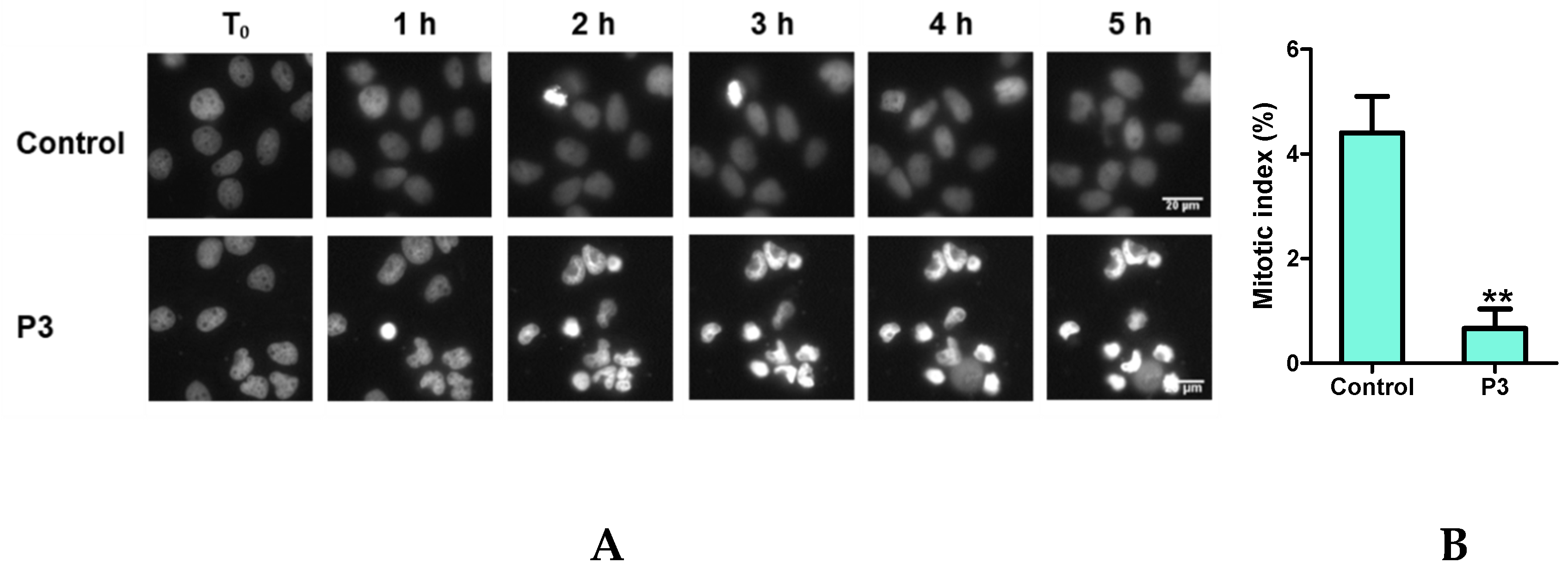
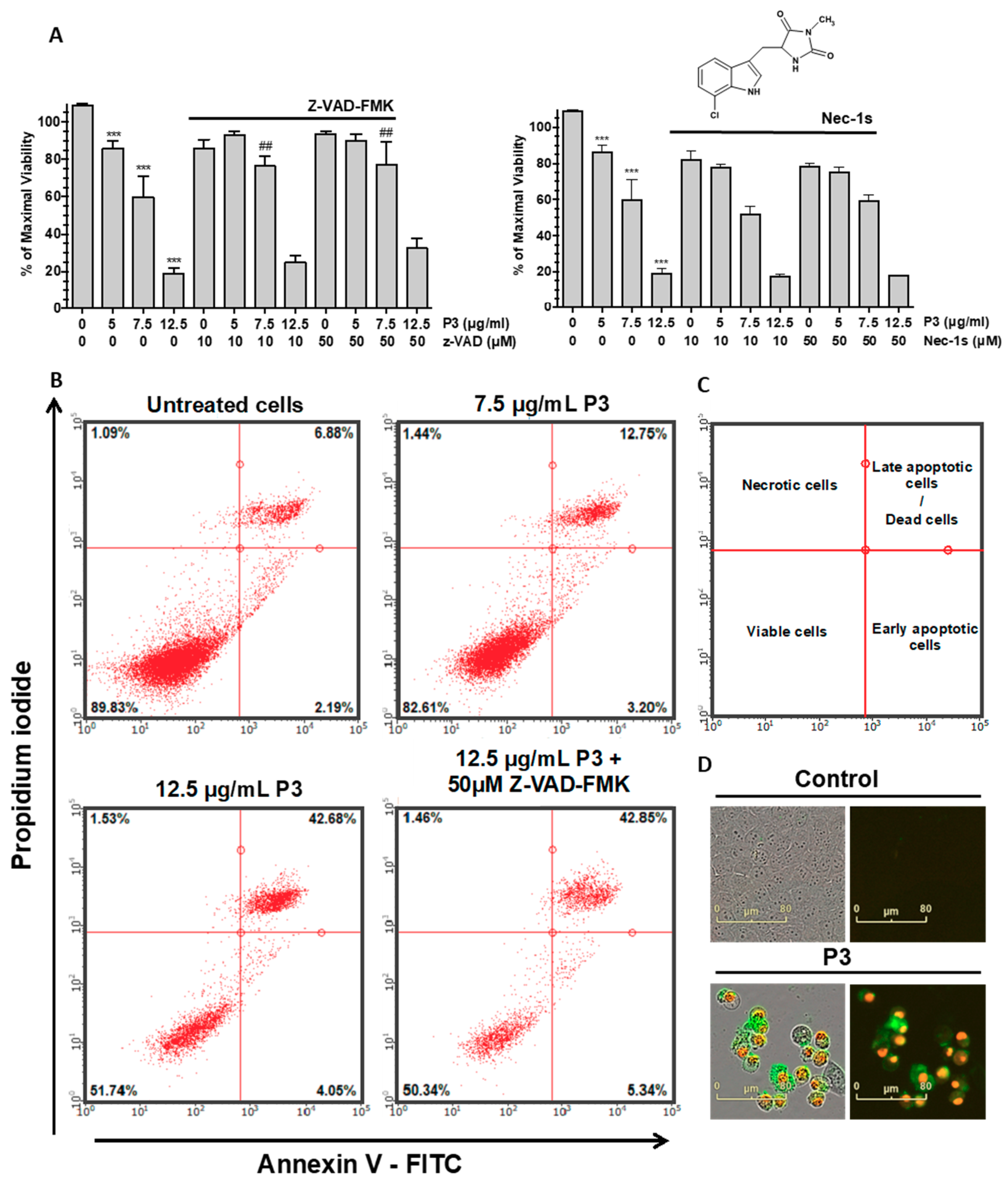
2.4. P3 Induces Apoptotic Death in Osteosarcoma U-2 OS Cell Line
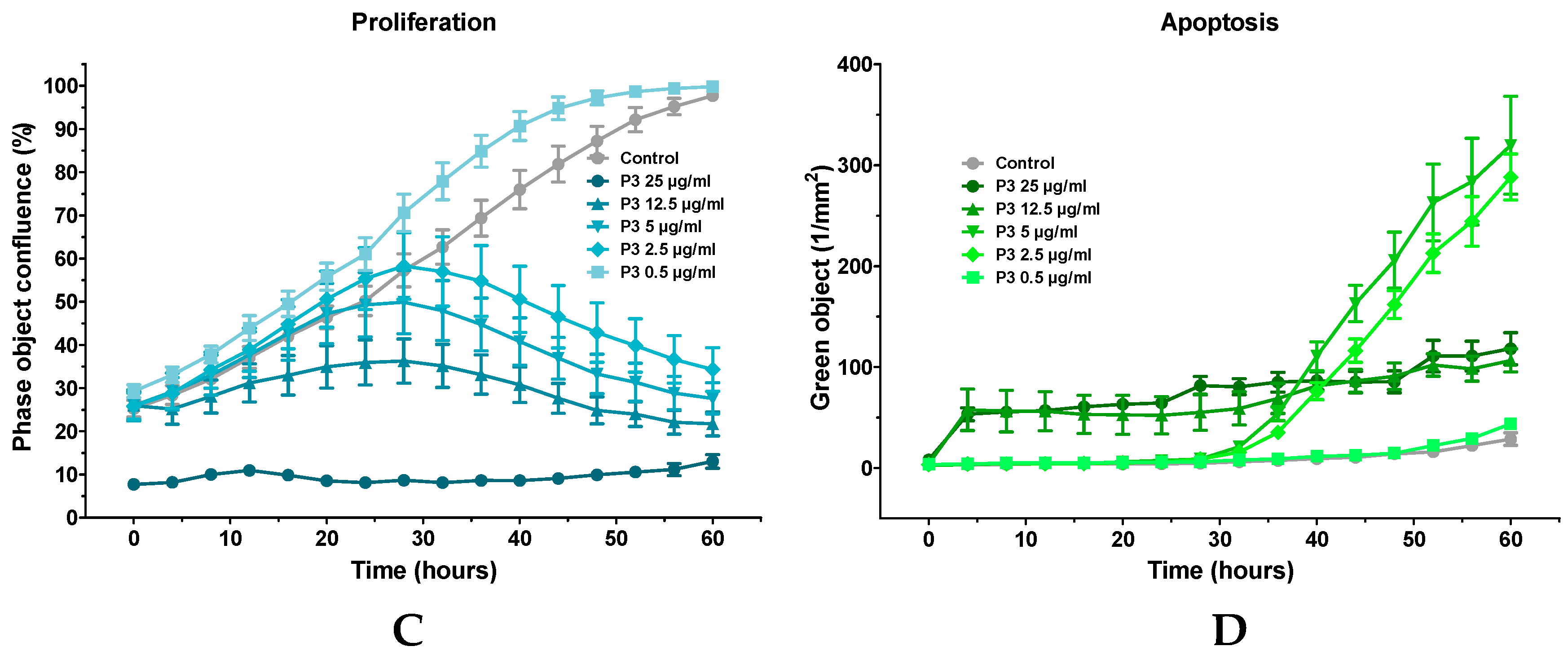
2.5. Characterization of P3-Induced Apoptotic Death in Osteosarcoma U-2 OS Cells
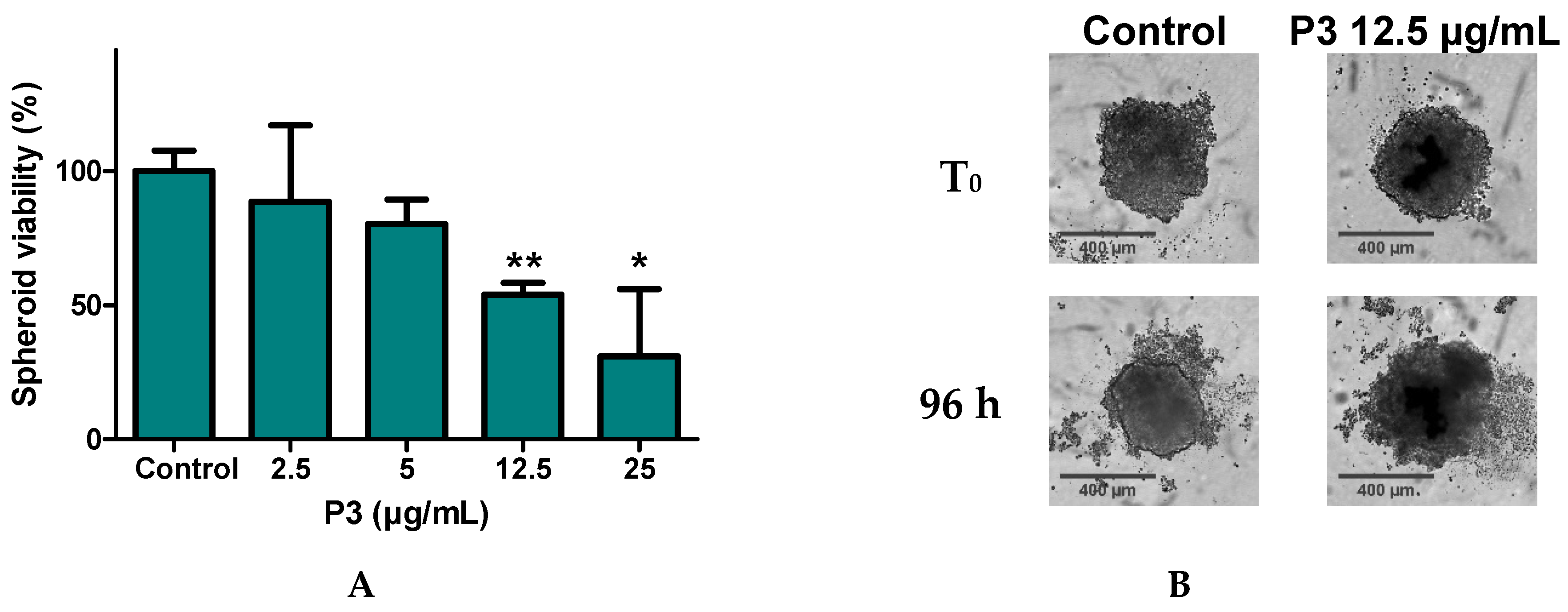
2.6. P3 Affects U-2 OS Spheroids Viability and Integrity
3. Discussion
4. Materials and Methods
4.1. Sample Material
4.2. Preparation of Natural Extracts and of P3 Pure Compound
4.3. Protein Kinase Assays
4.4. Cell Lines and Cell Culture
4.5. Cell Viability and LDH-Release Assays
4.6. Time-Lapse Assay
4.7. Live-Cell Imaging of Apoptosis in U-2 OS
4.8. Detection of Apoptosis by Flow Cytometry
4.9. Scratch Wound Assay to Evaluate the Cellular Migration and Proliferation
4.10. Spheroid Assay
