Investigation of the Possible Pharmacologically Active Forms of the Nicotinic Acetylcholine Receptor Agonist Anabaseine


Abstract
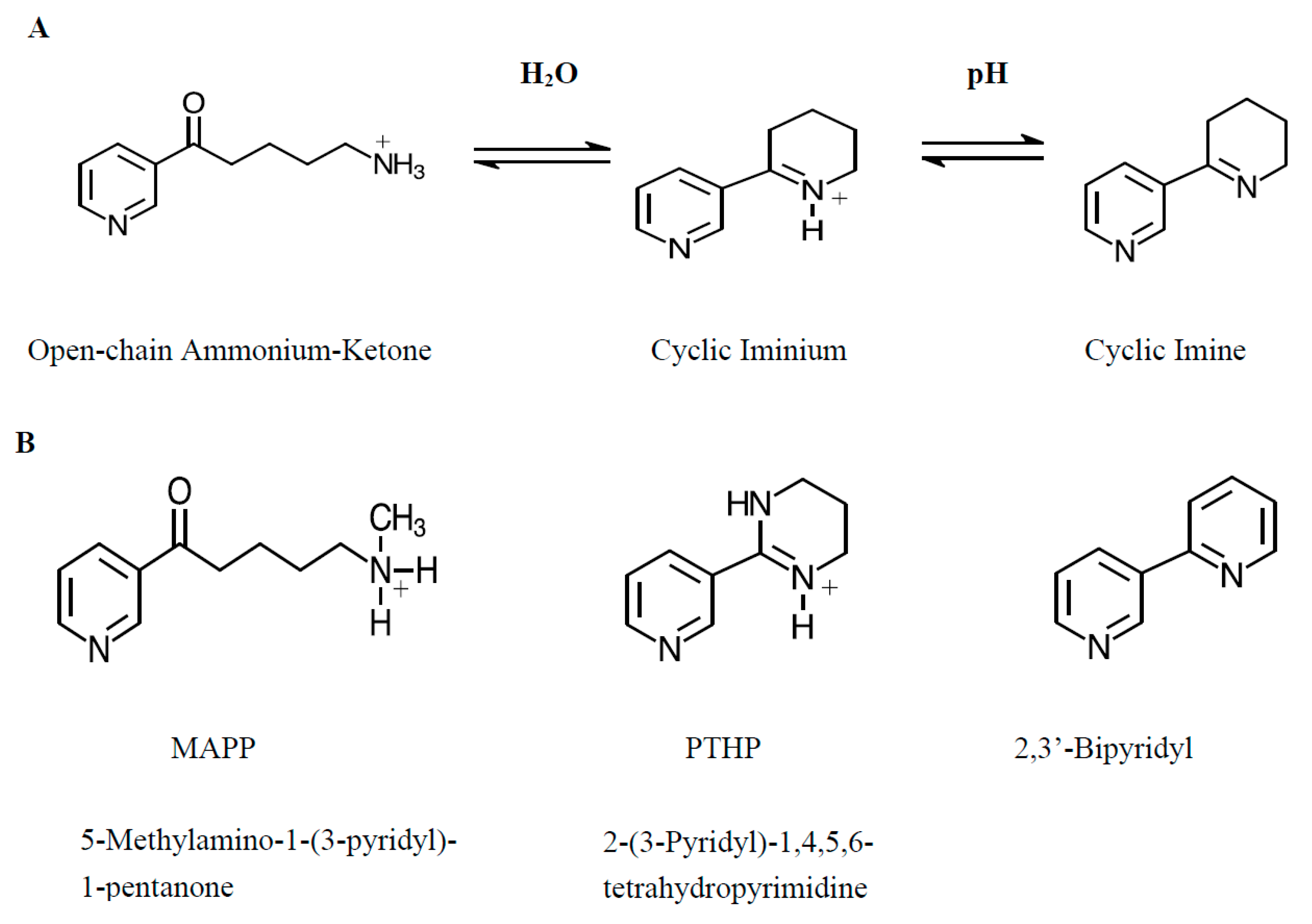
:1. Introduction
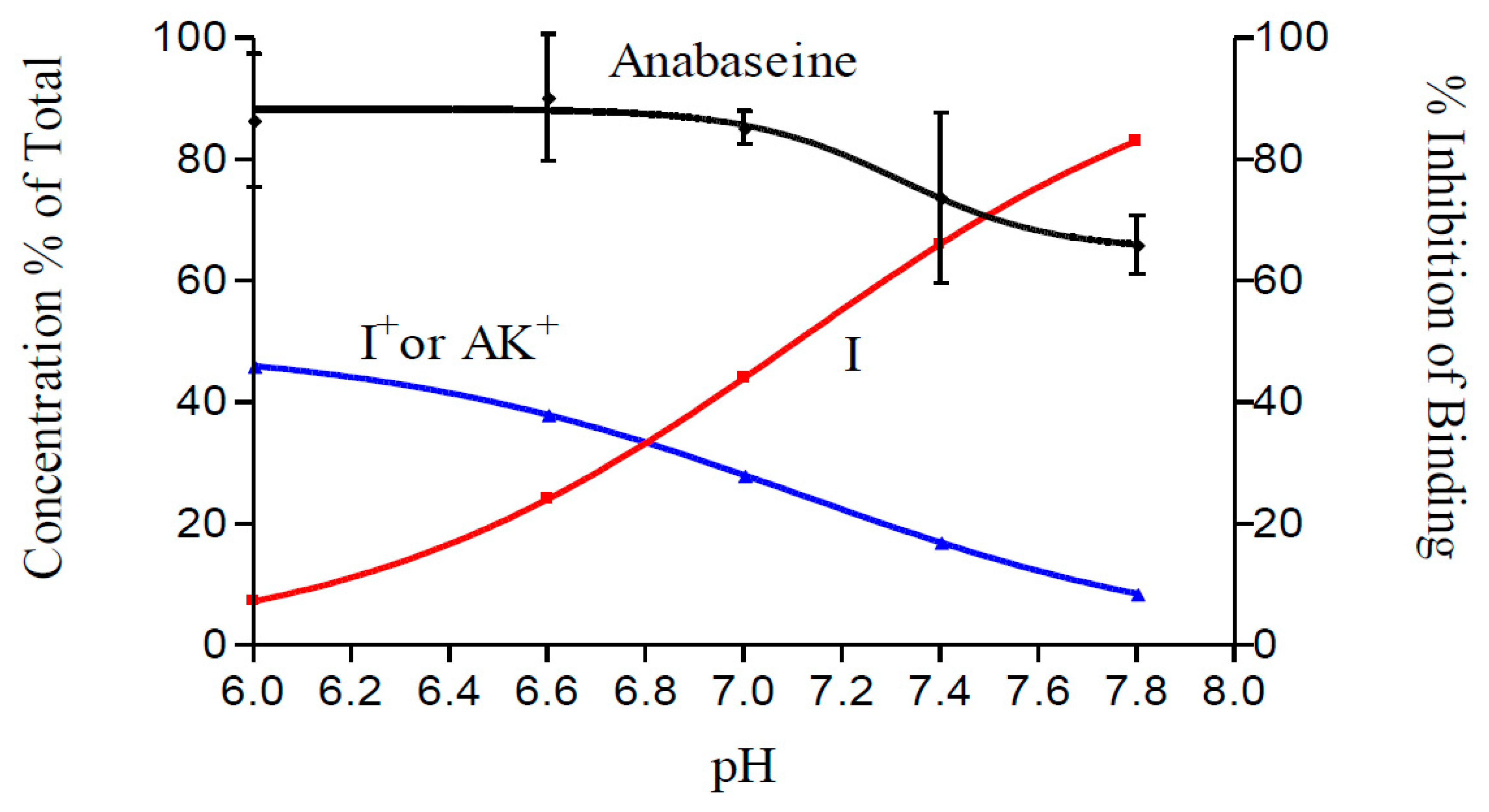
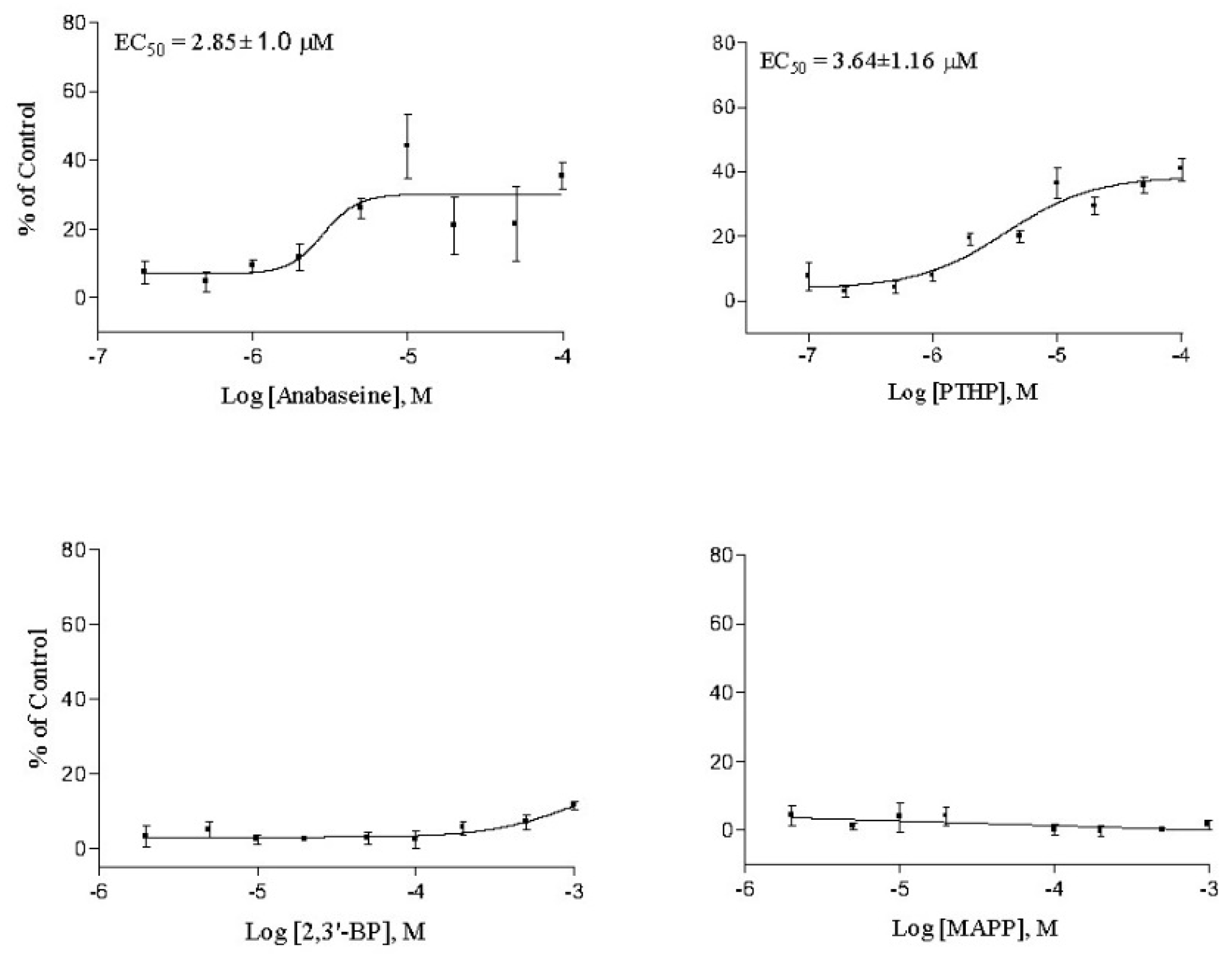
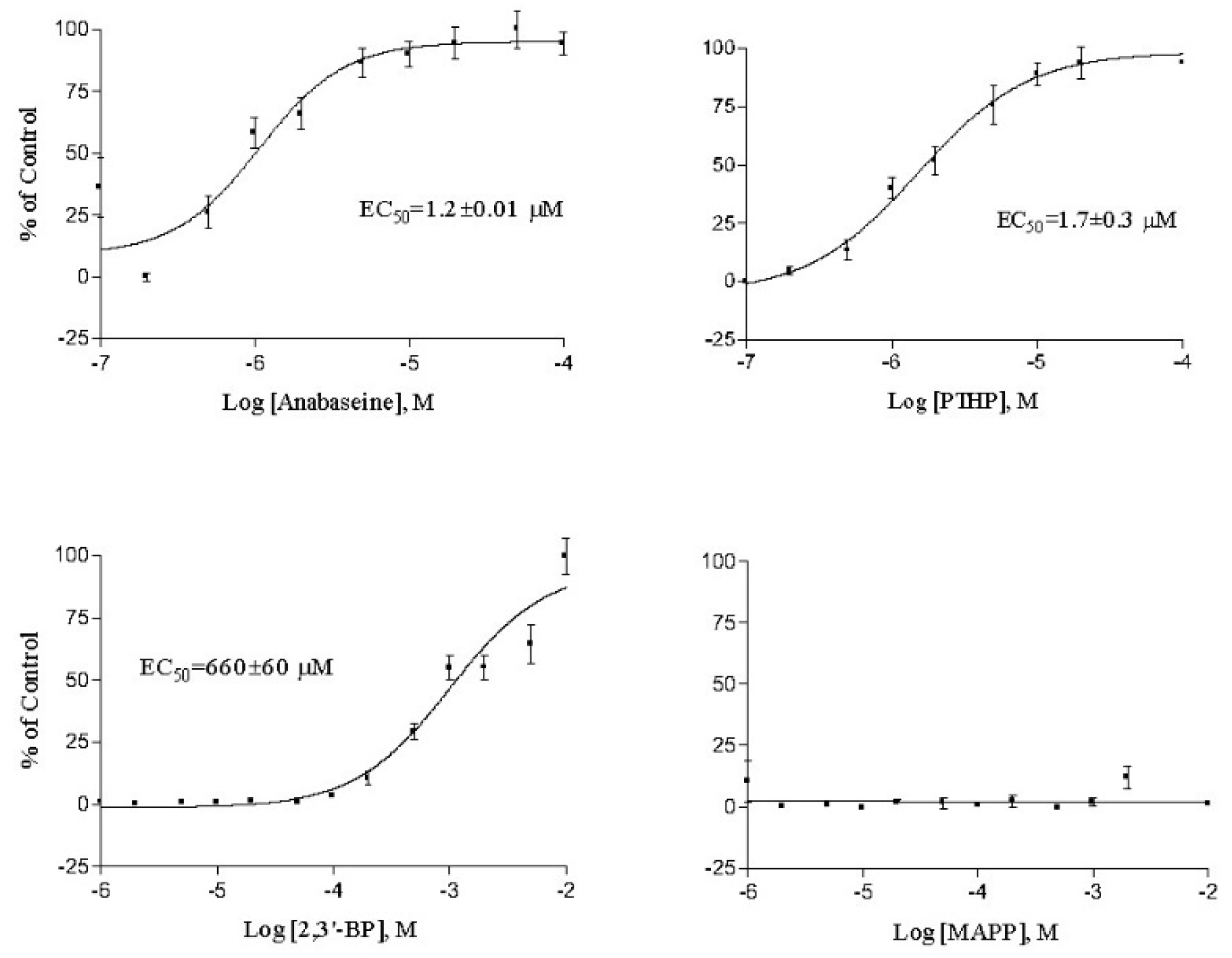
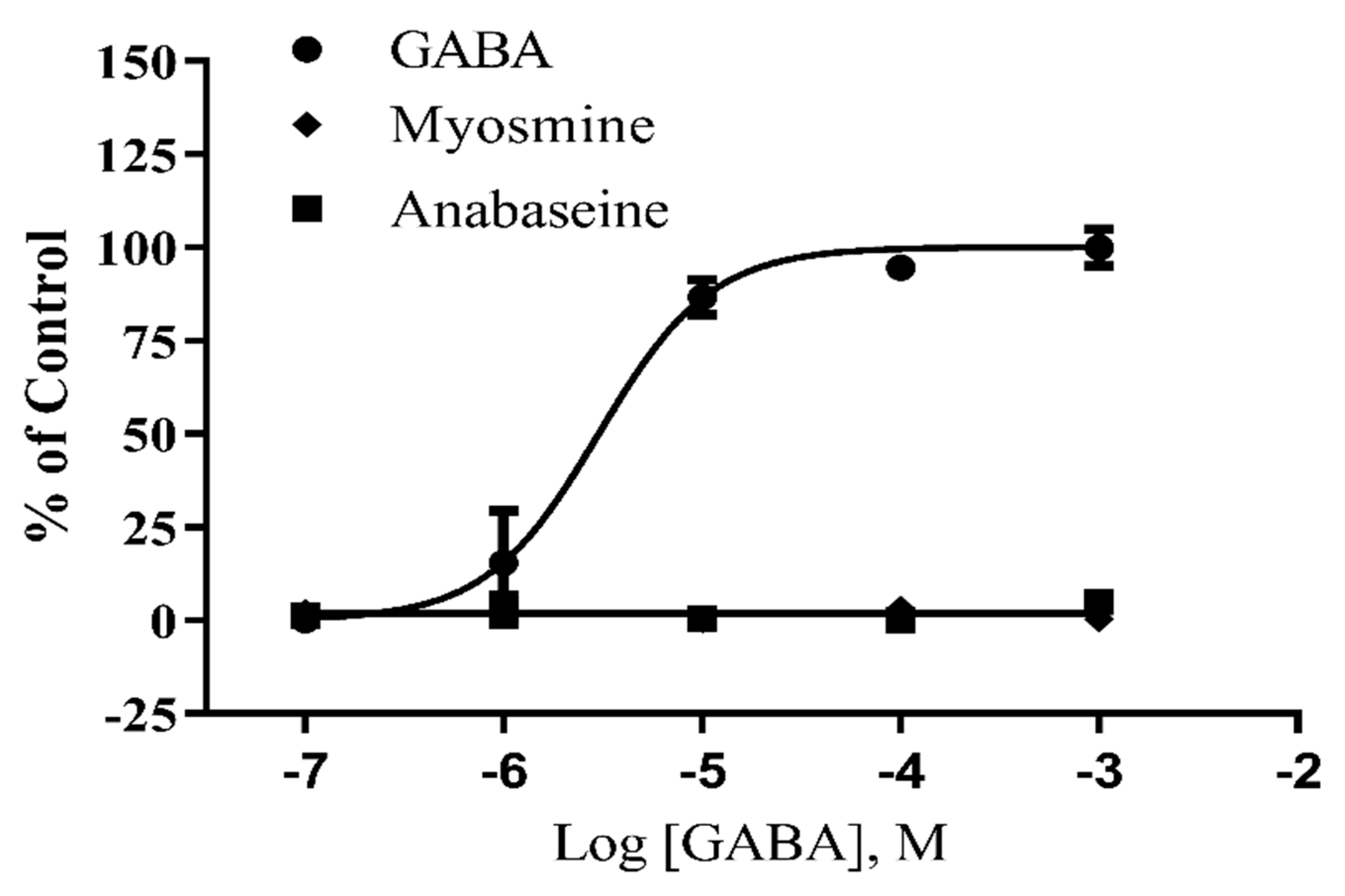
2. Results
3. Discussion
4. Materials and Methods
4.1. Compound Syntheses
4.2. Radioligand Binding Assays
4.3. Cell Culture
4.4. Nicotinic Receptor FlexStation Functional Assays
4.5. GABA Receptor Flexstation Funcional Assays
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bacq, Z.M. Les poisons des nemertiens. Bull. Cl. Sci. Acad. R. Belg. 1936, 22, 1072–1079. [Google Scholar]
- Bacq, Z.M. L’Amphiporine et La Némertine Poisons Des Vers Némertiens. Arch. Int. Physiol. 1937, 44, 190–204. [Google Scholar]
- King, H. Amphiporine, an active base from the marine worm Amphiporus lactifloreus. J. Chem. Soc. Lond. 1939, 1365. [Google Scholar]
- Kem, W.R. A Chemical Investigation of Nemertine Toxins. Ph.D. Thesis, University of Illinois at Urbana-Champaign, Champaign County, IL, USA, 1969; p. 116. [Google Scholar]
- Kem, W.R. A study of the occurrence of anabaseine in Paranemertes and other nemertines. Toxicon 1971, 9, 23–32. [Google Scholar] [CrossRef]
- Kem, W.R.; Abbott, B.C.; Coates, R.M. Isolation and structure of a hoplonemertine toxin. Toxicon 1971, 9, 15–22. [Google Scholar] [CrossRef]
- Wheeler, J.W.; Olubajo, O.; Storm, C.B. Anabaseine: Venom alkaloid of Aphaenogaster ants. Science 1981, 211, 1051–1052. [Google Scholar] [CrossRef]
- Kem, W.R.; Mahnir, V.M.; Papke, R.L.; Lingle, C.J. Anabaseine is a potent agonist on muscle and neuronal alpha-bungarotoxin-sensitive nicotinic receptors. J. Pharm. Exp. Ther. 1997, 283, 979–992. [Google Scholar]
- Kem, W.R. The brain alpha7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer’s disease: Studies with DMXBA (GTS-21). Behav. Brain Res. 2000, 113, 169–183. [Google Scholar] [CrossRef]
