1. Introduction
Gliotoxin (GT), which represents the most important epidithiodioxopiperazine (ETP) type fungal toxin, is considered a lead agent for anticancer drugs due to its potent cytotoxicity [
1,
2]. Since the first description by Weindling and Emerson as a metabolite from
Trichoderma lignorum [
3], gliotoxin and its derivatives have been discovered from various fungal species, such as bi(dethio)-10a-methylthio-3a-deoxy-3,3a-didehydrogliotoxin and 6-deoxy-5a,6-didehydrogliotoxin from
Aspergillus fumigatus [
2], bisdethiobis (methylthio) gliotoxin from
Gliocladium deliquescens [
4], and the dimeric eutypellizines N–S from
Eutypella sp [
5]. Furthermore, GT derivatives are well known for their diverse bioactivities, especially the cytotoxicity. Researchers who have focused on the anticancer mechanism of GT derivatives on different cancer cell lines indicated that the intramolecular disulfide bridge makes the most influential contribution to their activities [
6]. Nevertheless, to date, to the best of our knowledge, there is no relevant report about gliotoxin dimers which connect through inter-monomer disulfide bridge.
Deep-sea fungi, inhabiting the sediment or water at a depth over 1000 m below the surface, exhibit the potential to metabolize bioactive natural products with fantastic skeletons [
7]. Among them, over 50% show cytotoxicity against different human cancer cell lines [
7], for example, the cladosins I from
Cladosporium sphaerospermum can suppress HL-60 cell line with the IC
50 value of 2.8 μM [
8]. Our previous studies suggested that a strain of
Dichotomomyces cejpii FS110, isolated from deep-sea sediments from South China Sea, tended to produce novel cytotoxic gliotoxin derivatives while being cultured in PDB medium [
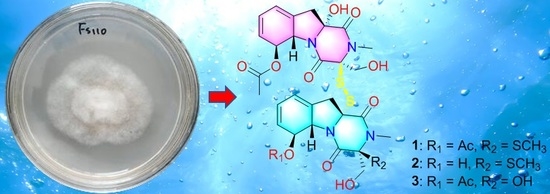
9]. In this study, further chemical and biological investigations of the extract from solid culture medium of this strain led to the discovery of three dimeric gliotoxin derivatives, dechdigliotoxins A–C (
1–3), which represented the first examples of gliotoxin dimers constructing an unprecedented exocyclic disulfide linkage, as well as four previously obtained monomers (
4–7). Herein, the details of the isolation, structure identification, and biological assay of the isolated compounds are discussed.
2. Results and Discussion
The solid fermentation product of the deep-sea-derived fungus
