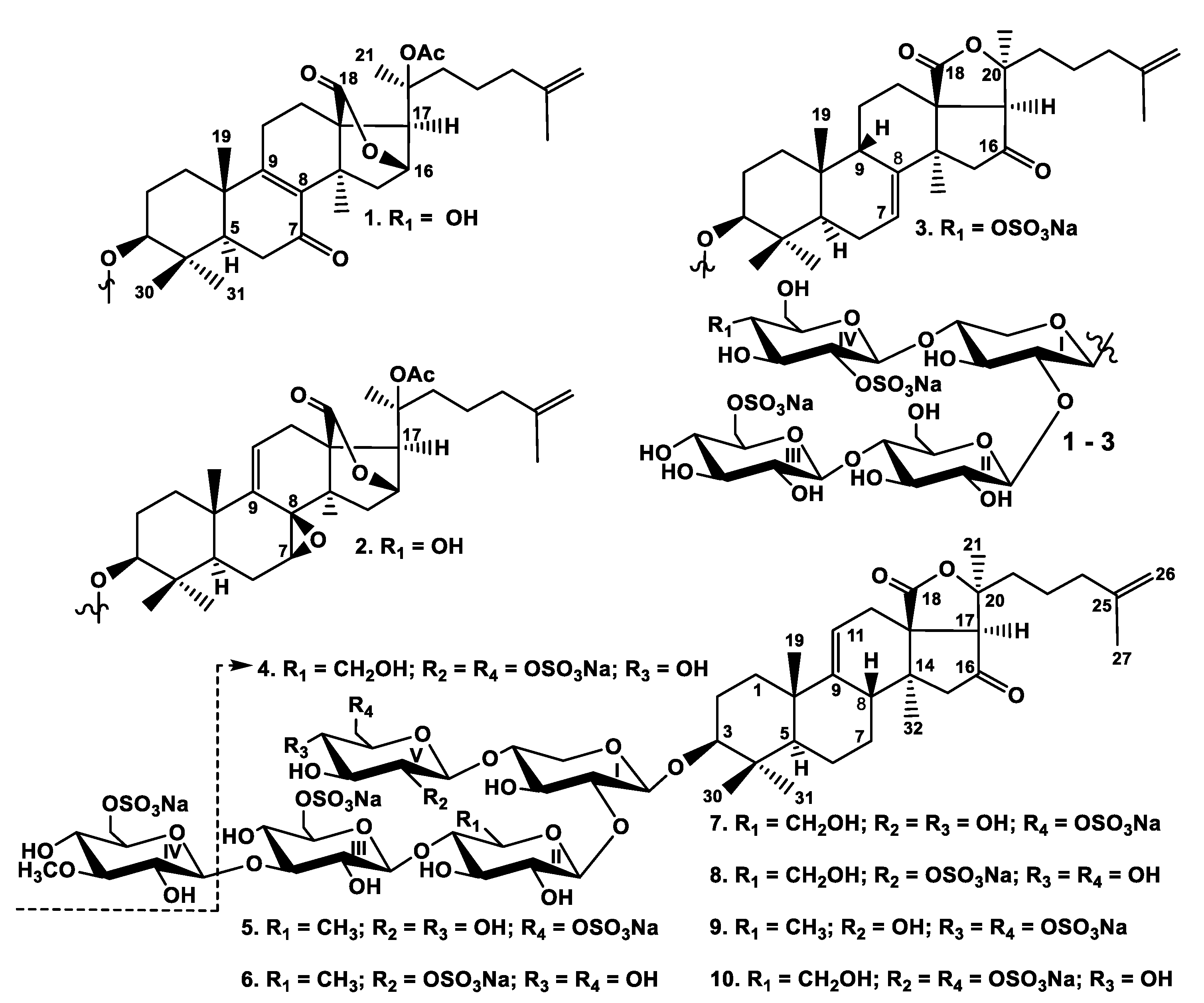
2.1. Structural Elucidation of the Glycosides
The initial stages of isolation of compounds
1–
10 were the same as for other glycosides from
P. fabricii and were described earlier [
10,
11,
12]. The individual glycosides were isolated by HPLC on reversed-phase columns to give psolusosides: B
1 (
1) (7,3 mg), B
2 (
2) (3.4 mg), J (
3) (4.8 mg), K (
4) (3.4 mg), L (
5) (60 mg), M (
6) (1.0 mg), N (
7) (8.8 mg), O (
8) (0.6 mg), P (
9) (8.5 mg), and Q (
10) (1.4 mg) (
Figure 1).
The
1H and
13C NMR spectra corresponding to the carbohydrate chains of psolusosides B
1 (
1) and B
2 (
2) were coincident to each other and to those of known psolusoside B [
12] showing the identity of their tetrasaccharide carbohydrate moieties branched by C-4 of the xylose unit and having two sulfate groups (
Table S1).