- Slavov, S.H.; Radzvilovits, M.; LeFrancois, S.; Stoyanova-Slavova, I.B.; Soti, F.; Kem, W.R.; Katritzky, A.R. A computational study of the binding of 3-(arylidene) anabaseines to two major brain nicotinic acetylcholine receptors and to the acetylcholine binding protein. Eur. J. Med. Chem. 2010, 45, 2433–2446. [Google Scholar] [CrossRef]
- Kem, W.; Soti, F.; Wildeboer, K.; LeFrancois, S.; MacDougall, K.; Wei, D.-Q.; Chou, K.-C.; Arias, H. The nemertine toxin anabaseine and its derivative DMXBA (GTS-21): Chemical and pharmacological properties. Mar. Drugs 2006, 4, 255–273. [Google Scholar] [CrossRef]
- Freedman, R.; Olincy, A.; Buchanan, R.W.; Harris, J.G.; Gold, J.M.; Johnson, L.; Allensworth, D.; Guzman-Bonilla, A.; Clement, B.; Ball, M.P.; et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am. J. Psychiatry 2008, 165, 1040–1047. [Google Scholar] [CrossRef]
- Kem, W.R.; Olincy, A.; Johnson, L.; Harris, J.; Wagner, B.D.; Buchanan, R.W.; Christians, U.; Freedman, R. Pharmacokinetic Limitations on Effects of an Alpha7-Nicotinic Receptor Agonist in Schizophrenia: Randomized Trial with an Extended-Release Formulation. Neuropsychopharmacology 2018, 43, 583–589. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Ochani, M.; Yang, L.H.; Gallowitsch-Puerta, M.; Ochani, K.; Lin, X.; Levi, J.; Parrish, W.R.; Rosas-Ballina, M.; Czura, C.J.; et al. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit. Care Med. 2007, 35, 1139–1144. [Google Scholar] [CrossRef]
- Kashiwagi, S.; Khan, M.A.; Yasuhara, S.; Goto, T.; Kem, W.R.; Tompkins, R.G.; Kaneki, M.; Martyn, J.A. Prevention of burn-induced inflammatory responses and muscle wasting by GTS-21, a specific agonist for α7 nicotinic acetylcholine receptors. Shock 2017, 47, 61–69. [Google Scholar] [CrossRef]