Dichotomomyces cejpii FS110 was extracted with methanol and then concentrated under reduced pressure to give an extract, which was further subjected to repeated column chromatography to afford compounds
1–7 (
Figure 1). The known compounds were identified as dichotocejpins C and B (
4 and
5, respectively), as well as gliotoxin and acetylgliotoxin (
6 and
7, respectively) by comparing their spectroscopic data with those previously isolated [
9].
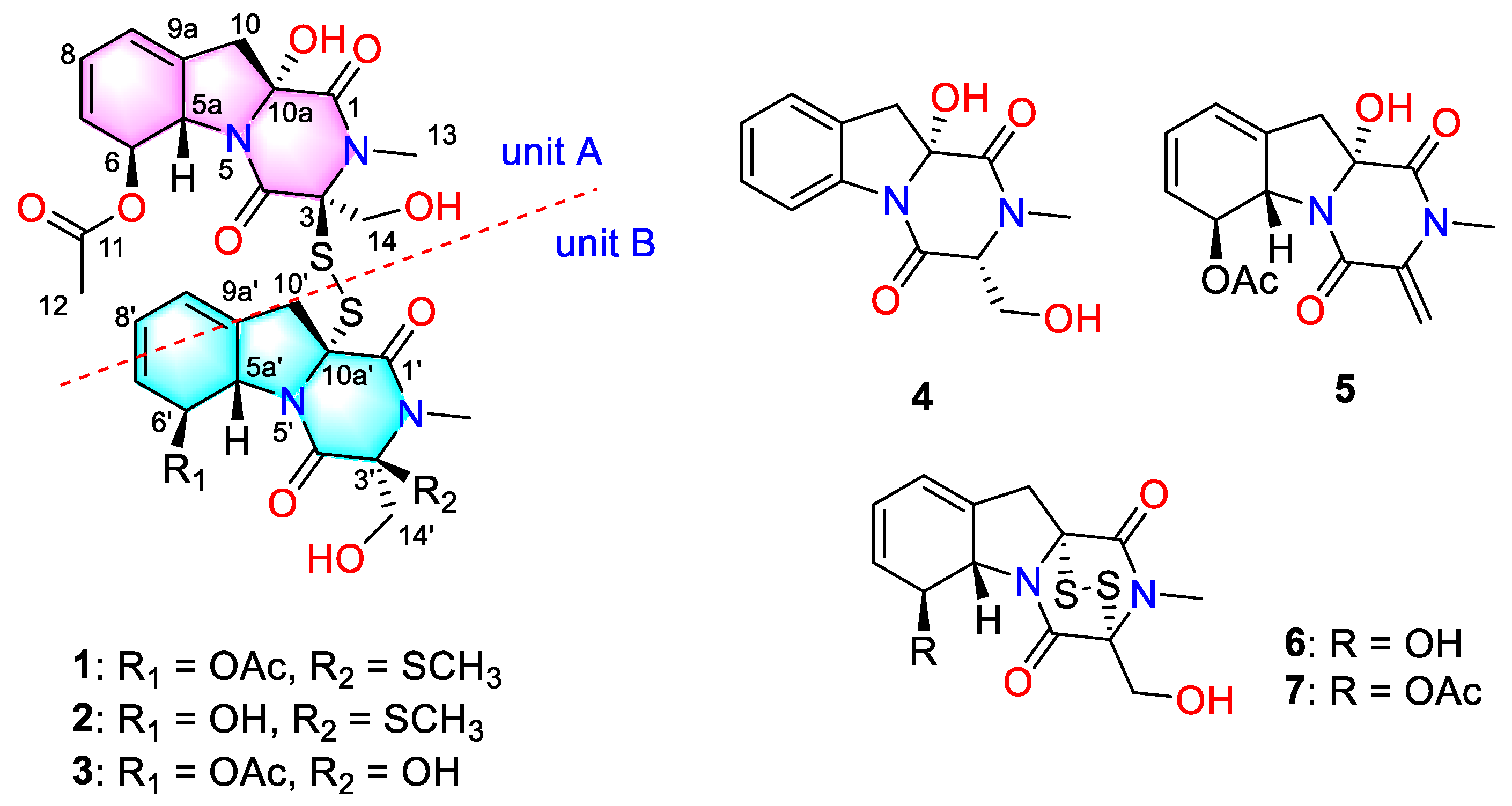
Dechdigliotoxin A (
1) was obtained as a colorless powder with molecular formula of C
31H
36N
4O
11S
3 established on the basis of the sodiated HRESIMS peak at m/z 759.1454 [M + Na]
+ (calcd 759.1440, C
31H
36N
4O
11S
3Na). The
1H NMR spectrum recorded in DMSO (
Table S1) gave the presence of a quaternary hydroxy groups at
δH 6.97 (10a-OH, s) and two primary alcohol hydroxy groups at
δH 5.28 (14-OH, t,
J = 5.8) and 5.40 (14′-OH, dd,
J = 6.5, 5.4); five methyls including two N-methyls at
δH 3.03 and 2.96 (13/13′-CH
3, s); and two hydroxymethyl groups at
δH 4.33/3.85 (14-CH
2, dd) and 4.05/3.61 (14′-CH
2, dd).
13C NMR, as well as HSQC spectra, revealed 31 signals, which could be divided into two sets, each containing five methines, two methyls, two methylene groups, and six quaternary carbons (including three carbonyl carbons). Comprehensive analysis of the 1D NMR and HRESIMS data suggested that dechdigliotoxin A was an unsymmetic dimeric derivative. The characteristic absorption band at 566 cm
−1 in IR spectrum indicated the existence of a disulfide bond structure feature.
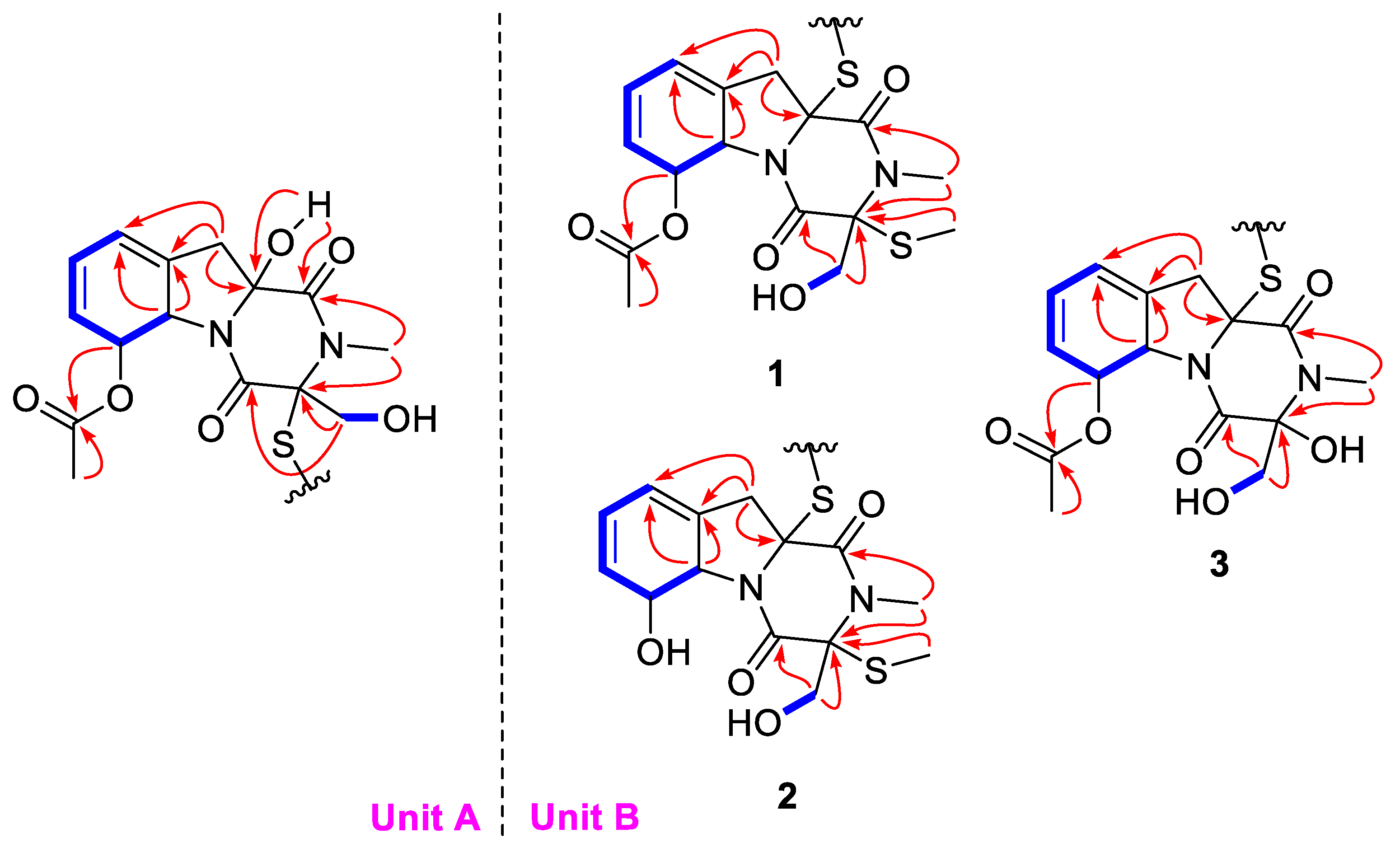
The complete structure was further elucidated by the COSY and HMBC correlations (
Figure 2). In unit A, the two independent coupling fragments were deduced by the
1H,
1H-COSY correlations of H-5a/H-6/H-7/H-8/H-9 and H
2-14/14-OH. The HMBC cross-peaks from 10a-OH to C-1, H
3-13 to C-1/C-3, as well as H
2-14 to C-3/C-4, indicated the presence of diketopiperazine moiety. Furthermore, the correlations from H-5a to C-9/C-9a and H
2-10 to C-5a/C-9/C-9a in HMBC spectrum constructed a gliotoxin derived skeleton of
1. The acetyl was linked to C-6 based on the detected correlation between H-6 to C-11. In unit B, the COSY and HMBC data indicated the same skeleton as that in unit A. The main difference was that an additional methyl was connected to C-3′ via a sulfur atom, which was evidenced by the chemical shifts and the only HMBC correlation from S-methyl to C-3′. Hence, the planar structure was deduced as shown.