The molecular formula of psolusoside B
1 (
1) was determined to be C
55H
82O
31S
2Na
2 from the [M
2Na − Na]
− ion peak at
m/z 1325.4164 (calc. 1325.4185) and [M
2Na − 2Na]
2− ion peak at
m/z 651.2157 (calc. 651.2146) in the (−)HR-ESI-MS. The signal of H-16 was observed as a broad singlet at δ
H 4.89 and the signal of H-17 was observed as a singlet at δ
H 2.97 in the
1H NMR spectrum of
1. These data as well as corresponding signals of carbons at δ
C 79.9 (C-16) and δ
C 58.8 (C-17) (
Table 1) were indicative for 18(16)-lactone moiety (
Table 1).
O-acetyl group (δ
C 170.9 (CH
3COO) and 21.6 (
CH
3COO) in the
13C NMR spectrum), attached to C-20, caused the deshielding of its signal to δ
C 83.8 in the same manner as in the spectrum of psolusoside B [
12]. The side chain of
1 was identical to that of psolusoside B due to the coincidence of those signals in the
1H and
13C NMR spectra. The signal at δ
C 199.3 corresponded to a keto-group adjacent to a double bond (the signals of olefinic carbons at δ
C 135.3 (C-8) and 169.0 (C-9)). The position of the keto-group was deduced as C-7 based on the correlations between H
2-6 (δ
H 2.42 and δ
H 2.29) and C-7 (δ
C 199.3) in the HMBC spectrum of
1. This was also corroborated by an isolated spin system between the doublet of doublets at δ
H 1.54 (H-5) and another doublet of doublets at δ
H 2.42 (H-6a) and the triplet at δ
H 2.29 (H-6b) observed in the
1H,
1H-COSY spectrum. The 8(9)-position of double bond was confirmed by the HMBC correlations H
3-32/C-8 and H
3-19/C-9. So, the aglycone of psolusoside B
1 (
1) is characterized by the unique combination of such structural features as 7-keto-8(9)-ene fragment and 18(16)-lactone.
The (−)ESI-MS/MS of 1 demonstrated the fragmentation of [M2Na − Na]− ion at m/z 1325.4. The peaks of fragment ions were observed at m/z 1265.4 [M2Na − Na − CH3COOH]−, 1145.4 [M2Na − Na − CH3COOH − NaHSO4]−, 1001.4 [M2Na − Na − CH3COOH − C6H10O8SNa (GlcSO3Na) + H]−, and 839.3 [M2Na − Na − CH3COOH − GlcSO3Na − Glc + H]− corroborating the structure of psolusoside B1 (1).
All these data indicate that psolusoside B1 (1) is 3β-O-{6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-[2-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-7-keto-20(S)-acetoxylanosta-8,25-diene-18(16)-lactone.
The molecular formula of psolusoside B
2 (
2) (C
55H
82O
31S
2Na
2) was determined to be the same as of
1 from the [M
2Na − Na]
− ion peak at
m/z 1325.4163 (calc. 1325.4185) and [M
2Na − 2Na]
2− ion peak at
m/z 651.2159 (calc. 651.2146) in the (−)HR-ESI-MS. In the
1H and
13C NMR spectra of the aglycone part of
2 the signals characteristic of 18(16)-lactone (δ
H 4.94 (brs, H-16), δ
H 3.01 (s, H-17), δ
C 79.3 (C-16), and δ
C 60.2 (C-17) as well as
O-acetylated C-20 (δ
H 2.05 (s, C
H3COO), δ
C 21.8 (
CH
3COO), δ
C 170.9(CH
3COO), and δ
C 83.9 (C-20)) were observed (
Table 2). The side chains in aglycones of
1 and
2 were identical to each other. The signal at δ
H 3.10 (d, 6.6, H-7) was assigned by the
1H,
1H-COSY spectrum where the protons H-5/H-6/H-7 formed an isolated spin system. The corresponding signal of C-7 at δ
C 56.2 was deduced by the HSQC spectrum of
2. The signal of quaternary C-8 assigned by the HMBC correlations H
3-32/C-8, H
2-6/C-8, and H-7/C-8 was deshielded to δ
C 59.6 in the
13C NMR spectrum. These data indicated the presence of an oxygen-bearing substituent at C-7 and C-8, which was supposed to be an 7,8-epoxide [
13] that correlated with the MS data. The olefinic broad doublet of doublets at δ
H 6.00 was assigned to H-11 due to its correlation with H
2-12 (δ
H 2.81 (dd, 5.2; 17.6, H-12a) and 2.60 (brdd, 2.3; 17.6, H-12b)) in the
1H,
1H-COSY spectrum. The signal at δ
C 122.7 corresponded to olefinic C-11 and was deduced by the HSQC spectrum. So, the double bond could occupy the 9(11)-position only. The signal of C-9 at δ
C 143.2 correlated in the HMBC spectrum with both δ
H 2.81 (H-12a) and 2.60 (H-12b) and the methyl singlet δ
H 1.13 (H
3-19).
The configuration of C-7 was established as (S) by the ROE-correlation H-7/H3-32 and was confirmed by the coupling pattern of H-7 (δH 3.10 (d, 6.6)), that coincided with the calculated coupling constant based on dihedral angle values in the optimized MM2 model of aglycone of psolusoside B2 (2) having H-7α-orientation and 8(R)-configuration. Thus, the aglycone of psolusoside B2 (2) has unprecedented 7(S),8(R)-epoxy-20(S)-acetoxylanosta-9(11),25-diene-18(16)-lactone structure.