- Schaller, S.J.; Nagashima, M.; Schönfelder, M.; Sasakawa, T.; Schulz, F.; Khan, M.A.S.; Kem, W.R.; Schneider, G.; Schlegel, J.; Lewald, H.; et al. GTS-21 attenuates loss of body mass, muscle mass, and function in rats having systemic inflammation with and without disuse atrophy. Pflüg. Arch. Eur. J. Physiol. 2018, 470, 1647–1657. [Google Scholar] [CrossRef]
- Suzuki, S.; Kawamata, J.; Matsushita, T.; Matsumura, A.; Hisahara, S.; Takata, K.; Kitamura, Y.; Kem, W.R.; Shimohama, S. 3-[(2,4-Dimethoxy) benzylidene]-anabaseine dihydrochloride prevents 6-hydroxydopamine-induced Parkinsonism neurodegeneration through α7 nicotinic acetylcholine receptor stimulation in rats. J. Neurosci. Res. 2012, 91, 462–471. [Google Scholar] [CrossRef]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T.; et al. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Aβ accumulation through suppression of neuronal γ-secretase activity and promotion of microglial amyloid-β phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar]
- Kem, W.R.; Soti, F. Alpha7 Nicotinic Receptor Selective Ligands. U.S. Patent 8,592,458, 14 December 2006. [Google Scholar]
- Kem, W.R.; Soti, F.; Xing, H. Bicyclic Tetrahydropyridyl Alpha7 Selective Agonists. U.S. Patent 9,150,558, 6 October 2015. [Google Scholar]
- Changeux, J.P. The nicotinic acetylcholine receptor: The founding father of the pentameric ligand-gated ion channel superfamily. J. Bio. Chem. 2012, 287, 40207–40215. [Google Scholar] [CrossRef]
- Brejc, K.; van Dijk, W.J.; Klaassen, R.V.; Schuurman, M.; van Der Oost, J.; Smit, A.B.; Sixma, T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar] [CrossRef]
- Barlow, R.B.; Hamilton, J.T. Effects of pH on the activity of nicotine and nicotine monomethiodide on the rat diaphragm preparation. Br. J. Pharmacol. Chemother. 1962, 18, 543–549. [Google Scholar] [CrossRef]
