Anticoagulant and Antithrombotic Properties in Vitro and in Vivo of a Novel Sulfated Polysaccharide from Marine Green Alga Monostroma nitidum

Abstract
:1. Introduction
2. Results and Discussion
2.1. Isolation and Chemical Composition of MS-1
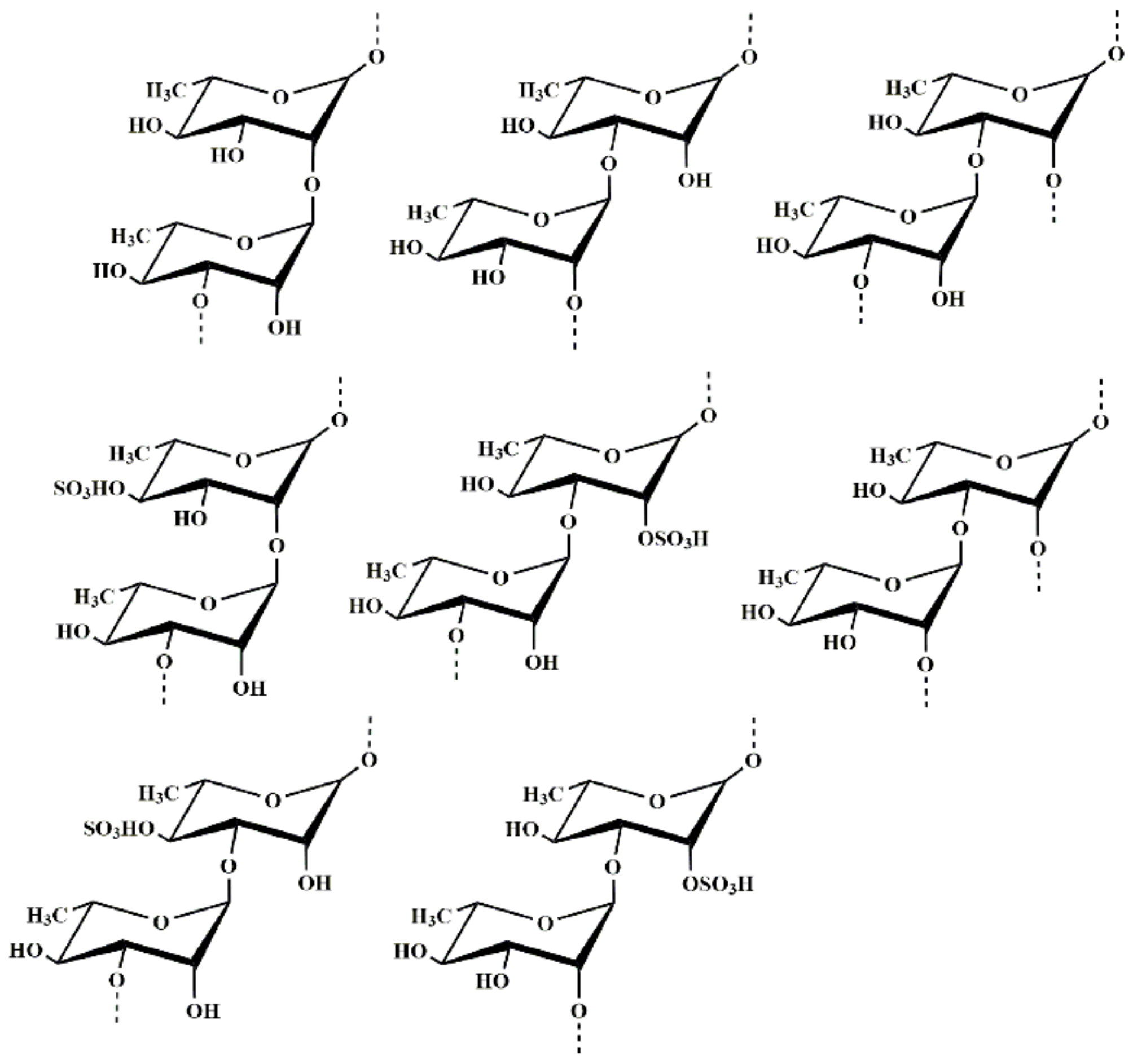
2.2. Structural Characteristics of MS-1
2.2.1. Methylation Analysis
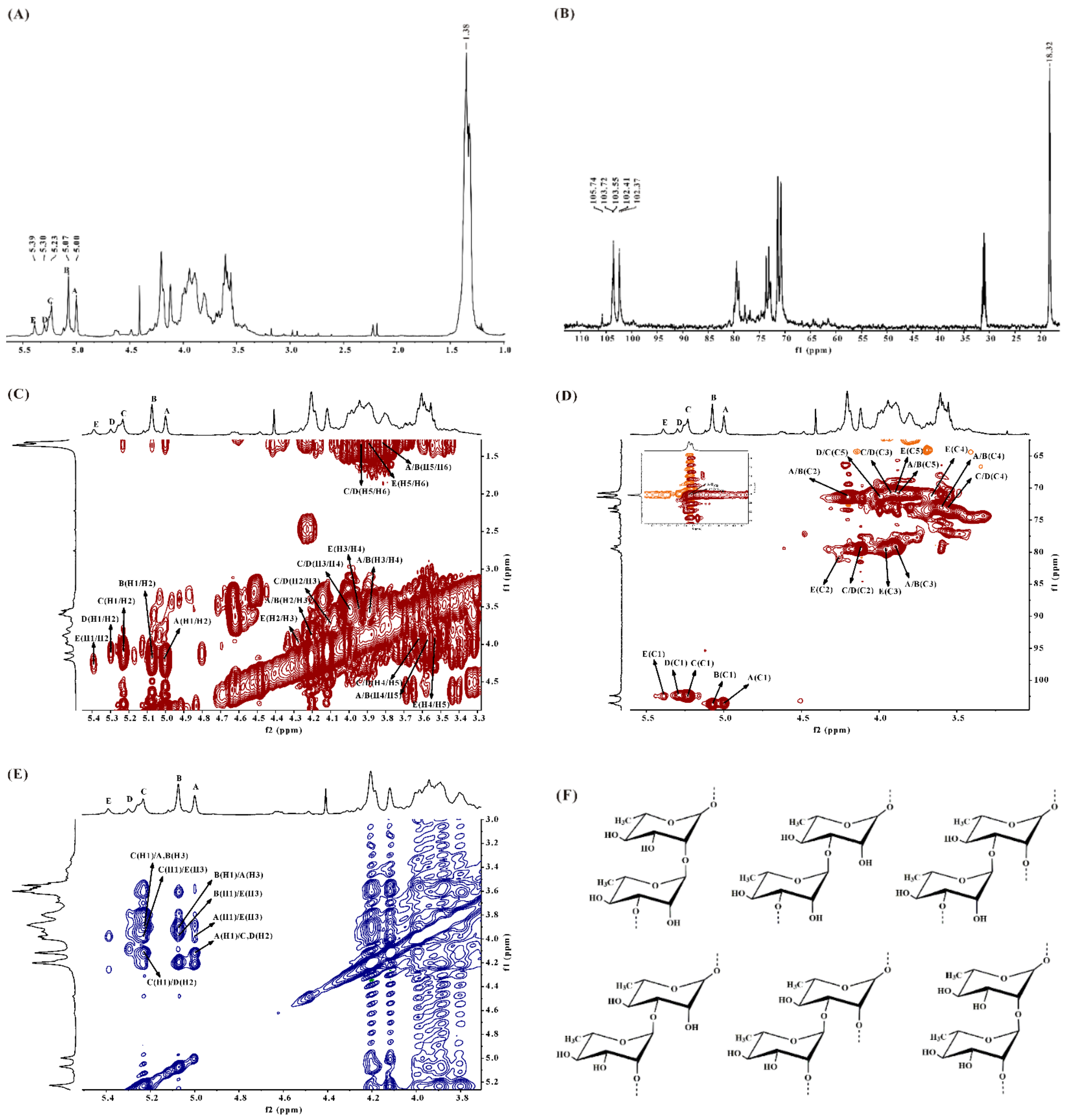
2.2.2. NMR Spectroscopy Analysis of DSMS-1
2.2.3. NMR Spectroscopy Analysis of MS-1
2.2.4. Hydrophilic Interaction Liquid Chromatography-Fourier-Transform Mass Spectrometry Analysis of MS-1-Derived Oligosaccharides
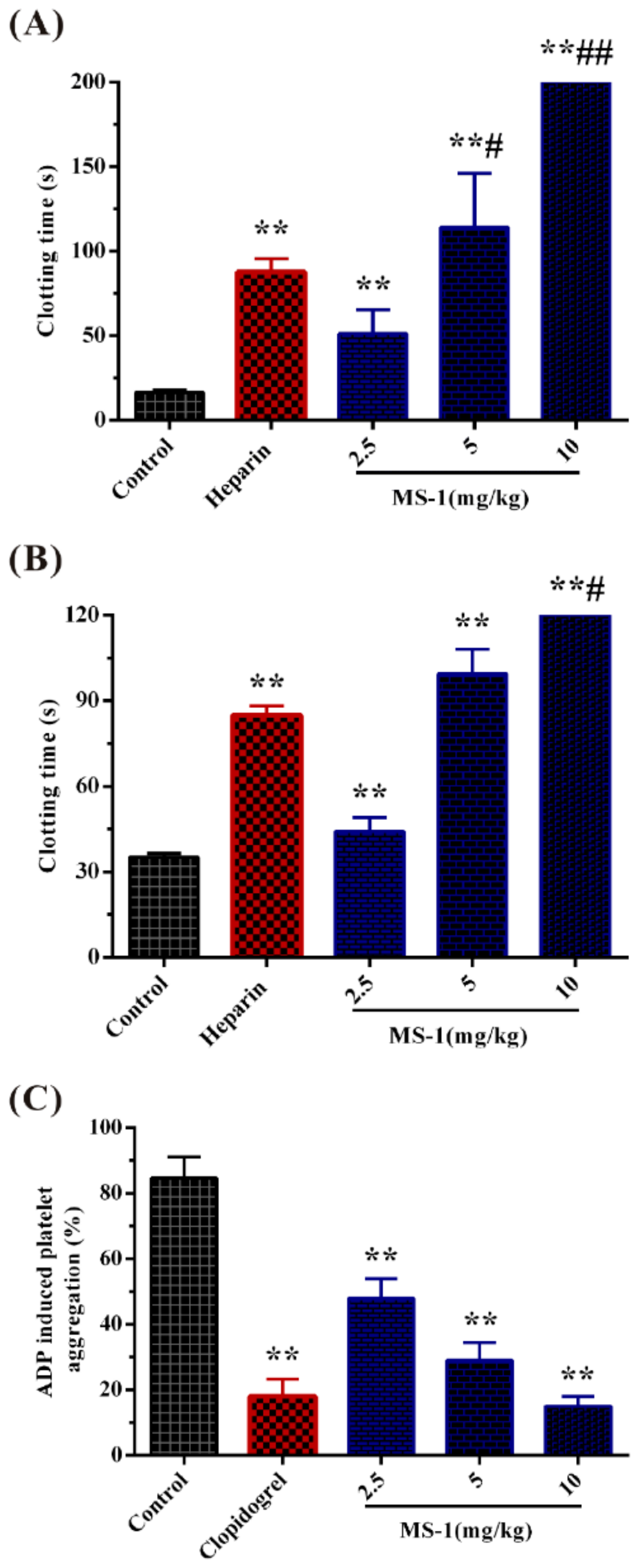
2.3. Anticoagulant Activity In Vitro and In Vivo of MS-1 and Its Platelet Aggregation
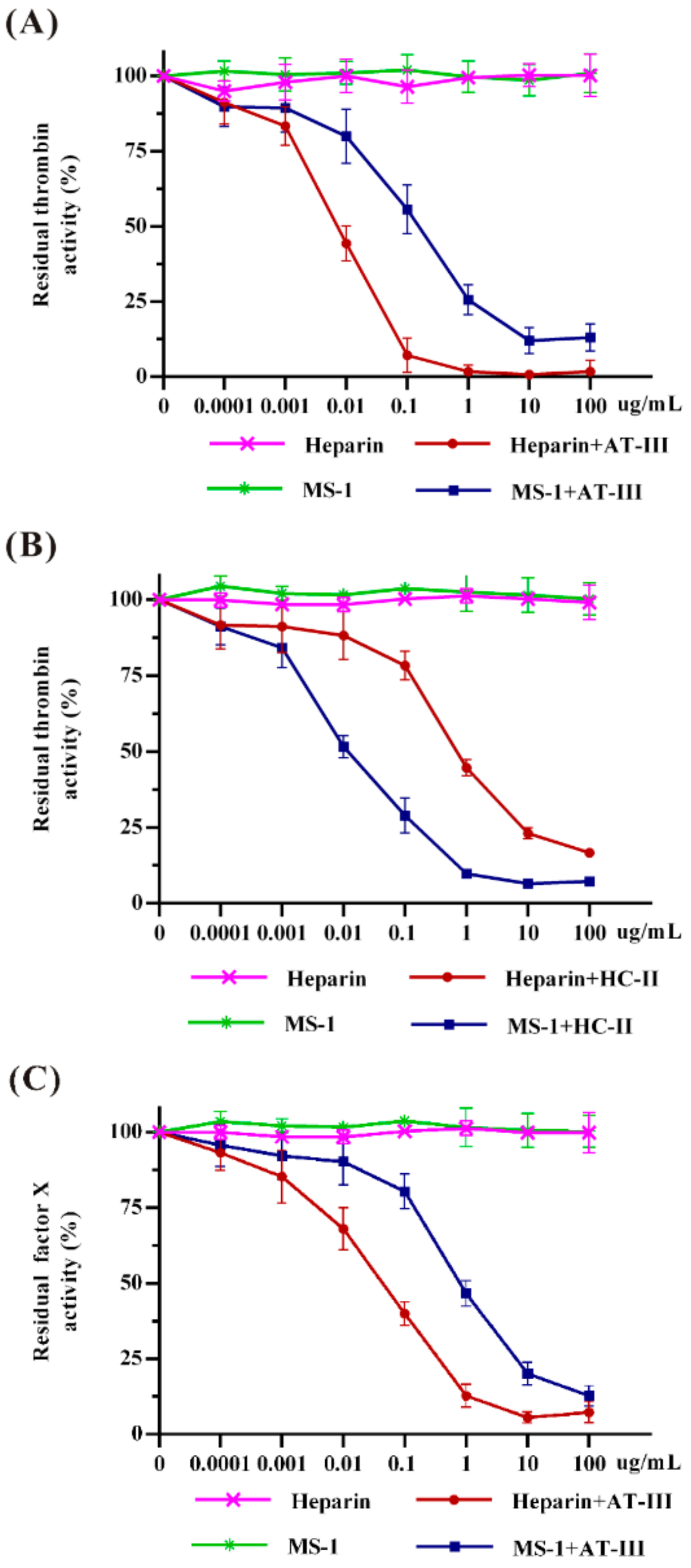
2.4. Effects of MS-1 on Thrombin and Factor Xa Activities Mediated by AT-III or HC-II
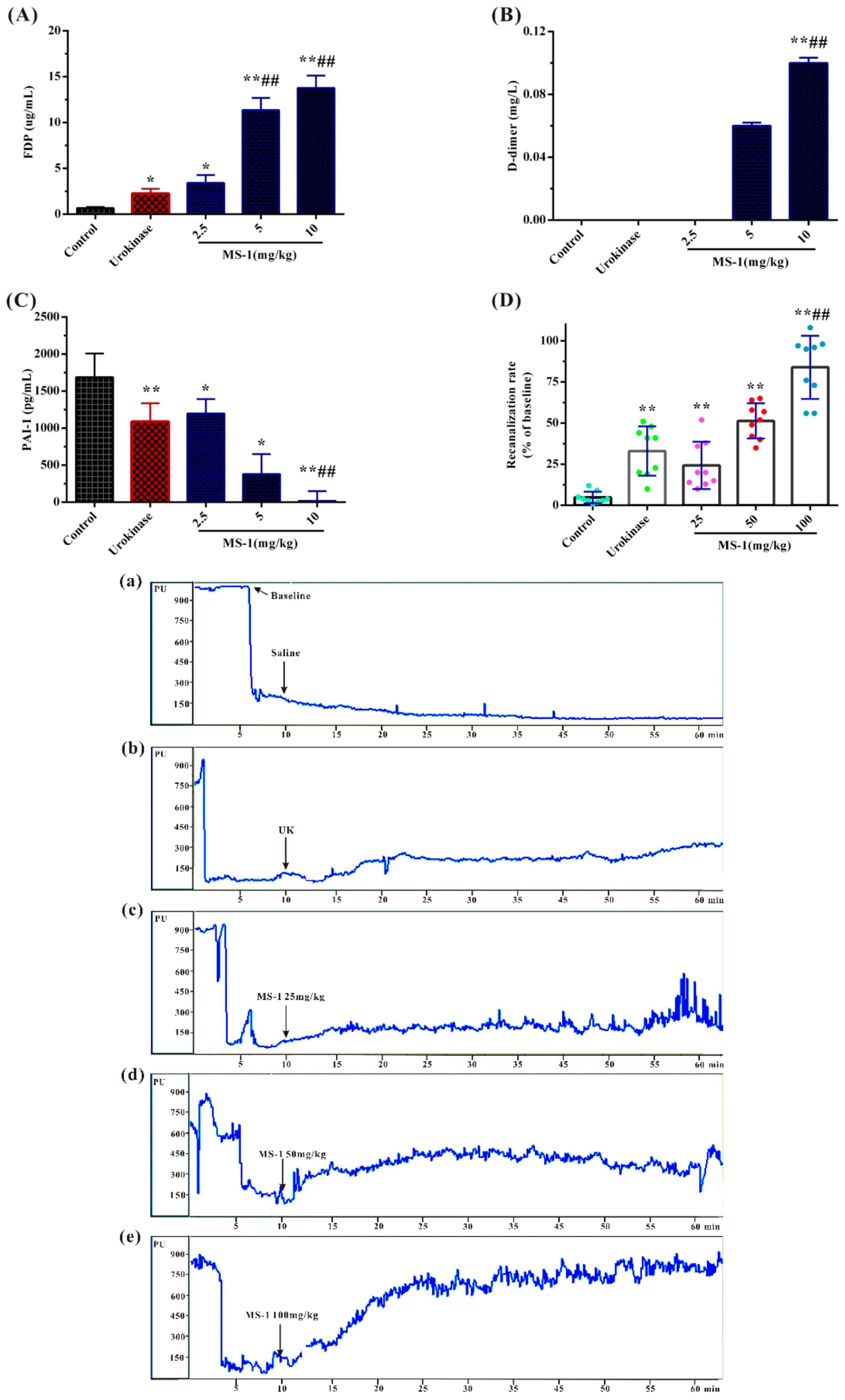
2.5. Fibrinolytic and Thrombolytic Activities of MS-1
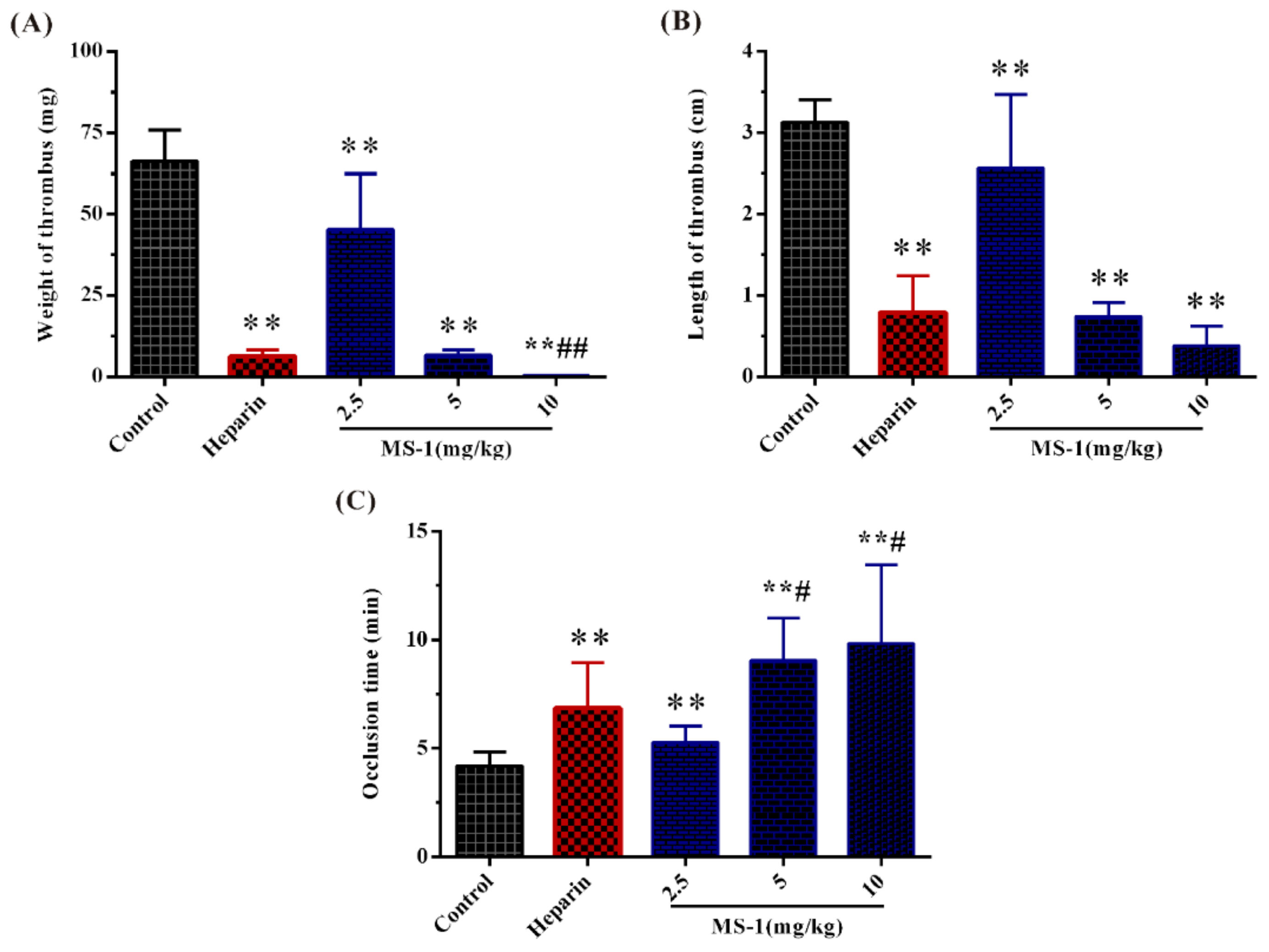
2.6. Antithrombotic Activities In Vitro and In Vivo of MS-1
3. Materials and Methods
3.1. Materials
3.2. Animals
3.3. Isolation and Purification of the Sulfated Polysaccharide
3.4. General Technique of Structural Characteristics
3.5. Spectroscopy Analysis
3.6. Profiling of MS-1-Derived Oligosaccharides by HILIC-FTMS
3.7. Assessment of Anticoagulant Activity and Platelet Aggregation
3.7.1. Anticoagulant Activity In Vitro
3.7.2. Anticoagulant Activity In Vivo and Platelet Aggregation
3.8. Effects of MS-1 on the Thrombin and Factor Xa Activities Mediated by AT-III or HC-II
3.9. Fibrinolytic and Thrombolytic Activities
3.9.1. Fibrinolytic Activity
3.9.2. FeCl3-Induced Carotid Artery Thrombosis
3.10. Assessment of Antithrombotic Activity
3.10.1. Experimental Thrombosis In Vitro
3.10.2. Carotid Artery Thrombosis In Vivo
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Akins, P.T.; Glenn, S.; Nemeth, P.M.; Derdeyn, C.P. Carotid artery thrombus associated with severe iron-deficiency anemia and thrombocytosis. Stroke 1996, 27, 1002–1005. [Google Scholar] [CrossRef]
- Weisel, J.W.; Stauffacher, C.V.; Bullitt, E.; Cohen, C. A model for fibrinogen: domains and sequence. Science 1985, 230, 1388–1391. [Google Scholar] [CrossRef] [PubMed]
- Raskob, G.E.; Angchaisuksiri, P.; Blanco, A.N.; Buller, H.; Gallus, A.; Hunt, B.J.; Hylek, E.M.; Kakkar, A.; Konstantinides, S.V.; Mccumber, M. Thrombosis: A major contributor to global disease burden. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- White, R.H. The epidemiology of venous thromboembolism. Thromb. Haemost. 2006, 21, 23–29. [Google Scholar] [CrossRef]