The (−)ESI-MS/MS of 2 showed the fragmentation of [M2Na − Na]− ion at m/z 1325.4. The peaks of fragment ions were observed at the same m/z values of 1265.4, 1145.4, 1001.4, and 839.3 as in the spectrum of 1, corroborating the identity of the carbohydrate chains of 1 and 2. Additionally, the fragment ion-peaks at m/z 535.1 [M2Na − Na − C32H45O6 (Agl) − C6H10O8SNa (GlcSO3Na)]− and 403 [M2Na − Na − C32H45O6 (Agl) − C6H10O8SNa (GlcSO3Na) − Xyl (C5H8O4)]− corresponding to the tri- and disaccharide fragments, were observed in the MS/MS spectrum of 2.
All these data indicate that psolusoside B2 (2) is 3β-O-{6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-[2-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-7(S),8(R)-epoxy-20(S)-acetoxylanosta-9(11),25-diene-18(16)-lactone.
The molecular formula of psolusoside J (
3) (C
53H
79O
32S
3Na
3) was determined from the [M
3Na − Na]
− ion peak at
m/z 1369.3485 (calc. 1369.3517), [M
3Na − 2Na]
2− ion peak at
m/z 673.1812 (calc. 673.1813), and [M
3Na − 3Na]
3− ion peak at
m/z 441.1248 (calc. 441.1244) in the (−)HR-ESI-MS. The
1H and
13C NMR spectra of the aglycone part of psolusoside J (
3) coincided with those of psolusoside H isolated earlier from
P. farbricii [
12] (
Table S2) indicating the identity of their holostane-type aglycones having 7(8)- and 25(26)-double bonds and 16-keto-group. This aglycone is common for the glycosides of sea cucumbers belonging to the orders Dendrochirotida and Aspidochirotida [
2,
12].
In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside J (
3) four characteristic doublets at δ
H 4.60–5.12 (
J = 7.3 − 8.1 Hz) and, corresponding to them, signals of anomeric carbons at δ
C 101.7–105.5 were indicative of a tetrasaccharide chain and
β-configurations of glycosidic bonds. The
13C NMR spectra of tetrasaccharide carbohydrate chain of
3 and those of
1 and
2 were quite different, while the
1H,
1H-COSY and 1D TOCSY spectra of
3 showed the signals of four isolated spin systems assigned to one xylose and three glucose residues as in psolusosides B [
12], B
1 (
1), and B
2 (
2). The positions of interglycosidic linkages were elucidated by the ROESY and HMBC spectra of
3 (
Table 3), where the correlations between H-1 of the xylose (Xyl1) and H-3 (C-3) of the aglycone, H-1 of the second residue (glucose, Glc2) and H-2 (C-2) of the xylose (Xyl1), H-1 of the third residue (glucose, Glc3) and H-4 (C-4) of the second residue (glucose, Glc2), H-1 of the fourth residue (glucose, Glc4) and H-4 (C-4) of the first residue (xylose, Xyl1) were observed, indicating the same architecture of sugar chains in
3 and
1 and
2. The comparison of the NMR spectra of
1 and
3 showed the coincidence of the signals of three monosaccharide residues corresponding to the linear part of the carbohydrate chain (residues I–III). The signals of terminal monosaccharide unit attached to C-4 of the first (Xyl1) unit, assigned by the
1H,
1H-COSY and 1D TOCSY spectra of
3 were indicative of a sulfated by C-2 glucose residue due to characteristic shifting effects observed in the
13C NMR spectrum: the signal of C-2 Glc4 was deshielded to δ
C 81.2 and the signal of C-1 Glc4 was shielded to δ
C 101.7 in comparison with the corresponding signals of the same sugar unit in the
13C NMR spectrum of psolusoside I isolated by us earlier [
12].
The δC of the signals of C-2 and C-1 of the fourth monosaccharide unit (Glc4) in the 13C NMR spectrum of psolusoside J (3) were very close to those in the 13C NMR spectrum of 1, corroborating the presence of a sulfate group at C-2 of this residue (Glc4). The correlations between H-2/H-3/H-4 in this monosaccharide residue, deduced by the 1H,1H-COSY spectrum of 3, indicated the signal of H-4 Glc4 at δH 4.90. The signal of the corresponding carbon (C-4 Glc4), deduced by the HSQC spectrum, was downshifted to δC 77.3 as compared with the same signal (C-4 Glc4) at δC 70.7 in the 13C NMR spectrum of 1. Actually, the signals at δC ~70.4–70.8 were absent and the signals of C-3 Glc4 and C-5 Glc4 were upshifted to δC 75.6 and 76.6, correspondingly, in the 13C NMR spectrum of 3 due to β-shifting effect of sulfate group, when compared with the corresponding signals in the 13C NMR spectrum of 1. Considering that (−)HR-ESI-MS indicated the presence of three sulfate groups as well as the NMR data, the attachment of the third sulfate group to C-4 of Glc4 was supposed. The signal at δC 62.4 (C-6 Glc4) was characteristic for carbons of non-sulfated hydroxy-methylene groups of glucopyranose residues and excluded the positioning of the third sulfate group at C-6 Glc4 that confirmed our supposition. Hence psolusoside J (3) is a trisulfated tetraoside with two sulfate groups attached to the same glucose residue. To the best our knowledge, this structural feature is first found in the glycosides.