- Hamilton, J.T. The influence of pH on the activity of nicotine at the neuromuscular junction. Can. J. Biochem. Physiol. 1963, 41, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Bartels, E.; Podleski, T.R. Action of nicotine on the electroplax and difference of potency between ionized and unionized forms. Biochim. Biophys. Acta 1964, 79, 511–520. [Google Scholar] [CrossRef]
- Palma, A.; Li, L.; Chen, X.; Pappone, P.; McNamee, M. Effects of pH on acetylcholine receptor function. J. Membr. Biol. 1991, 120, 67–73. [Google Scholar] [CrossRef]
- Abdrakhamanova, G.; Cleemann, L.; Lindstrom, J.; Morad, M. Differential modulation of β2 and β4subunits of human neuronal nicotinic acetylcholine receptors by acidification. Mol. Pharmacol. 2004, 66, 347–355. [Google Scholar] [CrossRef]
- Hutter, O.F.; Warner, A.E. The pH sensitivity of the chloride conductance of frog skeletal muscle. J. Physiol. 1967, 189, 403–425. [Google Scholar] [CrossRef] [Green Version]
- Mallart, A.; Trautmann, A. Ionic properties of the neuromuscular junction of the frog: Effects of denervation and pH. J. Physiol. 1973, 234, 553–567. [Google Scholar] [CrossRef]
- Yamamoto, I.; Kamimura, H.; Yamamoto, R.; Sakai, S.; Goda, M. Studies on nicotinoids as insecticides. Pt. I. Relation of structure to toxicity. Agric. Biol. Chem. 1962, 26, 709–716. [Google Scholar]
- Zoltewicz, J.A.; Bloom, L.B.; Kem, W.R. Quantitative determination of the ring-chain hydrolysis equilibrium constant for anabaseine and related tobacco alkaloids. J. Org. Chem. 1989, 54, 4462–4468. [Google Scholar] [CrossRef]
- Bloom, L.B. Influence of Solvent on the Ring-Chain Hydrolysis Equilibrium of Anabaseine and Syntheses of Anabaseine and Nicotine Analogs. Ph.D. Thesis, University of Florida, Gainesville, FL, USA, 1990; p. 207. [Google Scholar]
- Zoltewicz, J.A.; Bloom, L.B.; Kem, W.R. Hydrolysis of cholinergic anabaseine and N-methylanabaseine: Influence of cosolvents on the position of the ring-chain equilibrium-compensatory changes. Bioorg. Chem. 1990, 18, 395–412. [Google Scholar] [CrossRef]