- Liaw, P.C.Y.; Becker, D.L.; Stafford, A.R.; Fredenburgh, J.C.; Weitz, J.I. Molecular basis for the susceptibility of fibrin-bound thrombin to inactivation by heparin cofactor II in the presence of dermatan sulfate but not heparin. J. Biol. Chem. 2001, 276, 20959–20965. [Google Scholar] [CrossRef]
- Pan, J.; Qian, Y.; Zhou, X.D.; Pazandak, A.; Frazier, S.B.; Weiser, P.; Lu, H.; Zhang, L.J. Oversulfated chondroitin sulfate is not the sole contaminant in heparin. Nat. Biotechnol. 2010, 28, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Andriamanantoanina, H.; Rinaudo, M. Characterization of the alginates from five madagascan brown algae. Carbohydr. Polym. 2010, 82, 555–560. [Google Scholar] [CrossRef]
- Hu, Y.; Yu, G.L.; Zhao, X.X.; Wang, Y.F.; Sun, X.C.; Jiao, G.L.; Zhao, X.; Chai, W.G. Structural characterization of natural ideal 6-O-sulfated agarose from red alga Gloiopeltis furcata. Carbohydr. Polym. 2012, 89, 883–889. [Google Scholar] [CrossRef]
- Karnjanapratum, S.; You, S. Molecular characteristics of sulfated polysaccharides from Monostroma nitidum and their in vitro anticancer and immunomodulatory activities. Int. J. Biol. Macromol. 2011, 48, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Mou, R.; Zhang, Z.; Gao, N.; Lin, L.; Li, Z.; Wu, M.; Zhao, J. Structural analysis and anticoagulant activities of three highly regular fucan sulfates as novel intrinsic factor Xase inhibitors. Carbohydr. Polym. 2018, 195, 257–266. [Google Scholar] [CrossRef]
- Wu, M.; Wen, D.; Gao, N.; Xiao, C.; Yang, L.; Xu, L.; Wu, L.; Peng, W.; Jiang, J.; Zhao, J. Anticoagulant and antithrombotic evaluation of native fucosylated chondroitin sulfates and their derivatives as selective inhibitors of intrinsic factor Xase. Eur. J. Med. Chem. 2015, 92, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H.; Pereira, M.S.; Valente, A.P.; Tollefsen, D.M.; Pavão, M.S.; Mourão, P.A. Selective cleavage and anticoagulant activity of a sulfated fucan: Stereospecific removal of a 2-sulfate ester from the polysaccharide by mild acid hydrolysis, preparation of oligosaccharides, and heparin cofactor II-dependent anticoagulant activity. Glycobiology 2004, 15, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.A.G.; Queiroz, I.N.L.D.; Quinderé, A.L.G.; Vairo, B.C.; Mourão, P.A.D.S.; Benevides, N.M.B. An antithrombin-dependent sulfated polysaccharide isolated from the green alga Caulerpa cupressoides has in vivo anti- and prothrombotic effects. Cienc. Rural. 2011, 41, 634–639. [Google Scholar] [CrossRef]