The (−)ESI-MS/MS of 3 demonstrated the fragmentation of [M3Na − Na]− ion at m/z 1369.3. The peaks of fragment ions were observed at m/z 1249.4 [M3Na − Na − NaHSO4]−, 1105.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na)]−, 1003.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na) − NaSO3 + H]−, 841.4 [M3Na − Na − NaSO3 − GlcSO3Na − Glc + H]−, 403.0 [M3Na − Na − C30H43O4 (Agl) − C6H9O11S2Na2 (Glc(SO3Na)2) − Xyl (C5H8O4)]−, and 241.0 [M3Na − Na − C30H43O4 (Agl) − C6H9O11S2Na2 (Glc(SO3Na)2) − Xyl (C5H8O4) − Glc (C6H10O5)]−, corroborating the structure of psolusoside J (3).
All these data indicate that psolusoside J (3) is 3β-O-{6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-[2,4-O-sodium-disulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-ketoholosta-7,25-diene.
The
13C NMR spectra of the aglycone moieties of the glycosides
4–
10 were identical to each other (
Table S3) and to those of psolusosides E, F, and G containing 16-ketoholosta-9(11),25-dien-3β-ol as an aglycone, known earlier and frequently occurring in the glycosides of sea cucumbers [
12].
The molecular formula of psolusoside K (
4) was determined to be C
53H
79O
32S
3Na
3 from the [M
3Na − Na]
− ion peak at
m/z 1369.3485 (calc. 1369.3517), [M
3Na − 2Na]
2− ion peak at
m/z 673.1821 (calc. 673.1813), and [M
3Na − 3Na]
3− ion peak at
m/z 441.1255 (calc. 441.1244) in the (−)HR-ESI-MS and was coincident with the formula of psolusoside J (
3). In the
1H and
13C NMR spectra of the carbohydrate moiety of psolusoside K (
4) four characteristic doublets at δ
H 4.61–5.07 (
J = 7.2–8.4 Hz) and corresponding signals of anomeric carbons at δ
C 101.4–104.7 were indicative of a tetrasaccharide chain and
β-configurations of glycosidic bonds. The positions of interglycosidic linkages were elucidated by the ROESY and HMBC spectra of
4 (
Table 4) as described above indicating the presence of a tetrasaccharide carbohydrate chain branched by C-4 of the xylose residue (Xyl1). The monosaccharide composition of
4, deduced from the
1H,
1H-COSY and 1D TOCSY spectra, was the same as in glycosides
1–
3. The comparison of the
13C NMR spectra of trisulfated compounds
3 and
4 showed the coincidence of the signals corresponding to three monosaccharide residues (residues I–III in the formula) forming the linear part of the sugar chain. The signals of C-2 Glc4 at δ
C 80.3 and C-1 Glc4 at δ
C 101.4 in the
13C NMR spectrum of
4 were very close to those in the spectrum of
3 that indicated the attachment of a sulfate group to C-2 Glc4 in psolusoside K (
4). All of the signals of this monosaccharide residue were assigned using the
1H,
1H-COSY and 1D TOCSY spectra. The doublet at δ
H 5.00 and the doublet of doublets at δ
H 4.63 corresponded to the protons of the hydroxy-methylene group of the terminal glucose unit (H
2-6 Glc4) and were deshielded as compared with the corresponding signals in the
1H NMR spectrum of
3. The signal at δ
C 67.4 (C-6 Glc4) also indicated the presence of a sulfate group at C-6 of Glc4 in addition to another sulfate group at C-2 of Glc4. So, psolusoside K (
4) is an isomer of psolusoside J (
3) by the sulfate position and is the second glycoside from sea cucumbers that contains two sulfate groups bonded to the same monosaccharide residue.
The (−)ESI-MS/MS of 4 demonstrated the fragmentation of [M3Na − Na]− ion at m/z 1369.3. The peaks of fragment ions were observed at the same m/z: 1249.4 [M3Na − Na − NaHSO4]−, 1105.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na)]−, 1003.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na) − NaSO3 + H]−, 403.0 [M3Na − Na − C30H43O4 (Agl) − C6H9O11S2Na2 (Glc(SO3Na)2) − Xyl (C5H8O4)]−, and 241.0 [M3Na − Na − C30H43O4 (Agl) − C6H9O11S2Na2 (Glc(SO3Na)2) − Xyl (C5H8O4) − Glc (C6H10O5)]− as in the MS/MS of psolusoside J (3) corroborating their isomerism.
All these data indicate that psolusoside K (4) is 3β-O-{6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-[2,6-O-sodium-disulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-ketoholosta-9,25-diene.