- Krumholz, P. Structural studies on polynuclear pyridine compounds. J. Am. Chem. Soc. 1951, 73, 3487–3492. [Google Scholar] [CrossRef]
- Green, B.T.; Goulart, C.; Welch, K.D.; Pfister, J.A.; McCollum, I.; Gardner, D.R. The non-competitive blockade of GABAA receptors by an aqueous extract of water hemlock (Cicuta douglasii) tubers. Toxicon 2015, 108, 11–14. [Google Scholar] [CrossRef]
- Culver, P.; Fenical, W.; Taylor, P. Lophotoxin irreversibly inactivates the nicotinic acetylcholine receptor by preferential association at one of the two primary agonist sites. J. Biol. Chem. 1984, 259, 3763–3770. [Google Scholar]
- Tomizawa, M.; Casida, J.E. Molecular recognition of neonicotinoid insecticides: The determinants of life or death. Acc. Chem. Res. 2009, 42, 260–269. [Google Scholar] [CrossRef]
- Li, P.; Ann, J.; Akk, G. Activation and modulation of human alpha4beta2 nicotinic acetylcholine receptors by the neonicotinoids clothianidin and imidacloprid. J. Neurosci. Res. 2009, 89, 1295–1301. [Google Scholar] [CrossRef]
- Kaczanowska, K.; Camacho Hernandez, G.A.; Bendiks, L.; Kohs, L.; Cornejo-Bravo, J.M.; Harel, M.; Finn, M.G.; Taylor, P. Substituted 2-aminopyrimidines selective for α7-nicotinic acetylcholine receptor activation and association with acetylcholine binding proteins. J. Am. Chem. Soc. 2017, 139, 3676–3684. [Google Scholar] [CrossRef]
- Kem, W.R. Biochemistry of Nemertine Toxins. In Marine Pharmacognosy: Marine Biotoxins as Probes of Cellular Function; Martin, D.F., Padilla, G.M., Eds.; Academic Press: New York, NY, USA, 1973; pp. 37–84. [Google Scholar]
- Kem, W.R.; Mahnir, V.M.; Prokai, L.; Papke, R.L.; Cao, X.; LeFrancois, S.; Wildeboer, K.; Prokai-Tatrai, K.; Porter-Papke, J.; Soti, F. Hydroxy metabolites of the Alzheimer’s drug candidate 3-[(2,4-dimethoxy)benzylidene]-anabaseine dihydrochloride (GTS-21): Their molecular properties, interactions with brain nicotinic receptors, and brain penetration. Mol. Pharmacol. 2004, 6, 56–67. [Google Scholar] [CrossRef]