- Harada, N.; Maeda, M. Chemical structure of antithrombin-active rhamnan sulfate from Monostroma nitidum. Eur. J. Med. Chem. 1998, 62, 1647–1652. [Google Scholar]
- Lee, J.B.; Yamagaki, T.; Maeda, M.; Nakanishi, H. Rhamnan sulfate from cell walls of Monostroma latissimum. Phytochemistry 1998, 48, 921–925. [Google Scholar] [CrossRef]
- Lee, J.B.; Koizumi, S.; Hayashi, K.; Hayashi, T. Structure of rhamnan sulfate from the green alga Monostroma nitidum and its anti-herpetic effect. Carbohydr. Polym. 2010, 81, 572–577. [Google Scholar] [CrossRef]
- Qi, X.H.; Mao, W.J.; Gao, Y.; Chen, Y.; Chen, Y.L.; Zhao, C.Q.; Li, N.; Wang, C.Y.; Yan, M.X.; Lin, C.; et al. Chemical characteristic of an anticoagulant-active sulfated polysaccharide from Enteromorpha clathrata. Carbohydr. Polym. 2012, 90, 1804–1810. [Google Scholar] [CrossRef]
- Li, H.Y.; Mao, W.J.; Chen, Y.; Ren, S.M.; Qi, X.H.; Chen, Y.; Zhao, C.Q.; Li, N.; Wang, C.; Lin, C.; et al. Sequence analysis of the sulfated rhamno-oligosaccharides derived from a sulfated rhamnan. Carbohydr. Polym. 2012, 90, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Cassolato, J.E.F.; Noseda, M.D.; Pujol, C.A.; Pellizzari, F.M.; Damonte, E.B.; Duarte, M.E.R. Chemical structure and antiviral activity of the sulfated heterorhamnan isolated from the green seaweed Gayralia oxysperma. Carbohydr. Res. 2008, 343, 3085–3095. [Google Scholar] [CrossRef] [PubMed]
- Kovensky, J.; Cirelli, A.F. Chemical modification of glycosaminoglycans selective 2-sulfation of D-glucuronic acid units in heparan sulfate. Carbohydr. Res. 1997, 303, 119–122. [Google Scholar] [CrossRef]
- Li, G.; Steppich, J.; Wang, Z.; Sun, Y.; Xue, C.; Linhardt, R.J.; Li, L. Bottom-up low molecular weight heparin analysis using liquid chromatography-Fourier transform mass spectrometry for extensive characterization. Anal. Chem. 2014, 86, 6626–6632. [Google Scholar] [CrossRef]
- Maxwell, E.; Tan, Y.; Tan, Y.; Hu, H.; Benson, G.; Aizikov, K.; Conley, S.; Staples, G.O.; Slysz, G.W.; Smith, R.D.; et al. GlycReSoft: A software package for automated recognition of glycans from LC/MS data. PLoS ONE 2012, 7, e45474. [Google Scholar] [CrossRef]
- Anastyuk, S.D.; Shevchenko, N.M.; Nazarenko, E.L.; Dmitrenok, P.S.; Zvyagintseva, T.N. Structural analysis of a fucoidan from the brown alga Fucus evanescens by MALDI-TOF and tandem ESI mass spectrometry. Carbohydr. Res. 2009, 344, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Bérangère, T.; Jean-Yves, S.; Michael, M.; Marie-Pierre, G.; Régis, D. Differentiation of the fucoidan sulfated L-fucose isomers constituents by CE-ESIMS and molecular modeling. Carbohydr. Res. 2006, 341, 598–609. [Google Scholar]