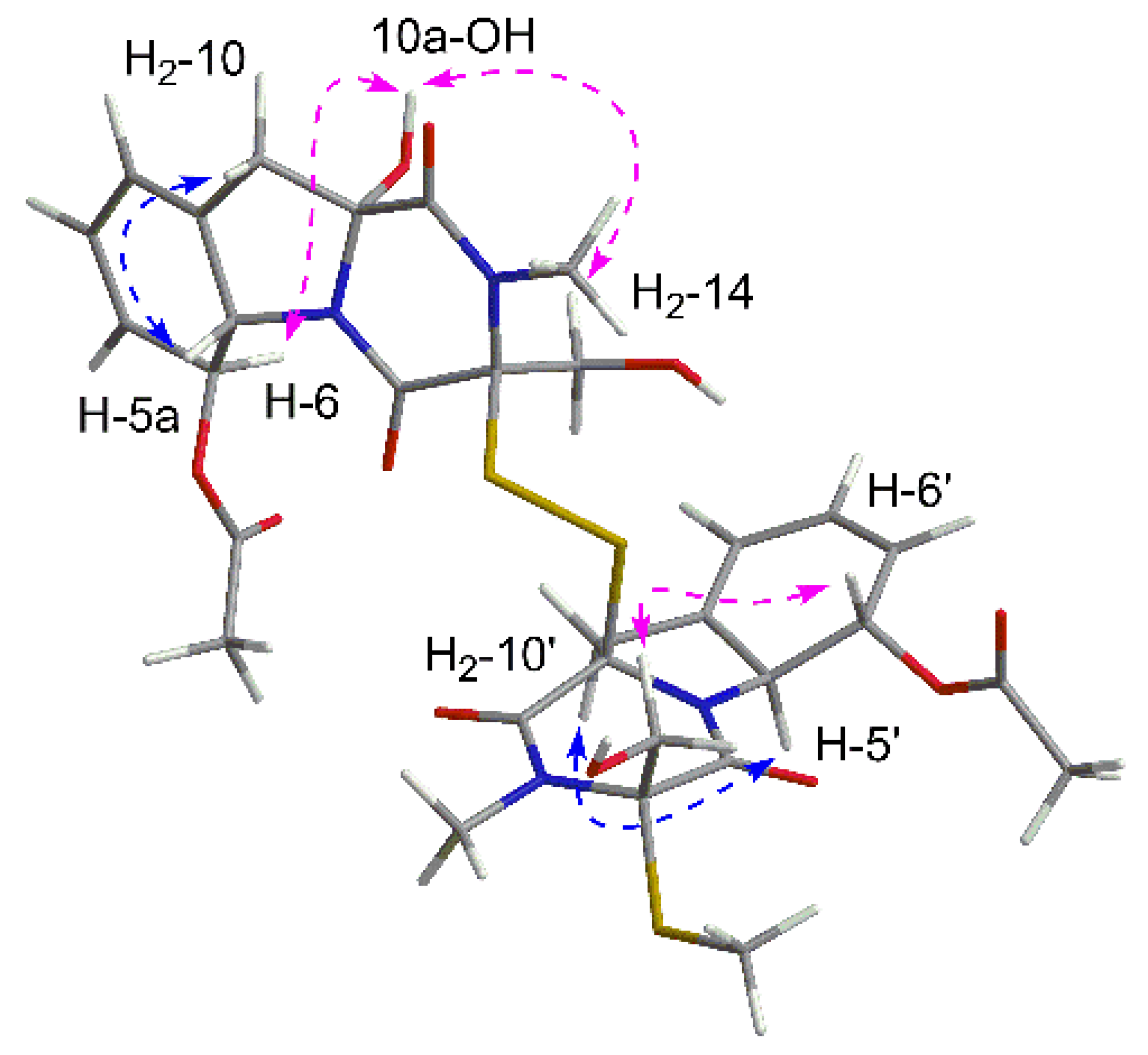
In order to elucidate the relative configuration of
1, the NOESY spectrum (
Figure 3) was recorded in DMSO. In unit A, the lack of correlations between H
2-10 and H
2-14 combined with the cross peaks of 10a-OH/H
2-14 suggested the same side of 10a-OH and the hydroxymethyl, which were assigned to be
α-oriatation. The NOE effect between H
2-10 and H-5a was detected, indicating the
β-oriatation of H-5a. Finally, the large coupling constant (
J = 14.3 Hz) and the correlation of H-6/10a-OH confirmed the trans configuration of H-5a and H-6. In unit B, the chirality of C-5a′, C-6′, C-10a′ could be easily deduced through NOE correlations and coupling constant, which were the same as those in unit A. Additionally, the correlations between H
2-14′ and H-6′ revealed the co-facial of them.