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baehrecke, E.H. How death shapes life during development. Nat. Rev. Mol. Cell Biol. 2002, 3, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Lockshin, R.A.; Williams, C.M. Programmed cell death—II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J. Insect Physiol. 1964, 10, 643–649. [Google Scholar] [CrossRef]
- Lockshin, R.A. Programmed cell death. Activation of lysis by a mechanism involving the synthesis of protein. J. Insect Physiol. 1969, 15, 1505–1516. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999, 6, 1028–1042. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Bergmann, W.; Burke, D.C. Contributions to the study of marine products. XXXIX. The nucleosides of sponges. III.1 spongothymidine and spongouridine2. J. Org. Chem. 1955, 20, 1501–1507. [Google Scholar] [CrossRef]
- Hussain, S.; Fareed, S.; Ansari, S.; Sajid Khan, M. Marine natural products: A lead for Anti-cancer. Indian J. Geo-Mar. Sciences 2012, 41, 27–39. [Google Scholar]
- Kijjoa, A.; Sawangwong, P. Drugs and Cosmetics from the Sea. Mar. Drugs 2004, 2, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, E.A. Regulated Cell Death Signaling Pathways and Marine Natural Products That Target Them. Mar. Drugs 2019, 17, 76. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Sawant, S.D.; Singh, P.P.; Vishwakarma, R.A. Kinase inhibitors of marine origin. Chem. Rev. 2013, 113, 6761–6815. [Google Scholar] [CrossRef]
- Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase inhibitors from marine sponges. Mar. Drugs 2011, 9, 2131–2154. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, N.; Zhang, T.; Zhang, B.; Sajeevan, T.P.; Joseph, V.; Armstrong, L.; He, S.; Yan, X.; Naman, C.B. A Systematic Review of Recently Reported Marine Derived Natural Product Kinase Inhibitors. Mar. Drugs 2019, 17, 493. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A curated, annotated and updated database of protein kinase inhibitors in clinical trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef]
- Kim, E.S. Midostaurin: First global approval. Drugs 2017, 77, 1251–1259. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, J.; Wlodkowic, D. Towards High-Throughput Chemobehavioural Phenomics in Neuropsychiatric Drug Discovery. Mar. Drugs 2019, 17, 340. [Google Scholar] [CrossRef] [PubMed]
- Crambe Tailliezi Vacelet & Boury-Esnault. 1982. Available online: https://inpn.mnhn.fr/espece/cd_nom/71341 (accessed on 14 January 2019).