- Li, H.Y.; Mao, W.J.; Zhang, X.L.; Qi, X.H.; Chen, Y.; Chen, Y.L.; Xu, J.; Zhao, C.Q.; Hou, Y.J.; Yang, Y.P. Structural characterization of an anticoagulant-active sulfated polysaccharide isolated from green alga Monostroma latissimum. Carbohydr. Polym. 2011, 85, 394–400. [Google Scholar] [CrossRef]
- Liu, X.; Hao, J.J.; He, X.X.; Wang, S.Y.; Cao, S.J.; Qin, L.; Mao, W.J. A rhamnan-type sulfated polysaccharide with novel structure from Monostroma angicava Kjellm (Chlorophyta) and its bioactivity. Carbohydr. Polym. 2017, 173, 732–748. [Google Scholar] [CrossRef]
- Li, N.; Liu, X.; He, X.X.; Wang, S.Y.; Cao, S.J.; Xia, Z.; Xian, H.L.; Qin, L.; Mao, W.J. Structure and anticoagulant property of a sulfated polysaccharide isolated from the green seaweed Monostroma angicava. Carbohydr. Polym. 2017, 159, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, M.; Robic, A. Structure and functional properties of Ulvan, a polysaccharide from green seaweeds. Biomacromolecules 2007, 8, 1765–1773. [Google Scholar] [CrossRef]
- Liu, J.; Pedersen, L.C. Anticogaulant heparan sulfate: structural specificity and biosynthesis. Appl. Microbiol. Biotechnol. 2007, 74, 263–272. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Hong, J.L.; Shu, P.; Ni, Y.J.; Qin, M.J. A new dicoumarin and anticoagulant activity from Viola yedoensis Makino. Fitoterapia 2009, 80, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.J.; Fang, F.; Li, H.Y.; Qi, X.H.; Sun, H.H.; Chen, Y.; Guo, S.D. Heparinoid-active two sulfated polysaccharides isolated from marine green algae Monostroma nitidum. Carbohydr. Polym. 2008, 74, 834–839. [Google Scholar] [CrossRef]
- Puri, R.N.; Colman, R.W. ADP induced blood platelet activation: a review. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 437–502. [Google Scholar]
- Souza, R.O.S.; Assreuy, A.M.S.; Madeira, J.C.; Chagas, F.D.S.; Parreiras, L.A.; Santos, G.R.C.; Mourão, P.A.S.; Pereira, M.G. Purified polysaccharides of Geoffroea spinosa barks have anticoagulant and antithrombotic activities devoid of hemorrhagic risks. Carbohydr Polym. 2015, 124, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.S.; Seto, S.W.; Krishna, S.M.; Sharma, S.; Jose, R.J.; Biros, E.; Wang, Y.; Morton, S.K.; Golledge, J. Parenteral administration of factor Xa/IIa inhibitors limits experimental aortic aneurysm and atherosclerosis. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Hayashi, T.; Lee, J.B.; Srisomporn, P.; Maeda, M.; Ozawa, T.; Sakuragawa, N. Inhibition of thrombin by sulfated polysaccharides isolated from green algae. Biochim. Biophys. Acta 2000, 1543, 86–94. [Google Scholar] [CrossRef]