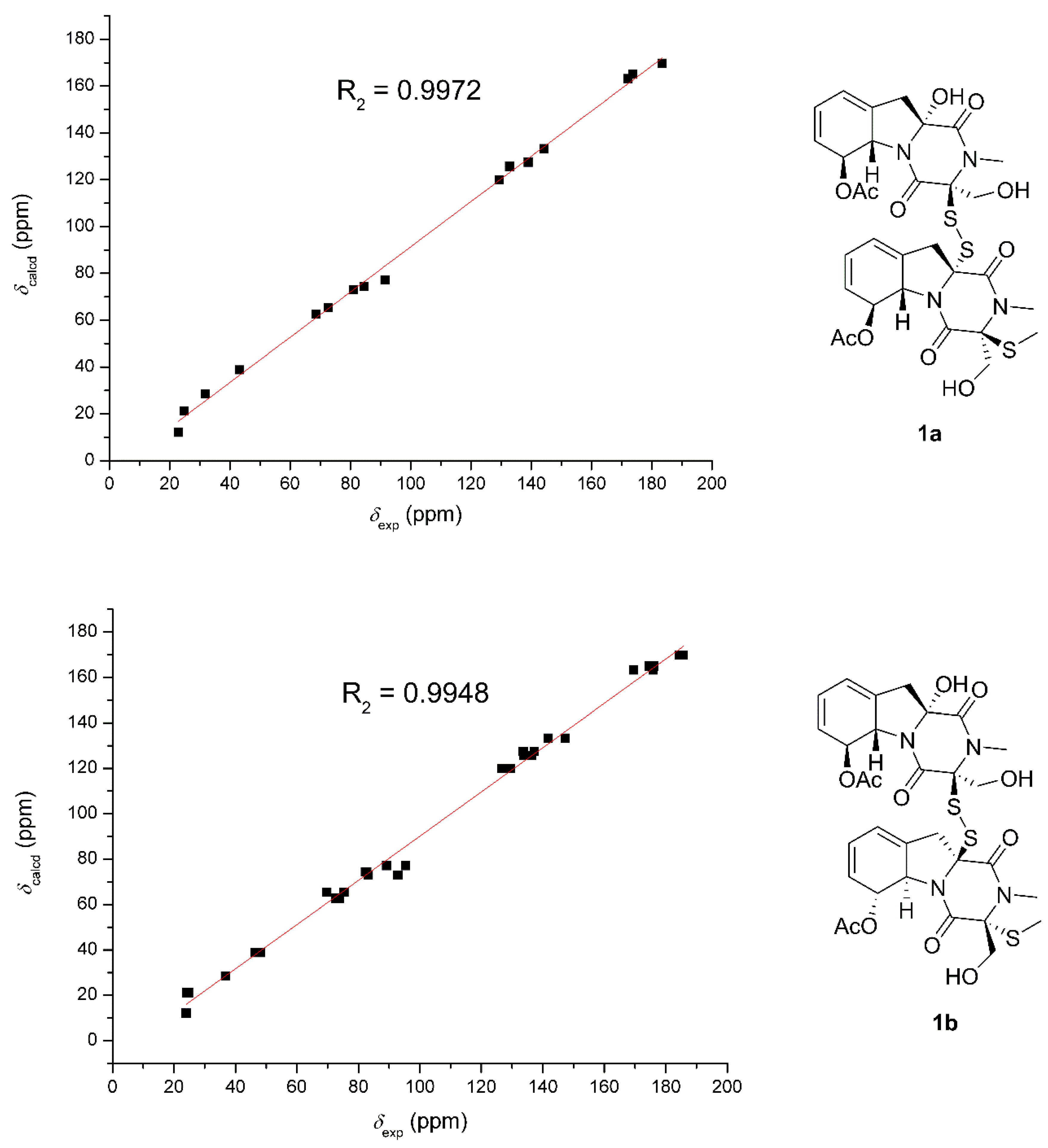
Because of the lack of convincible NOE correlations, the relative configuration between the two units could not be determined exactly. Hence, the theoretical
13C NMR chemical shifts calculations and the DP4+ probability simulation of two possible relative structures (
1a and
1b) were carried out. Conformers within the 5 kcal mol
−1 energy window were generated to the b3lyp/6-31+g(d,p) level. Optimized conformers with the Boltzmann distribution over 2% were chosen for NMR chemical shifts calculations in dimethyl sulphoxide at the mPW1PW91/6-311+g(d,p) level. The result (
Figure 4) indicated that
1a exhibited a better correlation coefficient (R
2: 0.9972) and mean absolute errors (MAE) value (9.5 ppm) as compared with those of
1b (R
2: 0.9948; MAE = 10.0 ppm). Moreover, DP4+ simulation suggested
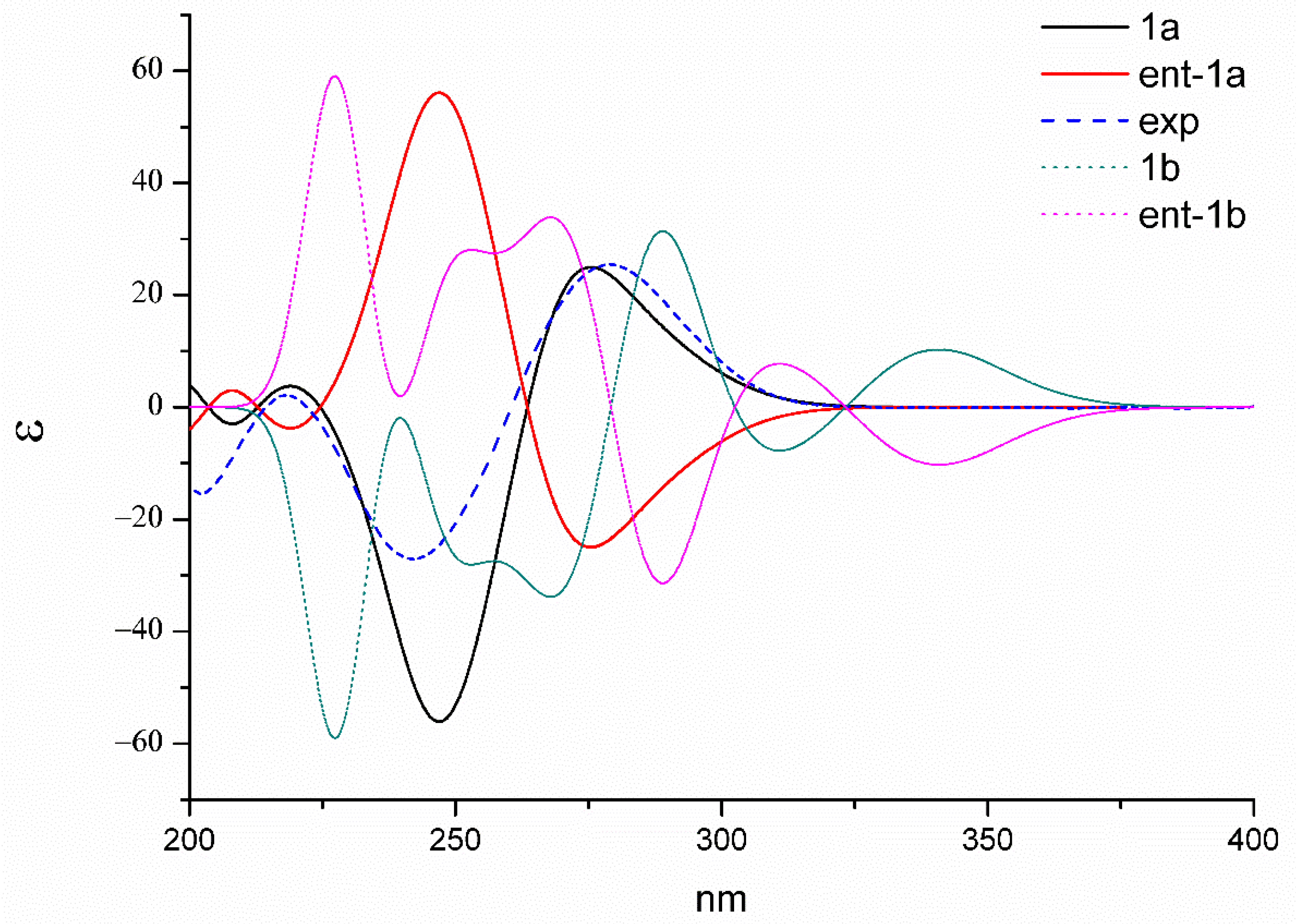