The molecular formula of psolusoside L (
5) (C
60H
91O
36S
3Na
3) was determined from the [M
3Na − Na]
− ion peak at
m/z 1529.4222 (calc. 1529.4253), [M
3Na − 2Na]
2− ion peak at
m/z 753.2190 (calc. 753.2180), and [M
3Na − 3Na]
3− ion peak at
m/z 494.4835 (calc. 494.4823) in the (−)HR-ESI-MS, indicating the presence of three sulfate groups. In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside L (
5) five characteristic doublets at δ
H 4.65–5.16 (
J = 6.9–8.1 Hz) and, corresponding to them, signals of anomeric carbons at δ
C 103.4–104.8 were indicative of a pentasaccharide chain and
β-configurations of glycosidic bonds (
Table 5). Analysis of the
1H,
1H-COSY and 1D TOCSY spectra of psolusoside L (
5) showed the presence of one xylose, one quinovose, two glucose, and one 3-
O-methylglucose residues. The presence of a quinovose residue was confirmed by the
1H and
13C NMR spectra demonstrating the characteristic doublet at δ
H 1.59 (H-6 Qui2) and the signal at δ
C 17.7 (C-6 Qui2). The positions of interglycosidic linkages and the consequence of monosaccharides in the chain of
5 were established by analysis of the ROESY and HMBC spectra (
Table 5) indicating the presence of branched pentasaccharide moiety with glucose, attached to C-4 Xyl1, and 3-
O-methylglucose, attached to C-3 Glc3, as terminal residues. The
13C NMR spectrum of
5 demonstrated three signals at δ
C 67.0, 67.5, and 67.6, corresponding to sulfated hydroxy-methylene groups of glucopyranose residues that indicated the sulfation of two glucose and 3-O-methylglucose units in the carbohydrate chain of
5.
The comparison of the
13C NMR spectrum of the sugar part of psolusoside L (
5) with those of known achlioniceosides A
1, A
2, and A
3, with identical carbohydrate chains, isolated earlier from the sea cucumber
Rhipidothuria racowitzai [
14] showed the coincidence of the signals of four monosaccharide residues in their spectra. The signals of terminal 3-
O-methylglucose residues of the novel and known compounds were different due to the absence of a sulfate group in this residue of known compounds. All these data indicated that psolusoside L (
5) is a pentaoside with a new trisulfated carbohydrate chain branched by C-4 Xyl1.
The (−)ESI-MS/MS of 5 demonstrated the fragmentation of [M3Na − Na]− ion at m/z 1529.4. The peaks of fragment ions were observed at m/z: 1409.5 [M3Na − Na − NaHSO4]−, 1265.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na)]−, 1131.5 [M3Na − Na − C7H12O9SNa (MeGlcSO3Na) − NaSO3]−, 665.1 [M3Na − Na − C30H43O4 (Agl) − C7H12O9SNa (MeGlcSO3Na) − NaSO3]−, and 519.0 [M3Na − Na − C30H43O4 (Agl) − C7H12O9SNa (MeGlcSO3Na) − C6H9O7SNa (GlcSO3Na)]−, confirming the structure of psolusoside L (5).
All these data indicate that psolusoside L (5) is 3β-O-{6-O-sodium-sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-ketoholosta-9(11),25-diene.
The molecular formula of psolusoside M (
6) was determined to be C
60H
91O
36S
3Na
3 from the ion peaks at
m/z 1529.4273 (calc. 1529.4253) [M
3Na − Na]
−, 753.2202 (calc. 753.2180) [M
3Na − 2Na]
2−, and 494.4844 (calc. 494.4823) [M
3Na − 3Na]
3− in the (−)HR-ESI-MS, indicating this glycoside to be an isomer of psolusoside L (
5). In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside M (
6) five characteristic doublets at δ
H 4.58–5.15 (
J = 7.1–8.5 Hz) and, corresponding to them, signals of anomeric carbons at δ
C 100.9–104.8, were indicative of a pentasaccharide chain and
β-configurations of glycosidic bonds (
Table 6). Analysis of the
1H,
1H-COSY and 1D TOCSY, ROESY, and HMBC spectra of psolusoside M (
6) showed the same monosaccharide composition and architecture of the carbohydrate chain as in
5. Actually, the comparison of their
13C NMR spectra showed the closeness of the signals corresponding to the monosaccharides from the first to the fourth. The differences of the
13C NMR spectra of compounds
6 and
5 were concerned with the terminal glucose residue (Glc5) connected to C-4 Xyl1. The characteristic signals at δ
C 100.9 (C-1 Glc5) and at δ
C 80.6 (C-2 Glc5) in the
13C NMR spectrum of
6 were very close to the corresponding signals in the spectra of the compounds
1–
4 indicating the presence of a sulfate group at C-2 Glc5 in the psolusoside M (
6). At the same time, the hydroxy-methylene group of this sugar was free from sulfation, since the signal of C-6 Glc5 was observed at δ
C 61.8. Two signals of sulfated hydroxy-methylene groups of the glucose (Glc3) and 3-
O-methylglucose (MeGlc4) residues were observed at δ
C 67.5 and 67.0 in the
13C NMR spectrum of
6. Therefore, psolusoside M (
6) is an isomer of psolusoside L (
5) by the sulfate group position.