- Vasconcelos, A.; Sucupira, I.; Guedes, A.; Queiroz, I.; Frattani, F.; Fonseca, R.; Pomin, V. Anticoagulant and antithrombotic properties of three structurally correlated sea urchin sulfated glycans and their low-molecular-weight derivatives. Mar. Drugs. 2018, 16, 304. [Google Scholar] [CrossRef]
- Melo, F.R.; Mourão, P.A. An algal sulfated galactan has an unusual dual effect on venous thrombosis due to activation of factor XII and inhibition of the coagulation proteases. Thromb. Haemost. 2008, 99, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Duan, X.; Feng, R.; Zhao, Z.; Feng, X.; Lu, Q.; Jin, Q.; Zhou, J.; Bao, J.; Jin, Z. Diagnostic implication of fibrin degradation products and D-dimer in aortic dissection. Sci. Rep. 2017, 7, 1–5. [Google Scholar] [CrossRef]
- Ay, C.; Vormittag, R.D. D-dimer and prothrombin fragment 1 + 2 predict venous thromboembolism in patients with cancer: results from the vienna cancer and thrombosis Study. J. Clin. Oncol. 2009, 27, 4124–4129. [Google Scholar] [CrossRef]
- Kline, J.A.; Hogg, M.M.; Courtney, D.M.; Miller, C.D.; Jones, A.E.; Smithline, H.A. D-dimer threshold increase with pretest probability unlikely for pulmonary embolism to decrease unnecessary computerized tomographic pulmonary angiography. J. Thromb. Haemost. 2012, 10, 572–581. [Google Scholar] [CrossRef]
- Bajou, K.; Herkenne, S.; Thijssen, V.L.; D’Amico, S.; Nguyen, N.Q.; Bouché, A.; Tabruyn, S.; Srahna, M.; Carabin, J.Y.; Nivelles, O. PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat. Med. 2014, 20, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Eitzman, D.; Westrick, R.Z.; Tyson, J.; Ginsburg, D. Plasminogen activator inhibitor-1 deficiency protects against atherosclerosis progression in the mouse carotid artery. Blood 2000, 96, 4212–4215. [Google Scholar] [PubMed]
- Zhu, Y.; Carmeliet, P.; Fay, W.P. Plasminogen activator inhibitor-1 is a major determinant of arterial thrombolysis resistance. Circulation 1999, 99, 3050–3055. [Google Scholar] [CrossRef]
- Dobrovolsky, A.B.; Titaeva, E.V. The fibrinolysis system: regulation of activity and physiologic functions of its main components. Biochemistry 2002, 67, 99–108. [Google Scholar]
- Cipriani, T.R.; Gracher, A.H.; de Souza, L.M.; Fonseca, R.J.; Belmiro, C.L.; Gorin, P.A.; Sassaki, G.L.; Iacomini, M. Influence of molecular weight of chemically sulfated citrus pectin fractions on their antithrombotic and bleeding effects. Thromb. Haemost. 2009, 101, 860–866. [Google Scholar] [CrossRef]