1a should be the true structure with 99.89% probability. Then, the absolute configuration of
1 was elucidated by performing a quantum chemical calculation of the ECD spectra at the b3lyp/6-311+g(d,p) level. The optimized conformers of both
1a and
1b were selected based on the previous NMR calculations. The results shown in
Figure 5 suggested that the calculated plot of
1a exhibited a better fit to the experimental spectrum. Thus, the absolute configuration was assigned as 3
S, 5a
S, 6
S, 10a
R, 3′
S, 5a′
S, 6′
S, and 10a′
R.
Dechdigliotoxin B (
2), which was obtained as a colorless powder, gave the molecular formula of C
29H
34N
4O
10S
3 based on the sodiated HRESIMS peak at m/z 717.1323 [M + Na]
+ (calcd 717.1335, C
29H
34N
4O
10S
3Na). The
1H and
13C NMR spectra recorded in methanol were nearly identical to those of
1 and the main difference was that the methyl signal
δH 2.00 (12′-Me) and the carbonyl carbon
δC 169.8 (C-11′) were absent. Moreover, the obvious upfield shifts of H-5a′ and H-6′ (from
δH-5a′ 5.18 and
δH-6′ 6.14 in
1 to
δH-5a′ 4.96 and
δH-6′ 4.86 in
2, respectively) were observed, suggesting that dechdigliotoxin B was a deacetylation product of
1 at C-6′. Further analysis of 2D NMR data confirmed the planar structure as shown (
Figure 2).
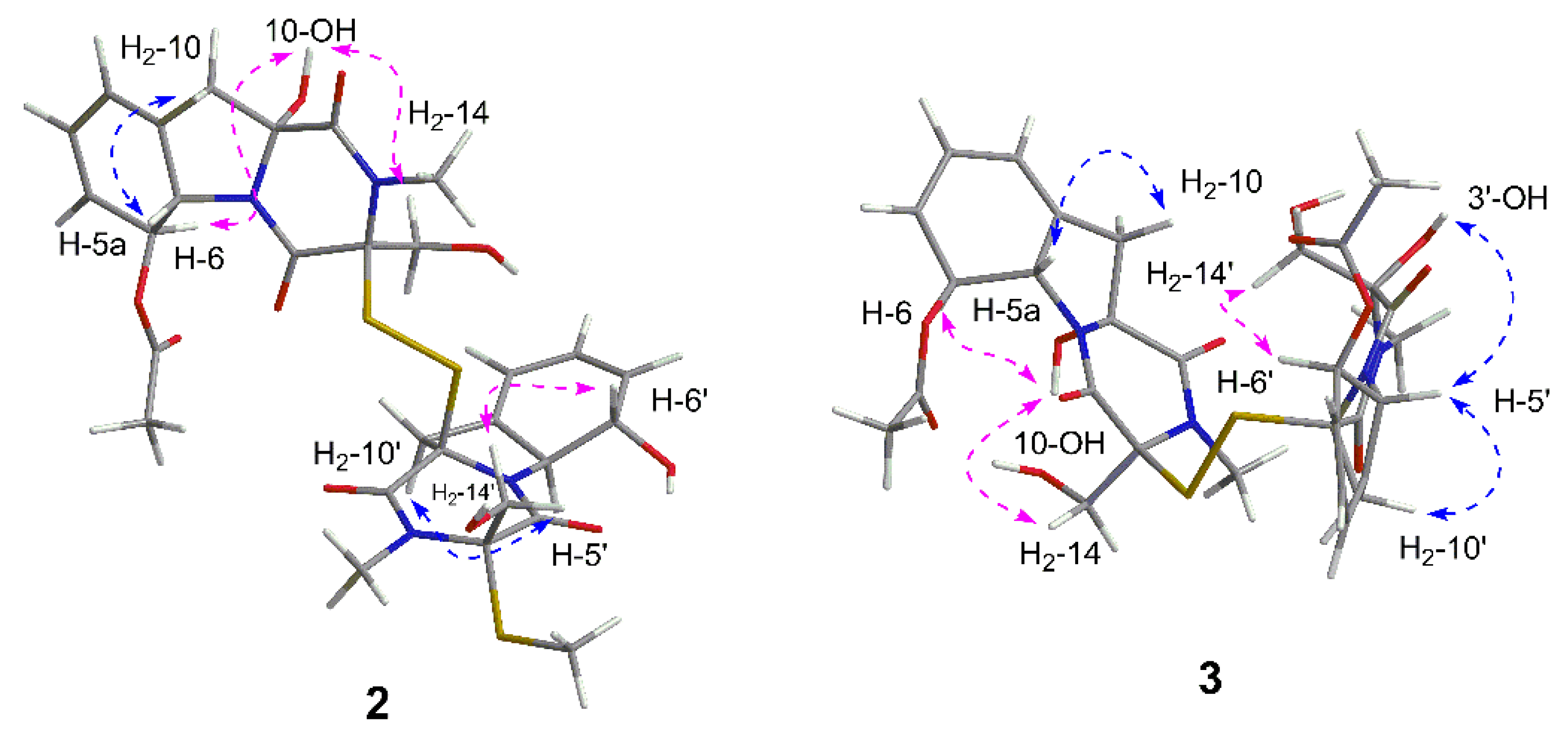
The relative configuration was elucidated based on the NOESY correlations recorded in dimethyl sulfoxide-
d6 (
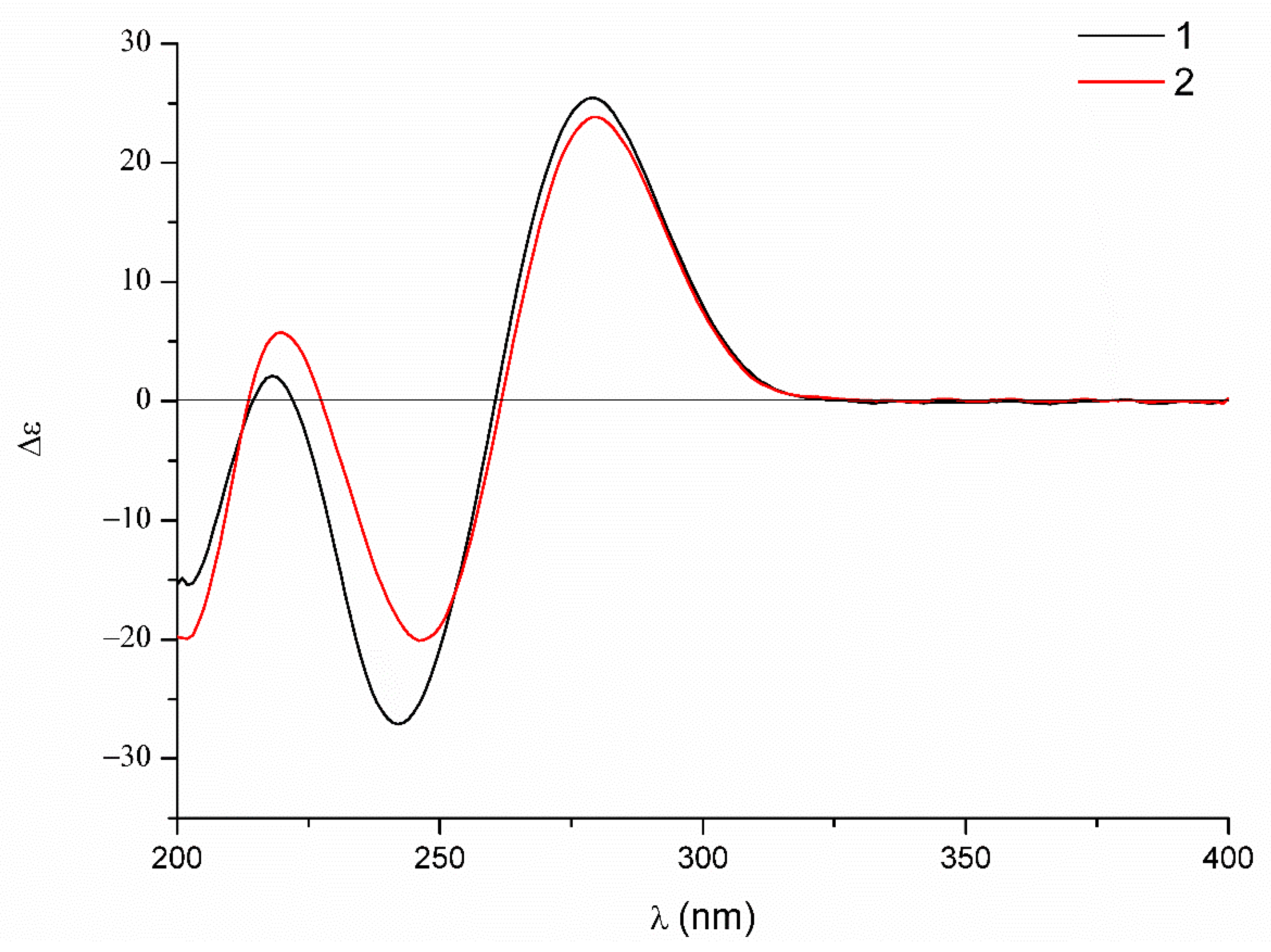
Figure 6) and the coupling constants, which were the same as those of dechdigliotoxin A. Additionally, the identical cotton effects in the experimental ECD spectra (
Figure 7) shared by
1 and
2 indicated the same absolute configuration of them. Hence, the complete structure of dechdigliotoxin B was deduced.