- Crambe Crambe (Schmidt, 1862). Available online: https://inpn.mnhn.fr/espece/cd_nom/71340 (accessed on 14 January 2019).
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Rogeau, D.; Amade, P. Crambines C1 and C2: Two Further Ichthyotoxic Guanidine Alkaloids from the Sponge Crambe crambe. J. Nat. Prod. 1992, 55, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic Guanidine Alkaloids from the Marine Sponge Crambe crambe and Ca++ Channel Blocker Activity of Crambescidin 816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef]
- Croué, J.; West, N.J.; Escande, M.-L.; Intertaglia, L.; Lebaron, P.; Suzuki, M.T. A single betaproteobacterium dominates the microbial community of the crambescidine-containing sponge Crambe crambe. Sci. Rep. 2013, 3, 2583. [Google Scholar] [CrossRef]
- Sfecci, E.; Lacour, T.; Amade, P.; Mehiri, M. Polycyclic Guanidine Alkaloids from Poecilosclerida Marine Sponges. Mar. Drugs 2016, 14, 77. [Google Scholar] [CrossRef]
- Butters, M.; Davies, C.D.; Elliott, M.C.; Hill-Cousins, J.; Kariuki, B.M.; Ooi, L.; Wood, J.L.; Wordingham, S.V. Synthesis and stereochemical determination of batzelladine C methyl ester. Org. Biomol. Chem. 2009, 7, 5001. [Google Scholar] [CrossRef]
- Görgün, G.; Calabrese, E.; Hideshima, T.; Ecsedy, J.; Perrone, G.; Mani, M.; Ikeda, H.; Bianchi, G.; Hu, Y.; Cirstea, D.; et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 2010, 115, 5202–5213. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Singh, K.; Mir, M.C.; Parker, Y.; Lindner, D.; Dreicer, R.; Ecsedy, J.A.; Zhang, Z.; Teh, B.T.; Almasan, A.; et al. The investigational Aurora kinase A inhibitor MLN8237 induces defects in cell viability and cell-cycle progression in malignant bladder cancer cells in vitro and in vivo. Clin. Cancer Res. 2013, 19, 1717–1728. [Google Scholar] [CrossRef]
- Agnese, V.; Bazan, V.; Fiorentino, F.P.; Fanale, D.; Badalamenti, G.; Colucci, G.; Adamo, V.; Santini, D.; Russo, A. The role of Aurora-A inhibitors in cancer therapy. Ann. Oncol. 2007, 18, 47–52. [Google Scholar] [CrossRef]
- Yeung, S.-C.J.; Gully, C.; Lee, M.-H. Aurora-B kinase inhibitors for cancer chemotherapy. Mini Rev. Med. Chem. 2008, 8, 1514–1525. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Linardopoulos, S. Aurora kinase inhibitors: current status and outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Fearnhead, H.O.; Cohen, G.M. An ICE-like protease is a common mediator of apoptosis induced by diverse stimuli in human monocytic THP.1 cells. FEBS Lett. 1995, 374, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Peng, W.; Liu, Y.; Yan, C.; Maki, J.; Degterev, A.; Yuan, J.; Shi, Y. Structural Basis of RIP1 Inhibition by Necrostatins. Structure 2013, 21, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, R.M.; Inch, W.R.; McCredie, J.A.; Kruuv, J. A multi-component radiation survival curve using an in vitro tumour model. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1970, 18, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of multicell spheroids in tissue culture as a model of nodular carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [PubMed]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Kayser, S. Midostaurin: A multiple tyrosine kinases inhibitor in acute myeloid leukemia and systemic mastocytosis. In Small Molecules in Hematology; Martens, U.M., Ed.; Recent Results in Cancer Research; Springer International Publishing: Cham, Switzerland, 2018; pp. 199–214. ISBN 978-3-319-91439-8. [Google Scholar]
- Schupp, P.; Eder, C.; Proksch, P.; Wray, V.; Schneider, B.; Herderich, M.; Paul, V. Staurosporine Derivatives from the Ascidian Eudistoma toealensis and Its Predatory Flatworm Pseudoceros sp. J. Nat. Prod. 1999, 62, 959–962. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Sakai, R.; Rinehart, K.L. Crambescidins: New antiviral and cytotoxic compounds from the sponge Crambe crambe. J. Org. Chem. 1991, 56, 5712–5715. [Google Scholar] [CrossRef]
- Olszewski, A.; Sato, K.; Aron, Z.D.; Cohen, F.; Harris, A.; McDougall, B.R.; Robinson, W.E.; Overman, L.E.; Weiss, G.A. Guanidine alkaloid analogs as inhibitors of HIV-1 Nef interactions with p53, actin, and p56lck. Proc. Natl. Acad. Sci. USA 2004, 101, 14079–14084. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Rodríguez, A.D.; Taglialatela-Scafati, O.; Fusetani, N. Marine Pharmacology in 2012–2013: Marine Compounds with Antibacterial, Antidiabetic, Antifungal, Anti-Inflammatory, Antiprotozoal, Antituberculosis, and Antiviral Activities; Affecting the Immune and Nervous Systems, and Other Miscellaneous Mechanisms of Action. Mar. Drugs 2017, 15, 273. [Google Scholar]
- Tischer, J.; Gergely, F. Anti-mitotic therapies in cancer. J. Cell Biol. 2019, 218, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Borner, C.; Monney, L. Apoptosis without caspases: An inefficient molecular guillotine? Cell Death Differ. 1999, 6, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Chiba, Y.; Tanioka, T.; Shimizu, N.; Shinozaki, S.; Yamada, M.; Kaneki, K.; Mori, S.; Araki, A.; Ito, H.; et al. NO donor induces Nec-1-inhibitable, but RIP1-independent, necrotic cell death in pancreatic β-cells. FEBS Lett. 2011, 585, 3058–3064. [Google Scholar] [CrossRef] [PubMed]
- Edrada-Ebel, R.; Jaspars, M. The 9th European Conference on Marine Natural Products. Mar. Drugs 2015, 13, 7150–7249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legrave, N.; Hamrouni-Buonomo, S.; Dufies, M.; Guérineau, V.; Vacelet, J.; Auberger, P.; Amade, P.; Mehiri, M. Nepheliosyne B, a New Polyacetylenic Acid from the New Caledonian Marine Sponge Niphates sp. Mar. Drugs 2013, 11, 2282–2292. [Google Scholar] [CrossRef] [Green Version]