The (−)ESI-MS/MS of 6 demonstrated the fragmentation of [M3Na − Na]− ion at m/z 1529.4. The peaks of fragment ions were observed at the same m/z: 1409.5 [M3Na − Na − NaHSO4]−, 1265.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na)]−, 1131.5 [M3Na − Na − C7H12O9SNa (MeGlcSO3Na) − NaSO3]−, 665.1 [M3Na − Na − C30H43O4 (Agl) − C7H12O9SNa (MeGlcSO3Na) − NaSO3]−, and 519.0 [M3Na − Na − C30H43O4 (Agl) − C7H12O9SNa (MeGlcSO3Na) − C6H9O7SNa (GlcSO3Na)]− as in the MS/MS spectrum of glycoside 5.
All these data indicate that psolusoside M (6) is 3β-O-{6-O-sodium-sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[2-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-ketoholosta-9(11),25-diene.
The molecular formula of psolusoside N (
7) was determined to be C
60H
91O
37S
3Na
3 from the [M
3Na − Na]
− ion peak at
m/z 1545.4171 (calc. 1545.4202), [M
3Na − 2Na]
2− ion peak at
m/z 761.2164 (calc. 761.2155), and [M
3Na − 3Na]
3− ion peak at
m/z 499.8151 (calc. 499.8139) in the (−)HR-ESI-MS. In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside N (
7) five characteristic doublets at δ
H 4.67–5.12 (
J = 6.8–8.3 Hz) and, corresponding to them, signals of anomeric carbons at δ
C 103.3–104.7 were indicative of a pentasaccharide chain and
β-configurations of glycosidic bonds (
Table 7). Analysis of the
1H,
1H-COSY and 1D TOCSY spectra of psolusoside N (
7) showed the presence of one xylose, three glucose, and one 3-
O-methylglucose residues. The positions of interglycosidic linkages and the consequence of monosaccharides in the carbohydrate chain of
7 were established in the same manner as for
1–
6 (
Table 7) indicating the presence of branched pentasaccharide moiety having the same architecture as in compounds
5 and
6. The comparison of the
13C NMR spectra of
7 and
5 showed the closeness of the signals of all the monosaccharide residues except for the signals assigned to the second sugar units in their chains. Actually, in the
1H and
13C NMR spectra of
7, the signals characteristic of quinovose residue were absent but two doublets of doublets at δ
H 4.95 (H-6a Glc2) and at δ
H 4.75 (H-6b Glc2) and the signal at δ
C 61.0 (C-6 Glc2), assigned to hydroxy-methylene group of glucopyranose moiety, were detected. These data indicated the replacement of quinovose by the glucose residue in the second position of a carbohydrate chain in psolusoside N (
7) as compared with psolusoside L (
5). Three sulfate groups were supposed to attach the C-6 of two glucose and 3-
O-methylglucose residues due to the signals at δ
C 67.4, 67.5, and 66.9 observed in the spectrum of
7. The carbohydrate chain of psolusoside N (
7) is the first found in the glycosides from holothurians.
The (−)ESI-MS/MS of 7 demonstrated the fragmentation of [M3Na − Na]− ion at m/z 1545.4. The peaks of fragment ions were observed at m/z: 1425.5 [M3Na − Na − NaHSO4]−, 1281.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na)]−, 1147.5 [M3Na − Na − C7H12O9SNa (MeGlcSO3Na) − NaSO3]−, 1003.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na) − C7H12O8SNa (MeGlcSO3Na) + H]−, 681.1 [M3Na − Na − C30H43O4 (Agl) − C7H12O9SNa (MeGlcSO3Na) − NaSO3 + H]−, and 519.0 [M3Na − Na − C30H43O4 (Agl) − C7H12O9SNa (MeGlcSO3Na) − C6H9O7SNa (GlcSO3Na)]−, corroborating the structure of psolusoside N (7).
All these data indicate that psolusoside N (7) is 3β-O-{6-O-sodium-sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-[6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-ketoholosta-9(11),25-diene.
The molecular formula of psolusoside O (8) was established as the same (C60H91O37S3Na3) as compound 7 from the [M3Na − Na]− ion peak at m/z 1545.4197 (calc. 1545.4202), [M3Na − 2Na]2− ion peak at m/z 761.2171 (calc. 761.2155), and [M3Na − 3Na]3− ion peak at m/z 499.8155 (calc. 499.8139) in the (−)HR-ESI-MS.
In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside O (
8), five characteristic doublets at δ
H 4.60–5.12 (
J = 7.0–8.6 Hz) and, corresponding to them, signals of anomeric carbons at δ
C 101.0–104.8 indicated a pentasaccharide carbohydrate chain and
β-configurations of glycosidic bonds (
Table 8).