Dechdigliotoxin C (
3) was isolated as a colorless powder, of which the molecular formula was deduced as C
30H
34N
4O
12S
2 on the basis of the sodiated ion peak at m/z 729.1519 [M + Na]
+ (calcd 717.1615, C
30H
34N
4O
12S
2Na) in HRESIMS. The similar 1D NMR data (
Table 1) and the characteristic IR absorption band at 555 cm
−1 as compared to those of
1 suggested that they shared the same framework except the substituent at C-3′. The absence of S-methyl (
δH 2.11;
δC 13.0) and the downfield shift of C-3′ (
δC 88.2) indicated that the SMe in unit B was replaced by a hydroxy group. The further HMBC correlations (
Figure 2) from H
3-13′ to C-1′/C-3′ and from H
2-14′ to C-3′/C-4′ confirmed the planar structure of dechdigliotoxin C. The relative configurations of unit A and B were elucidated to be the same as those of
1 and
2 on the basis of NOESY correlation measured in dimethyl sulfoxide-
d6 (
Figure 6).
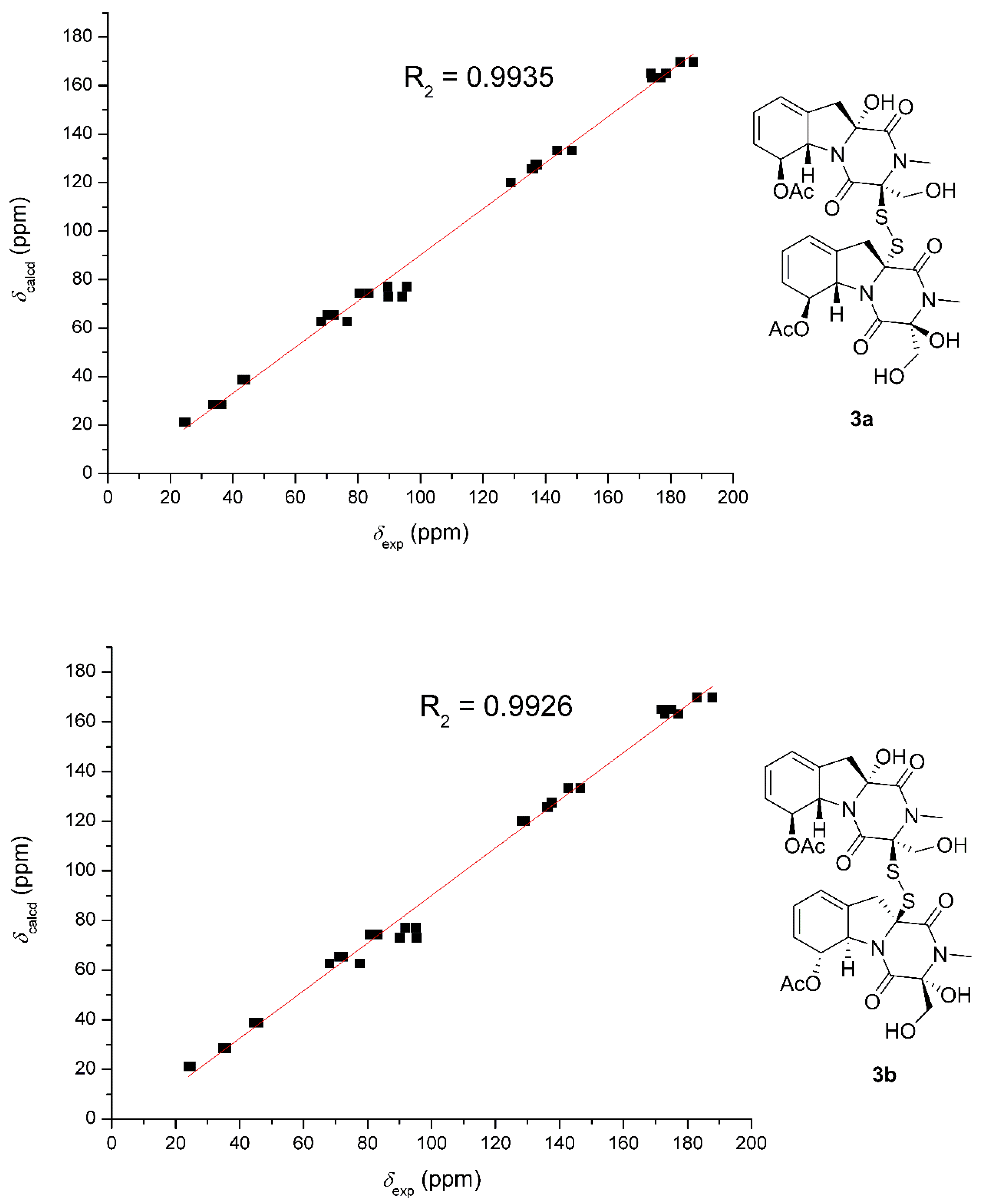
The relative structure between the two monomers were determined based on the
13C NMR chemical shift calculations and DP4+ simulation (
Figure 8). The method used was the same as those adopted in
1. The R
2 of
3a was 0.9935, which was slightly better than that of
3b (0.9926). DP4+ (chemical shift of carbon data) suggested
3a should be the likely candidate with 99.5% probability. Finally, the identical experimental ECD spectra suggested the same absolute configuration of
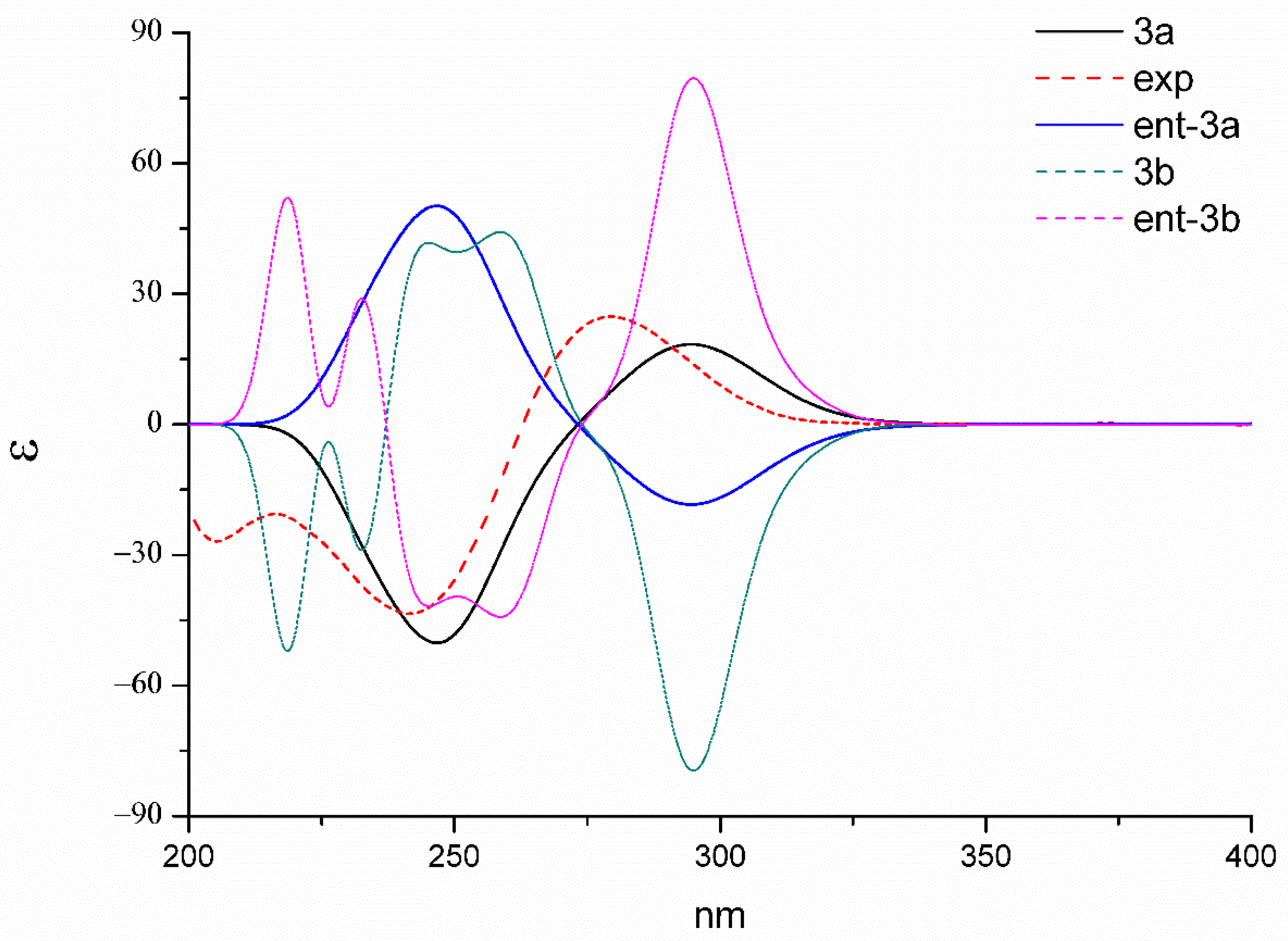
3. In order to further confirm the stereochemistry of dechdigliotoxin C, the theoretical ECD spectra of the two enantiomers (
3a and ent-
3a) were also calculated at the b3lyp/6-311+g(d,p) level (
Figure 9). The experimental spectrum exhibited an excellent fit to the theoretical plot of
3, which confirmed the absolute configuration.
Cyclic phenylalanylserine cyclo (L-Pshe-L-Ser) is a known biosynthetic precursor of the epidithiodioxopiperazine gliotoxin [