- Gracher, A.H.P.; Cipriani, T.R.; Carbonero, E.R.; Gorin, P.A.J.; Marcello, I. Antithrombin and heparin cofactor II-mediated inactivation of alpha-thrombin by a synthetic, sulfated mannogalactan. Thromb. Haemost. 2010, 126, 180–187. [Google Scholar]
- Martinichen-Herrero, J.C.; Carbonero, E.R.; Gorin, P.A.J.; Iacomini, M. Anticoagulant and antithrombotic activity of a sulfate obtained from a glucan component of the lichen Parmotrema mantiqueirense Hale. Carbohydr. Polym. 2005, 60, 7–13. [Google Scholar] [CrossRef]
- Dubois, M.E.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F. Colorimetric method for determination of sugars and related substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]
- Li, N.; Mao, W.J.; Yan, M.X.; Liu, X.; Xia, Z.; Wang, S.Y.; Xiao, B.; Chen, C.L.; Zhang, L.F.; Cao, S.J. Structural characterization and anticoagulant activity of a sulfated polysaccharide from the green alga Codium divaricatum. Carbohydr. Polym. 2015, 121, 175–182. [Google Scholar] [CrossRef]
- Therho, T.T.; Hartiala, K. Method for determination of the sulfate content of glycosaminoglycans. Anal. Biochem. 1971, 41, 471–476. [Google Scholar] [CrossRef]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakashima, T.; Ueda, T.; Tomii, K.; Kouno, I. Facilediscrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 2007, 55, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Falshaw, R.; Furneaux, R.H. Structural analysis of carrageenans from thetetrasporic stages of the red algae, Gigartina lanceata and Gigartina chapmanii (Gigartinaceae, Rhodophyta). Carbohydr. Res. 1998, 307, 325–331. [Google Scholar] [CrossRef]
- Sims, I.M.; Carnachan, S.M.; Bell, T.J.; Hinkley, S.F.R. Methylation analysis of polysaccharides: Technical advice. Carbohyd. Polym. 2018, 188, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shingel, K.I. Determination of structural peculiarities of dexran, pullulan and gamma-irradiated pullulan by Fourier-transform IR spectroscopy. Carbohydr. Res. 2002, 337, 1445–1451. [Google Scholar] [CrossRef]
- Li, H.Y.; Mao, W.J.; Hou, Y.J.; Gao, Y.; Qi, X.H.; Zhao, C.Q.; Chen, Y.; Chen, Y.L.; Li, N. Preparation, structure and anticoagulant activity of a low molecular weight fraction produced by mild acid hydrolysis of sulfated rhamnan from Monostroma latissimum. Bioresour. Technol. 2012, 114, 414–418. [Google Scholar] [CrossRef]