- Zoltewicz, J.A.; Prokai-Tatrai, K.; Bloom, L.B.; Kem, W.R. Long range transmission of polar effects of cholinergic 3-arylideneanabaseines, Conformations calculated by molecular modelling. Heterocycles 1993, 35, 171–179. [Google Scholar] [CrossRef]
- Mallipeddi, P.L.; Pedersen, S.E.; Briggs, J.M. Interactions of acetylcholine binding site residues contributing to nicotinc acetylcholine receptor gating: Role of residues Y93, Y190, K145 and D200. J. Mol. Gr. Model. 2013, 44, 145–154. [Google Scholar] [CrossRef]
- Hibbs, R.E.; Sulzenbacher, G.; Shi, J.; Talley, T.T.; Kem, W.R.; Marchot, P.; Taylor, P.; Bourne, Y. Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal α7 nicotinic acetylcholine receptor. EMBO J. 2009, 28, 3040–3051. [Google Scholar] [CrossRef]
- Brimblecombe, R.E.; Hunt, R.R.; Rickard, R.L.; Taylor, J.V. The synthesis and pharmacology of some 1,4,5,6-tetrahydropyrimidines. Br. J. Pharmacol. 1969, 37, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Upshall, D.G. Correlation of chick embryo teratogenicity with the nicotinic activity of a series of tetrahydropyrimidines. Teratology 1972, 5, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.N.; Walker, R.J.; Newton, L.C. Actions of some muscarinic and nicotinic agonists on the Retzius cells of the leech. Comp. Gen. Pharmacol. 1971, 2, 106–117. [Google Scholar] [CrossRef]
- Smith, C.R.; Richardson, C.H.; Shepard, H.H. Neonicotine and certain other derivatives of the dipyridyls as insecticides. J. Econ. Entomol. 1930, 23, 863–870. [Google Scholar] [CrossRef]
- Kem, W.R.; Scott, K.N.; Duncan, J.H. Hoplonemertine worms—A new source of pyridine neurotoxins. Experientia 1976, 32, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Kem, W.R.; Soti, F.; Rittschof, D. Inhibition of barnacle larval settlement and crustacean toxicity of some hoplonemertine pyridyl alkaloids. Biomol. Eng. 2003, 20, 355–361. [Google Scholar] [CrossRef]