Analysis of the
1H,
1H-COSY and 1D TOCSY spectra of psolusoside O (
8) showed the same monosaccharide composition and positions of interglycosidic linkages as in the carbohydrate chain of compound
7 (
Table 8). The coincidence of the molecular formulae of
8 and
7 and the presence of three-charged ions in the (−)HR-ESI-MS of
8 indicated their difference in the position of a sulfate group. Really, the signals of monosaccharide residues from the first to the fourth were almost coincident in their
13C NMR spectra. The characteristic signals at δ
C 101.0 and δ
C 80.6 indicated the bonding of a sulfate group to C-2 of a terminal residue which glycosylates C-4 Xyl1. Analysis of the
1H,
1H-COSY and 1D TOCSY spectra of
8 showed this unit is a glucose (Glc5). Indeed, the comparison of the
13C NMR spectra of
8 and
6 revealed their difference only in the signals of the second monosaccharide unit and the coincidence of the signals of the remaining ones. All these data indicate psolusoside O (
8) has new trisulfated carbohydrate chain with the sulfate groups attached to C-6 of the third (Glc3), to C-6 of the fourth (MeGlc), and to C-2 of the fifth (Glc5) monosaccharide residues.
The (−)ESI-MS/MS of 8 demonstrated the fragmentation of [M3Na − Na]− with peaks of fragment ions, observed at m/z 1425.5 [M3Na − Na − NaHSO4]−, 1281.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na)]−, 1161.5 [M3Na − Na − C6H9O8SNa (GlcSO3Na) − NaHSO4]−, 1147.5 [M3Na − Na − C7H12O9SNa (MeGlcSO3Na) − NaSO3]−, 1003.4 [M3Na − Na − C6H9O8SNa (GlcSO3Na) − C7H12O8SNa (MeGlcSO3Na) + H]−, and 681.1 [M3Na − Na − C30H43O4 (Agl) − C7H12O9SNa (MeGlcSO3Na) − NaSO3 + H]−, 519.0 [M3Na − Na − C30H43O4 (Agl) − C7H12O9SNa (MeGlcSO3Na) − C6H9O7SNa (GlcSO3Na)]−, corroborating the isomerism of psolusosides O (8) and N (7).
All these data indicate that psolusoside O (8) is 3β-O-{6-O-sodium-sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-[2-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-ketoholosta-9(11),25-diene.
The molecular formula of psolusoside P (
9) was determined to be C
60H
90O
39S
4Na
4 from the [M
4Na − Na]
− ion peak at
m/z 1631.3598 (calc. 1631.3641), [M
4Na − 2Na]
2− ion peak at
m/z 804.1879 (calc. 804.1874), [M
4Na − 3Na]
3− ion peak at
m/z 528.4628 (calc. 528.4619), and [M
4Na − 4Na]
4− ion peak at
m/z 390.6001 (calc. 390.5991) in the (−)HR-ESI-MS indicating the presence of four sulfate groups. In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside P (
9), five characteristic doublets at δ
H 4.66–5.16 (
J = 7.2–8.3 Hz) and corresponding signals of anomeric carbons at δ
C 103.1–104.8 were indicative of a pentasaccharide chain and
β-configurations of glycosidic bonds (
Table 9). Analysis of the
1H,
1H-COSY and 1D TOCSY spectra of psolusoside P (
9) showed the presence of one xylose, one quinovose, two glucose, and one 3-
O-methylglucose residues. The positions of interglycosidic linkages and the consequence of monosaccharides in the chain of
9 established by the ROESY and HMBC spectra were the same as in the glycosides
5 and
6 (
Table 9). The comparison of the
13C NMR spectra of the compounds
9 and
5 showed the coincidence of the signals corresponding to the monosaccharides from the first to the fourth indicating their identity in these glycosides. The signals of the fifth terminal sugar residue assigned by the
1H,
1H-COSY and 1D TOCSY spectra corresponded to the glucose residue sulfated by C-6 (the signal at δ
C 67.9 (C-6 Glc5)). Thus, three sulfate groups were positioned at C-6 of 3-
O-methylglucose (MeGlc4) and C-6 of two glucose residues (Glc3 and Glc5) in the carbohydrate chain of psolusoside P (
9). The position of the fourth sulfate group at C-4 Glc5 was established by the comparison of the
13C NMR spectra of psolusosides P (
9) and L (
5). The signal of C-4 Glc5, deduced by the
1H,
1H-COSY spectrum of
9, was deshielded to δ
C 77.1 due to α-shifting effect of the sulfate group, as compared with the corresponding signal in the
13C NMR spectrum of
5 observed at δ
C 70.7. Oppositely, the signal of C-5 Glc5 was shielded to δ
C 73.7 in the spectrum of
9 due to the β-shifting effect of the sulfate group as compared with the spectrum of
5 (δ
C 75.65 (C-5 Glc5)). So, psolusoside P (
9) is the first case of triterpene glycoside having four sulfate groups, in that two of them were connected to one monosaccharide residue.