- Hogan, J.D.; Klein, J.A.; Wu, J.; Chopra, P.; Boons, G.J.; Carvalho, L.; Lin, C.; Zaia, J. Software for peak finding and elemental composition assignment for glycosaminoglycan tandem mass spectra. Mol. Cell. Proteomics 2018, 17, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Y.; Cai, C.; Chang, Y.; Zhang, F.; Linhardt, R.J.; Xue, C.; Li, G.; Yu, G. A novel structural fucosylated chondroitin sulfate from Holothuria mexicana and its effects on growth factors binding and anticoagulation. Carbohyd. Polym. 2018, 181, 1160–1168. [Google Scholar] [CrossRef]
- Xin, M.; Ren, L.; Sun, Y.; Li, H.H.; Guan, H.S.; He, X.X.; Li, C.X. Anticoagulant and antithrombotic activities of low-molecular-weight propylene glycol alginate sodium sulfate (PSS). Eur. J. Med. Chem. 2016, 114, 33–40. [Google Scholar] [CrossRef]
- Colliec, S.; Fischer, A.M.; Tapon-Bretaudiere, J.; Boisson, C.; Durand, P.; Jozefonvicz, J. Anticoagulant properties of a fucoidan fraction. Thromb. Res. 1991, 64, 143–154. [Google Scholar] [CrossRef]
- Markus, H.S.; Droste, D.W.; Manfred, K.; Vincent, L.; Lees, K.R.; Mario, S.; E Bernd, R. Dual antiplatelet therapy with clopidogrel and aspirin in symptomatic carotid stenosis evaluated using doppler embolic signal detection: The clopidogrel and aspirin for reduction of emboli in symptomatic carotid stenosis (CARESS) trial. Circulation 2005, 111, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Ulusal, B.G.; Cheng, M.H.; Wei, F.C.; Ho-Asjoe, M.; Song, D. Breast reconstruction using the entire transverse abdominal adipocutaneous flap based on unilateral superficial or deep inferior epigastric vessels. Plast. Reconstr. Surg. 2006, 117, 1395–1403. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methylated Alditol Acetate | Linkage Pattern | Molar Percent Ratio | |
|---|---|---|---|
| MS-1 | DSMS-1 | ||
| 1,5-Di-O-acetyl-2,3,4-tri-O-methyl-rhamnitol | Rhap-(1→ | 3.53 | 3.91 |
| 1,5-Di-O-acetyl-2,3,4-tri-O-methyl-xylitol | Xylp-(1→ | 0.68 | 0.62 |
| 1,2,5-Tri-O-acetyl-3,4-di-O-methyl-rhamnitol | →2)-Rhap-(1→ | 22.68 | 30.84 |
| 1,3,5-Tri-O-acetyl-2,4-di-O-methyl-rhamnitol | →3)-Rhap-(1→ | 30.02 | 45.17 |
| 1,5-Di-O-acetyl-2,3,4,6-tetra-O-methyl-glucitol | Glcp-(1→ | 1.03 | 1.13 |
| 1,4,5-Tri-O-acetyl-2,3-tri-O-methyl-xylitol | →4)-Xylp-(1→ | 1.38 | 1.72 |
| 1,3,4,5-Tetra-O-acetyl-2-O-methyl-rhamnitol | →3,4)-Rhap-(1→ | 9.27 | 1.13 |
| 1,2,3,5-Tetra-O-acetyl-4-O-methyl-rhamnitol | →2,3)-Rhap-(1→ | 20.87 | 12.10 |
| 1,2,4,5-Tetra-O-acetyl-3-O-methyl-rhamnitol | →2,4)-Rhap-(1→ | 7.16 | 0.57 |
| 1,6,5-Tri-O-acetyl-2,3,4-tri-O-methyl-glucitol | →6)-Glcp-(1→ | 1.42 | 1.17 |
| 1,4,5-Tri-O-acetyl-2,3,6-tri-O-methyl-glucitol | →4)-Glcp-(1→ | 1.86 | 1.64 |