10], however, the obtained new metabolites, dechdigliotoxins A–C, in this study, which exhibited a converse chirality at C-3′ as compared with those reported gliotoxin analogues, were suspected to be generated from unusual L-Phe and D-Ser. The possible biosynthesis pathway was shown in
Scheme 1. The cyclo-phenylalanyl-serine (L-Phe-D-Ser) was constructed through the dipeptidase. Then, the hydroxylation of
α-protons of the two amino acid were performed with retention of the chirality of C-3′ and C-10a′ (intermediate i), which would metabolize
4 and
5. The glutathione-S-transferase should be the key enzyme contributed to the sulfuriztion at C-3′ or/and C-10a′, giving intermediates
ii and
iii. Followed by the monooxygenase and O-methyltransferase, the two precursor units A and B were formed from intermediates
i and
ii. Finally, the formation of disulfide bond between the two units which constructed
1–
3 was catalyzed by oxidoreductase. Since the isolated dimers (
1–
3) and the monomer (
5, of which the absolute configuration was confirmed by X-ray diffraction, no. CCDC 1491672,
Figure S28) shared the same precursor of intermediate
i, the stereochemistry could be further evidenced by the same biogenetic consistency.
As it is known that gliotoxin exhibits strong cytotoxicity against cancer cell lines, as well as the normal cell lines, hence, the dimeric analogues
1–
7 were evaluated for their cytotoxic effects against SF-268, MCF-7, HepG-2, and NCI-H460 cancer cell lines using cisplatin as a positive control. However, none of them showed effects against the tested cell lines except for the gliotoxin (
6) and acetyl-gliotoxin (
7) (
Table 1). According to the primary structure-activity relationship, we speculated that when the intramolecular disulfide bridge was deficient or converted to an “intermolecular-like” in the dimer, the cytotoxicity would reduce significantly. This would offer a new idea to reduce the toxicity of gliotoxin derivatives toward the normal cell lines. On the other hand, more bioactivities, not limited to the cytotoxicity, would be screened for the new dimers
1–
3 in the future.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}