The (−)ESI-MS/MS of 9 demonstrated the fragmentation of [M4Na − Na]− ion at m/z 1631.4. The peaks of fragment ions were observed at m/z: 1265.4 [M4Na − Na − C6H8O11S2Na2 (Glc(SO3Na)2)]−, 1233.4 [M4Na − Na − C7H12O9SNa (MeGlcSO3Na) − NaSO3]−, 1145.5 [M4Na − Na − C6H9O11S2Na2 (Glc(SO3Na)2 − NaSO4]−, 1089.4 [M4Na − Na − C7H12O9SNa (MeGlcSO3Na) − C6H8O7SNa (GlcSO3Na)]−, 969.4 [M4Na − Na − C7H12O9SNa (MeGlcSO3Na) − C6H8O7SNa (GlcSO3Na) − NaHSO4]−, and 943.3 [M4Na − Na − C7H12O9SNa (MeGlcSO3Na) − C6H8O7SNa (GlcSO3Na) − C6H10O4 (Qui)]− corroborating the structure of carbohydrate chain of psolusoside P (9).
All these data indicate that psolusoside P (9) is 3β-O-{6-O-sodium-sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-[4,6-O-sodium-disulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-ketoholosta-9(11),25-diene.
The molecular formula of psolusoside Q (
10) was determined to be C
60H
90O
40S
4Na
4 from the [M
4Na − Na]
− ion peak at
m/z 1647.3544 (calc. 1647.3590), [M
4Na − 2Na]
2− ion peak at
m/z 812.1854 (calc. 812.1849), [M
4Na − 3Na]
3− ion peak at
m/z 533.7944 (calc. 533.7935), and [M
4Na − 4Na]
4− ion peak at
m/z 394.5989 (calc. 394.5978) in the (−)HR-ESI-MS demonstrating the presence of four sulfate groups. In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside Q (
10), five characteristic doublets at δ
H 4.61–5.12 (
J = 6.7–8.4 Hz) and, corresponding to them, signals of anomeric carbons at δ
C 101.5–104.8, were indicative of a pentasaccharide chain and
β-configurations of glycosidic bonds (
Table 10). The molecular weights of tetrasulfated psolusosides P (
9) and Q (
10) differed by 16
amu in HR-ESI-MS that along with the absence of the signals corresponding to the quinovose residue in the NMR spectra of
10 indicated the presence of a glucose residue in the second position of its carbohydrate chain. Actually, the coincidence of the signals of monosaccharide residues from the first to the fourth the
13C NMR spectra of psolusosides Q (
10), N (
7), and O (
8) confirmed this supposition. Analysis of the
1H,
1H-COSY, 1D TOCSY, ROESY, and HMBC spectra of psolusoside Q (
10) showed the same monosaccharide composition and the consequence of monosaccharides in the chain of
10 as in psolusosides N (
7) and O (
8) (
Table 10). The characteristic signals at δ
C 101.5 (C-1 Glc5) and δ
C 80.3 (C-2 Glc5) indicated attachment of a sulfate group to C-2 of the fifth residue (Glc5) in the sugar part of
10. The signal of C-6 Glc5 was assigned by the HSQC spectrum of
10, demonstrating the correlation of the both doublet at δ
H 5.02 (H-6a Glc5) and doublet of doublets at δ
H 4.64 (H-6b Glc5) with the corresponding resonance at δ
C 67.5 that indicated the presence of an additional sulfate group at C-6 Glc5 in psolusoside Q (
10). All these data show that psolusoside Q (
10) has a new carbohydrate chain with four sulfate groups, in that two of them are attached to C-2 and C-6 of the same (Glc5) residue.
The (−)ESI-MS/MS of 10 demonstrated the fragmentation of [M4Na − Na]− ion at m/z 1647.4. The peaks of fragment ions were observed at m/z 1527.4 [M4Na − Na − NaHSO4]−, 1281.4 [M4Na − Na − C6H8O11S2Na2 (Glc(SO3Na)2)]−, 1161.5 [M4Na − Na − C6H8O11S2Na2 (Glc(SO3Na)2) − NaHSO4]−, 1003.4 [M4Na − Na − C6H8O11S2Na2 (Glc(SO3Na)2) − C7H11O8SNa (MeGlcSO3Na)]−, 681.1 [M4Na − Na − C30H43O4 (Agl) − C6H9O11S2Na2 (Glc(SO3Na)2) − C5H8O4 (Xyl)]−, and 519.0 [M4Na − Na − C30H43O4 (Agl) − C6H9O11S2Na2 (Glc(SO3Na)2) − C5H8O4 (Xyl) − C6H10O5 (Glc)]− corroborating the sequence of monosaccharide residues in psolusoside Q (10).
All these data indicate that psolusoside Q (10) is 3β-O-{6-O-sodium-sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-[2,6-O-sodium-disulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16-ketoholosta-9(11),25-diene.
Thus, highly polar tetrasulfated glycosides are first discovered in sea cucumbers. Although polysulfated polysaccharides are common biopolymers of marine macrophytes and invertebrates, low molecular weight metabolites, containing several sulfate groups are extremely rare. So far, trisulfated natural compounds such as steroid glycosides were found only in sponges [
15,
16,
17] and trisulfated triterpene glycosides, in some representatives of the class Holothuroidea [
18,
19].



 ,
, 

{kind=link}