- Stierlin, E.; Azoulay, S.; Massi, L.; Fernandez, X.; Michel, T. Cosmetic potentials of Prunus domestica L. leaves. J. Sci. Food Agric. 2018, 98, 726–736. [Google Scholar] [CrossRef]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and Homogeneous ADP Monitoring Assay for Kinases. ASSAY Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef]
- Raynal, P.; Pollard, H.B. Annexins: The problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim. et Biophys. Acta (BBA)—Rev. Biomembr. 1994, 1197, 63–93. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin, V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extract | Marine organism | Hs_CDK2/CyclinA | Hs_CDK5/p25 | Hs_CDK9/CyclinT | Hs_Haspin | Hs_PIM1 | Rn_DYRK1A | Hs_CK1ε | Hs_GSK3α | Hs_GSK3β | Hs_RIPK3 | Hs_AuroraB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E1 | Ascidium Aplidium sp. | 81 | ≥100 | 100 | 63 | ≥100 | 97 | 94 | ≥100 | 76 | 87 | 99 |
| E2 | Ascidium Cystodytes sp. | 68 | 58 | 52 | 25 | 62 | 29 | 89 | 54 | 70 | 77 | 83 |
| E3 | Ascidium Halocynthia papillosa | 85 | 73 | 81 | 65 | 90 | 95 | ≥100 | 91 | 97 | 91 | 100 |
| E4 | Ascidium Polysyncraton sp. | 69 | 82 | ≥100 | 94 | 92 | ≥100 | ≥100 | ≥100 | 88 | 86 | 90 |
| E5 | Sponge Acanthella acuta | 100 | ≥100 | 83 | 86 | ≥100 | 87 | 78 | ≥100 | 69 | ≥100 | ≥100 |
| E6 | Sponge Agelas oroides | 71 | 74 | 46 | 26 | 67 | 19 | 46 | 23 | 35 | 80 | 69 |
| E7 | Sponge Axinella polypoides | ≥100 | ≥100 | 87 | 84 | ≥100 | 81 | 77 | ≥100 | 67 | ≥100 | ≥100 |
| E8 | Sponge Axinella sp. | 98 | ≥100 | 91 | 93 | 87 | 63 | 82 | 68 | 71 | 87 | ≥100 |
| E9 | Sponge Axinyssa sp. | 71 | 63 | 80 | 96 | ≥100 | 51 | ≥100 | 96 | 88 | 96 | 83 |
| E10 | Sponge Cacospongia sp. | ≥100 | ≥100 | 71 | 79 | 100 | ≥100 | 92 | 88 | 68 | 93 | 96 |
| E11 | Sponge Cliona viridis | 64 | ≥100 | 94 | ≥100 | ≥100 | 85 | 98 | 100 | 85 | 95 | 100 |
| E12 | Sponge Crambe crambe | 66 | 62 | 47 | ≥100 | 16 | 37 | 19 | 84 | ≥100 | ≥100 | 79 |
| E13 | Sponge Crambe tailliezi | 77 | 57 | 48 | 78 | 54 | 27 | 22 | 21 | 28 | 87 | 8 |
| E14 | Sponge Haliclona mediterranea | 91 | ≥100 | 89 | 87 | ≥100 | 61 | 96 | 84 | ≥100 | 97 | 100 |
| E15 | Sponge Hemimycale columella | ≥100 | ≥100 | ≥100 | 84 | ≥100 | 100 | ≥100 | 92 | 62 | 91 | 94 |
| E16 | Sponge Hexadella sp. | 98 | 65 | 7 | 17 | 63 | 4 | 2 | 2 | 10 | 65 | 61 |
| E17 | Sponge Ircinia oros | 40 | ≥100 | 30 | 34 | ≥100 | 26 | 17 | 13 | 9 | 72 | 91 |
| E18 | Sponge Ircinia variabilis | 100 | 92 | 58 | 42 | 75 | 50 | ≥100 | 41 | 37 | 88 | 94 |
| E19 | Sponge Oscarella sp. | 66 | ≥100 | 96 | 89 | 92 | ≥100 | ≥100 | ≥100 | ≥100 | 97 | 98 |
| E20 | Sponge Phorbas topsenti | ≥100 | ≥100 | 91 | 77 | ≥100 | 75 | 88 | 93 | 69 | 98 | ≥100 |
| E21 | Sponge Pleraplesila spinifera | ≥100 | ≥100 | 78 | 79 | ≥100 | ≥100 | ≥100 | 98 | 54 | 90 | 87 |
| E22 | Sponge Pseudaxinyssa sp. | 96 | ≥100 | 31 | 12 | ≥100 | 35 | 1 | 5 | 25 | 84 | ≥100 |