| Rhamnosyl Residues | Chemical Shift (ppm) | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| A →3)-α-l-Rhap-(1→ | 1H | 5.07 | 4.20 | 3.96 | 3.59 | 4.10 | 1.43 |
| 13C | 103.32 | 71.36 | 79.31 | 73.16 | 70.69 | 18.39 | |
| B →2)-α-l-Rhap-(1→ | 1H | 5.23 | 4.28 | 4.00 | 3.59 | 4.14 | 1.38 |
| 13C | 101.85 | 79.78 | 70.72 | 73.19 | 70.79 | 18.39 | |
| C →2)-α-l-Rhap(4SO4)-(1→ | 1H | 5.25 | 4.30 | 4.12 | 4.39 | 4.10 | 1.43 |
| 13C | 101.85 | 79.78 | 70.86 | 80.39 | 70.74 | 18.39 | |
| D →2,3)-α-l-Rhap-(1→ | 1H | 5.39 | 4.26 | 4.09 | 3.60 | 4.00 | 1.38 |
| 13C | 101.31 | 79.30 | 80.46 | 73.19 | 70.67 | 18.39 | |
| E →3)-α-l-Rhap(4SO4)-(1→ | 1H | 5.45 | 4.25 | 4.00 | 4.31 | 4.02 | 1.35 |
| 13C | 100.40 | 71.46 | 79.35 | 81.99 | 70.69 | 18.39 | |
| F →3)-α-l-Rhap(2SO4)-(1→ | 1H | 5.50 | 4.70 | 4.11 | 3.58 | 3.98 | 1.35 |
| 13C | 100.76 | 78.18 | 77.42 | 73.16 | 70.51 | 18.39 | |
| Clotting Time a (s) | Sample | Concentration (μg/mL) | ||||
|---|---|---|---|---|---|---|
| 0 | 5 | 10 | 25 | 50 | ||
| APTT | MS-1 | 40.3 ± 1.1 | 59.2 ± 0.5 * | 97.8 ± 5.0 ** | >200 ** | - |
| Heparin | 127.8 ± 12.9 ** | >200 ** | - | - | ||
| TT | MS-1 | 14.5 ± 0.3 | 17.1 ± 0.2 | 19.4 ± 1.5 * | 57.4 ± 3.6 ** | >120 ** |
| Heparin | 70.3 ± 0.9 ** | >120 ** | - | - | ||
| PT | MS-1 | 13.9 ± 0.7 | 12.8 ± 0.1 | 13.3 ± 0.2 | 15.5 ± 0.5 | 19.3 ± 0.8 |
| Heparin | 17.4 ± 4.4 * | 33.6 ± 1.7 ** | 81.3 ± 2.2 ** | >120 ** | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, S.; He, X.; Qin, L.; He, M.; Yang, Y.; Liu, Z.; Mao, W. Anticoagulant and Antithrombotic Properties in Vitro and in Vivo of a Novel Sulfated Polysaccharide from Marine Green Alga Monostroma nitidum. Mar. Drugs 2019, 17, 247. https://doi.org/10.3390/md17040247
Cao S, He X, Qin L, He M, Yang Y, Liu Z, Mao W. Anticoagulant and Antithrombotic Properties in Vitro and in Vivo of a Novel Sulfated Polysaccharide from Marine Green Alga Monostroma nitidum. Marine Drugs. 2019; 17(4):247. https://doi.org/10.3390/md17040247
Chicago/Turabian StyleCao, Sujian, Xiaoxi He, Ling Qin, Meijia He, Yajing Yang, Zhichun Liu, and Wenjun Mao. 2019. "Anticoagulant and Antithrombotic Properties in Vitro and in Vivo of a Novel Sulfated Polysaccharide from Marine Green Alga Monostroma nitidum" Marine Drugs 17, no. 4: 247. https://doi.org/10.3390/md17040247
APA StyleCao, S., He, X., Qin, L., He, M., Yang, Y., Liu, Z., & Mao, W. (2019). Anticoagulant and Antithrombotic Properties in Vitro and in Vivo of a Novel Sulfated Polysaccharide from Marine Green Alga Monostroma nitidum. Marine Drugs, 17(4), 247. https://doi.org/10.3390/md17040247