Marine-Derived Anticancer Agents: Clinical Benefits, Innovative Mechanisms, and New Targets

 ,
, 

 ,
, 
 , and
, and 
Abstract
:1. An Overview on Fifty Years of Marine-Derived Drug Discovery
2. Marine Chemotherapeutic Pipeline and Clinical Benefits
2.1. Licensed Drugs
2.2. Agents in Clinical Trials
3. New Routes in Oncological Research—Innovative Mechanisms and New Molecular Targets
4. Additional Technological Improvements
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bergmann, W.; Burke, D.C. Contributions to the study of marine products. XXXIX. The nucleosides of sponges. III. Spongothymidine and spongouridine. J. Org. Chem. 1955, 11, 1501–1507. [Google Scholar] [CrossRef]
- Burnett, A.; Wetzler, M.; Lowenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.R. Academic drug discovery: current status and prospects. Expert Opin. Drug Discov. 2015, 10, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Feher, M.; Schmidt, J.M. Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef]
- Gomes, N.G.M.; Pereira, D.M.; Valentão, P.; Andrade, P.B. Hybrid MS/NMR methods on the prioritization of natural products: applications in drug discovery. J. Pharm. Biomed. Anal. 2018, 147, 234–249. [Google Scholar] [CrossRef]
- Liu, Z.; Delavan, B.; Roberts, R.; Tong, W. Lessons learned from two decades of anticancer drugs. Trends Pharmacol. Sci. 2017, 38, 852–872. [Google Scholar] [CrossRef]
- Gomes, N.G.M.; Dasari, R.; Chandra, S.; Kiss, R.; Kornienko, A. Marine invertebrate metabolites with anticancer activities: solutions to the “supply problem”. Mar. Drugs 2016, 14, 98. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Talley, R.W.; Vaitkevicius, V.K. Megaloblastosis produced by a cytosine antagonist: 1-β-ᴅ-arabinofuranosylcytosine. Blood 1963, 21, 352–362. [Google Scholar] [PubMed]
- Frei, E.; Freireich, E.J.; Gehan, E.; Pinkel, D.; Holland, J.F.; Selawry, O.; Haurani, F.; Spurr, C.L.; Hayes, D.M.; James, G.W.; et al. Studies of sequential and combination antimetabolite therapy in acute leukemia: 6–mercaptopurine and methotrexate. Blood 1961, 18, 431. [Google Scholar]
- Howard, J.P.; Albo, V.; Newton, W.A., Jr. Cytosine arabinoside: Results of a cooperative study in acute childhood leukemia. Cancer 1968, 21, 341–345. [Google Scholar] [CrossRef]
- Lichtman, M.A. A historical perspective on the development of the cytarabine (7 days) and daunorubicin (3 days) treatment regimen for acute myelogenous leukemia: 2013 the 40th anniversary of 7+3. Blood Cells Mol. Dis. 2013, 50, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Glantz, M.J.; LaFollette, S.; Jaeckle, K.A.; Shapiro, W.; Swinnen, L.; Rozental, J.R.; Phuphanich, S.; Rogers, L.R.; Gutheil, J.C.; Batchelor, T.; et al. Randomized trial of a slow-release versus a standard formulation of cytarabine for the intrathecal treatment of lymphomatous meningitis. J. Clin. Oncol. 1999, 17, 3110–3116. [Google Scholar] [CrossRef] [PubMed]
- Glantz, M.J.; Jaeckle, K.A.; Chamberlain, M.C.; Phuphanich, S.; Recht, L.; Swinnen, L.J.; Maria, B.; LaFollette, S.; Schumann, G.B.; Cole, B.F.; et al. A randomized controlled trial comparing intrathecal sustained–release cytarabine (DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from solid tumors. Clin. Cancer Res. 1999, 5, 3394–3402. [Google Scholar] [PubMed]
- Jaeckle, K.A.; Batchelor, T.; O’Day, S.J.; Phuphanich, S.; New, P.; Lesser, G.; Cohn, A.; Gilbert, M.; Aiken, R.; Heros, D.; et al. An open label trial of sustained–release cytarabine (DepoCytTM) for the intrathecal treatment of solid tumor neoplastic meningitis. J. Neurooncol. 2002, 57, 231–239. [Google Scholar] [CrossRef]
- Mack, F.; Baumert, B.G.; Schäfer, N.; Hattingen, E.; Scheffler, B.; Herrlinger, U.; Glas, M. Therapy of leptomeningeal metastasis in solid tumors. Cancer Treat. Rev. 2016, 43, 83–91. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Cytarabine. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=cytarabine&cntry=&state=&city=&dist=&Search=Search (accessed on 15 March 2019).