- Stricker, S.A.; Cloney, R.A. The ultrastructure of venom-producing cells in Paranemertes peregrina (Nemertea, Hoplonemertea). J. Morphol. 1983, 177, 89–107. [Google Scholar] [CrossRef]
- Schaefer, F.C.; Peters, G.A. Base-catalyzed reaction of nitriles with alcohols. A convenient route to imidates and amidine salts. J. Org. Chem. 1960, 26, 412. [Google Scholar] [CrossRef]
- Brown, D.J.; Evans, R.F. Hydropyrimidines. Part II. A new general synthesis of substituted 1,4,5,6-tetrahydropyridines. J. Chem. Soc. 1962, 4039. [Google Scholar] [CrossRef]
- Boksa, P.; Quirion, R. [3H]-methyl-carbamylcholine, a new radioligand specific for nicotinic acetylcholine receptors in brain. Eur. J. Pharmacol. 1987, 139, 323–333. [Google Scholar] [CrossRef]
- Kuryatov, A.; Luo, J.; Cooper, J.; Lindstrom, J. Nicotine acts as a pharmacological chaperone to upregulate human alpha4 beta2 acetylcholine receptors. Mol. Pharmacol. 2005, 68, 1839–1851. [Google Scholar] [CrossRef]
- Fitch, R.W.; Xiao, Y.; Kellar, K.J.; Daly, J.W. Membrane potential fluorescence: A rapid and highly sensitive assay for nicotinic receptor channel function. Proc. Natl. Acad. Sci. USA 2003, 100, 4909–4914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.; Sei, Y.; Skolnick, P. Stable expression of type I gamma-aminobutyric acidA/benzodiazepine receptors in a transfected cell line. Mol. Pharmacol. 1992, 42, 996–1003. [Google Scholar] [PubMed]
- Green, B.T.; Lee, S.T.; Panter, K.E.; Welch, K.D.; Cook, D.; Pfister, J.A.; Kem, W.R. Actions of piperidine alkaloid teratogens at fetal nicotinic acetylcholine receptors. Neurotoxicol. Teratol. 2010, 32, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Wildeboer, K.; Panter, K.E.; Kem, W.R.; Gardner, D.R.; Molyneux, R.J.; Chang, S.-W.T.; Soti, F.; Pfister, J.A. Relative toxicities and neuromuscular nicotinic receptor agonistic potencies of anabaseine enantiomers and anabaseine. Neurotoxicol. Teratol. 2006, 28, 220–228. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neuronal nAChR | Neuromuscular nAChR | ||||