| E23 | Sponge Reniera fulva | 65 | ≥100 | ≥100 | 61 | ≥100 | 83 | ≥100 | 100 | 58 | ≥100 | 99 |
| E24 | Sponge Reniera mucosa | 88 | 86 | 28 | 25 | 96 | 50 | 17 | 14 | 45 | 64 | 43 |
| E25 | Sponge Reniera sarai | ≥100 | 76 | 36 | 11 | 50 | 42 | 42 | 28 | 51 | 86 | 86 |
| E26 | Sponge Sarcotragus foetidus | 60 | ≥100 | 66 | 14 | 79 | 29 | 21 | 13 | 4 | 92 | 97 |
| E27 | Sponge Sarcotragus spinosulus | 65 | ≥100 | 62 | 35 | 40 | 6 | 7 | 8 | 12 | 86 | 91 |
| Extract/Compound | Marine organism | IC50 (µg/mL) | |
|---|---|---|---|
| Aurora A | Aurora B | ||
| E12 | Sponge Crambe crambe | N/A | 124.9 |
| E13 | Sponge Crambe tailliezi | 14.7 | 7.51 |
| P3 | Sponge Crambe tailliezi | 7.58 | 2.63 |
| Cell type | Cell line | Tissue | EC50 (µg/mL) ± SD |
|---|---|---|---|
| Leukemia | A3 | T lymphocyte | 7.0 ± 0.6 |
| Solid malignancies | U-2 OS | Bone | 6.6 ± 0.2 * |
| SH-SY5Y | Bone marrow | 8.9 ± 0.2 | |
| U-87 | Brain | 10.6 ± 0.5 | |
| Hep G-2 | Liver | 11.8 ± 1.6 | |
| MCF-7 | Breast | 12.2 ± 1.6 | |
| AsPC-1 | Pancreas | 12.4 ± 0.6 | |
| HT-29 | Colon | 20.4 ± 0.7 | |
| PANC-1 | Pancreas | 22.4 ± 1.4 | |
| Non-malignant | hTERT RPE-1 | Retina, eye | 7.2 ± 0.4 |
| HEK-293 | Kidney | 11.2 ± 0.6 | |
| RC-124 | Kidney | 13.3 ± 0.8 | |
| HT-22 | Brain (mouse) | 14.0 ± 1.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.-N.-D.; Feizbakhsh, O.; Sfecci, E.; Baratte, B.; Delehouzé, C.; Garcia, A.; Moulin, C.; Colas, P.; Ruchaud, S.; Mehiri, M.; et al. Kinase-Based Screening of Marine Natural Extracts Leads to the Identification of a Cytotoxic High Molecular Weight Metabolite from the Mediterranean Sponge Crambe tailliezi. Mar. Drugs 2019, 17, 569. https://doi.org/10.3390/md17100569
Nguyen T-N-D, Feizbakhsh O, Sfecci E, Baratte B, Delehouzé C, Garcia A, Moulin C, Colas P, Ruchaud S, Mehiri M, et al. Kinase-Based Screening of Marine Natural Extracts Leads to the Identification of a Cytotoxic High Molecular Weight Metabolite from the Mediterranean Sponge Crambe tailliezi. Marine Drugs. 2019; 17(10):569. https://doi.org/10.3390/md17100569
Chicago/Turabian StyleNguyen, Thi-Ngoc-Dung, Omid Feizbakhsh, Estelle Sfecci, Blandine Baratte, Claire Delehouzé, Adrien Garcia, Corentin Moulin, Pierre Colas, Sandrine Ruchaud, Mohamed Mehiri, and et al. 2019. "Kinase-Based Screening of Marine Natural Extracts Leads to the Identification of a Cytotoxic High Molecular Weight Metabolite from the Mediterranean Sponge Crambe tailliezi" Marine Drugs 17, no. 10: 569. https://doi.org/10.3390/md17100569
APA StyleNguyen, T. -N. -D., Feizbakhsh, O., Sfecci, E., Baratte, B., Delehouzé, C., Garcia, A., Moulin, C., Colas, P., Ruchaud, S., Mehiri, M., & Bach, S. (2019). Kinase-Based Screening of Marine Natural Extracts Leads to the Identification of a Cytotoxic High Molecular Weight Metabolite from the Mediterranean Sponge Crambe tailliezi. Marine Drugs, 17(10), 569. https://doi.org/10.3390/md17100569