|---|---|---|---|---|---|
| Human | Rat | Rat | Human | Torpedo | |
| Compound | α4β2 | α4β | α7 | TE671 | Electric Organ |
| μM: | EC50 | Ki 1 | Ki 2 | EC50 | IC50 3 |
| Anabaseine | 2.85 ± 1.0 | 0.096 ± 0.01 | 1.87 ± 0.10 | 1.2 ± 0.0 | 0.29 ± 0.18 |
| PTHP | 3.64 ± 1.2 | 0.38 ± 0.07 | 1.54 ± 0.76 | 1.7 ± 0.3 | 0.47 ± 0.01 |
| 2,3′-Bipyridyl | >1000 | >50 | >2300 | 660 ± 60 | 343 ± 48 |
| MAPP | >2000 | 10 | >2000 | >1000 | >500 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrud, K.; Xing, H.; Gabrielsen, B.; Bloom, L.; Mahnir, V.; Lee, S.; Green, B.T.; Lindstrom, J.; Kem, W. Investigation of the Possible Pharmacologically Active Forms of the Nicotinic Acetylcholine Receptor Agonist Anabaseine. Mar. Drugs 2019, 17, 614. https://doi.org/10.3390/md17110614
Andrud K, Xing H, Gabrielsen B, Bloom L, Mahnir V, Lee S, Green BT, Lindstrom J, Kem W. Investigation of the Possible Pharmacologically Active Forms of the Nicotinic Acetylcholine Receptor Agonist Anabaseine. Marine Drugs. 2019; 17(11):614. https://doi.org/10.3390/md17110614
Chicago/Turabian StyleAndrud, Kristin, Hong Xing, Bjarne Gabrielsen, Linda Bloom, Vladimir Mahnir, Stephen Lee, Benedict T. Green, Jon Lindstrom, and William Kem. 2019. "Investigation of the Possible Pharmacologically Active Forms of the Nicotinic Acetylcholine Receptor Agonist Anabaseine" Marine Drugs 17, no. 11: 614. https://doi.org/10.3390/md17110614
APA StyleAndrud, K., Xing, H., Gabrielsen, B., Bloom, L., Mahnir, V., Lee, S., Green, B. T., Lindstrom, J., & Kem, W. (2019). Investigation of the Possible Pharmacologically Active Forms of the Nicotinic Acetylcholine Receptor Agonist Anabaseine. Marine Drugs, 17(11), 614. https://doi.org/10.3390/md17110614