- European Union Clinical Trials Register. Cytarabine. Available online: https://www.clinicaltrialsregister.eu/ctr–search/search?query=cytarabine (accessed on 15 March 2019).
- Rinerhart, K.L.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.; Sun, F.; Li, H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumour agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Samuels, B.L.; Chawla, S.; Patel, S.; von Mehren, M.; Hamm, J.; Kaiser, P.E.; Schuetze, S.; Li, J.; Aymes, A.; Demetri, G.D. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: Results of a worldwide expanded access program study. Ann. Oncol. 2013, 24, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Blessing, J.A.; Street, D.G.; Muller, C.Y.; Burke, J.J.; Hensley, M.L. A phase II evaluation of trabectedin in the treatment of advanced, persistent, or recurrent uterine leiomyosarcoma: A gynecologic oncology group study. Gynecol. Oncol. 2012, 124, 48–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasner, C.N.; Poveda, A.; Herzoq, T.J.; Vermorken, J.B.; Kaye, S.B.; Nieto, A.; Claret, P.L.; Park, Y.C.; Parekh, T.; Monk, B.J. Patient–reported outcomes in relapsed ovarian cancer: results from a randomized Phase III study of trabectedin with pegylated liposomal doxorubicin (PLD) versus PLD alone. Gynecol. Oncol. 2012, 127, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Ghatage, P.; Parekh, T.; Henitz, E.; Knoblauch, R.; Matos-Pita, A.S.; Nieto, A.; Park, Y.C.; Cheng, P.S.; Li, W.; et al. Effect of BRCA1 and XPG mutations on treatment response to trabectedin and pegylated liposomal doxorubicin in patients with advanced ovarian cancer: Exploratory analysis of the phase 3 OVA-301 study. Ann. Oncol. 2015, 26, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Fetterly, G.J.; Owen, J.S.; Stuyckens, K.; Passarell, J.A.; Zannikos, P.; Soto-Matos, A.; Izquierdo, M.A.; Perez–Ruixo, J.J. Semimechanistic pharmacokinetic/pharmacodynamic model for hepatoprotective effect of dexamethasone on transient transaminitis after trabectedin (ET-743) treatment. Cancer Chemother. Pharmacol. 2008, 62, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Chawla, S.P.; von Mehren, M.; Ritch, P.; Baker, L.H.; Blay, J.Y.; Hande, K.R.; Keohan, M.L.; Samuels, B.L.; Schuetze, S.; et al. Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase II study of two different schedules. J. Clin. Oncol. 2009, 27, 4188–4196. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Uemura, D. Halichondrins—Antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Cortés, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Diéras, V.; Delozier, T.; et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): A phase 3 open–label randomized study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Pean, E.; Klaar, S.; Berglund, E.G.; Salmonson, T.; Borregaard, J.; Hofland, K.F.; Ersbøll, J.; Abadie, E.; Giuliani, R.; Pignatti, F. The European Medicines Agency review of eribulin for the treatment of patients with locally advanced or metastatic breast cancer: summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Clin. Cancer Res. 2012, 18, 4491–4497. [Google Scholar] [CrossRef]
- Aseyev, O.; Ribeiro, J.M.; Cardoso, F. Review on the clinical use of eribulin mesylate for the treatment of breast cancer. Expert Opin. Pharmacother. 2016, 17, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Devriese, L.A.; Witteveen, P.O.; Wanders, J.; Law, J.; Edwards, G.; Reyderman, L.; Copalu, W.; Peng, F.; Marchetti, S.; Beijnen, J.H.; et al. Pharmacokinetics of eribulin mesylate in patient with solid tumours receiving repeated oral rifampicin. Br. J. Clin. Pharmacol. 2013, 75, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Taur, J.-S.; DesJardins, C.S.; Schuck, E.L.; Wong, Y.N. Interactions between the chemotherapeutic agent eribulin mesylate (E7389) and P–glycoprotein in CF–1 abcb1a–deficient mice and Caco–2 cells. Xenobiotica 2011, 41, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Huang, W.; Fan, W. Emerging therapies for breast cancer. J. Hematol. Oncol. 2017, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.; Savulsky, C.; Aktan, G.; Savulsky, C.; Olivo, M.; Aktan, G.; Kaufman, P.A.; Xing, D.; Almonte, A.; Misir, S.; et al. Phase 1b/2 study to evaluate eribulin mesylate in combination with pembrolizumab in patients with metastatic triple–negative breast cancer. Cancer Res. 2018, 78, PD6-13. [Google Scholar] [CrossRef]
- Schöffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open–label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. caC10–vcMMAE, an anti–CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN–35) in patients with relapsed or refractory systemic anaplastic large cell lymphoma: results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef]
- De Claro, R.A.; McGinn, K.; Kwitkowski, V.; Bullock, J.; Khandelwal, A.; Habtemariam, B.; Ouyang, Y.; Saber, H.; Lee, K.; Koti, K.; et al. U.S. Food and Drug Administration approval summary: Brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin. Cancer Res. 2012, 18, 5845–5849. [Google Scholar] [CrossRef] [PubMed]
- Gravanis, I.; Tzogani, K.; van Hennik, P.; de Graeff, P.; Schmitt, P.; Mueller-Berghaus, J.; Salmonson, T.; Gisselbrecht, C.; Laane, E.; Bergmann, L.; et al. The European Medicines Agency review of brentuximab vedotin (Adcetris) for the treatment of adult patients with relapsed or refractory CD30+ Hodgkin lymphoma or systemic anaplastic large cell lymphoma: summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Oncologist 2016, 21, 102–109. [Google Scholar] [PubMed]
- Bonthapally, V.; Yang, H.; Ayyagari, R.; Tan, R.D.; Cai, S.; Wu, E.; Gautam, A.; Chi, A.; Huebner, D. Brentuximab vedotin compared with other therapies in relapsed/refractory Hodgkin lymphoma post ASCT: Median overall survival meta-analysis. Curr. Med. Res. Opin. 2015, 31, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Chen, R.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Connors, J.M.; Engert, A.; Larsen, E.K.; Chi, X.; et al. Durable remissions in a pivotal phase 2 study of brentuximab vedotin in relapsed or refractory Hodgkin lymphoma. Blood 2015, 125, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Pro, B.; Advani, R.H.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.A.; Connors, J.M.; et al. Three-year survival results from an ongoing phase 2 study of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2013, 122, 1809. [Google Scholar]
- Han, T.H.; Gopal, A.K.; Ramchandren, R.; Gov, A.; Chen, R.; Matous, J.V.; Cooper, M.; Grove, L.E.; Alley, S.C.; Lynch, C.M.; et al. CYP3A-mediated drug-drug interaction potential and excretion of brentuximab vedotin, an antibody-drug conjugate, in patients with CD30-positive hematologic malignancies. J. Clin. Pharmacol. 2013, 53, 866–877. [Google Scholar] [CrossRef]
- Fanale, M.A.; Horwitz, S.M.; Forero-Torres, A.; Bartlett, N.L.; Advani, R.H.; Pro, B.; Chen, R.W.; Davies, A.; Illidge, T.; Huebner, D.; et al. Brentuximab vedotin in the front–line treatment of patients with CD30+ peripheral T–cell lymphomas: Results of a phase I study. J. Clin. Oncol. 2014, 32, 3137–3143. [Google Scholar] [CrossRef]
- Younes, A. Brentuximab vedotin for the treatment of patients with Hodgkin lymphoma. Hematol. Oncol. Clin. N. Am. 2014, 28, 27–32. [Google Scholar] [CrossRef]
- Deng, C.; Pan, B.; O’Connor, O.A. Brentixumab vedotin. Clin. Cancer Res. 2013, 19, 22–27. [Google Scholar] [CrossRef]
- Illés, Á.; Jóna, Á.; Miltényi, Z. Brentuximab vedotin for treating Hodgkin’s lymphoma: An analysis of pharmacology and clinical efficacy. Expert Opin. Drug Metab. Toxicol. 2015, 11, 451–459. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Keifer, P.A.; Wilson, G.R.; Perun, T.J.; Sakai, R.; Thompson, A.G.; Stroh, J.G.; Shield, L.S.; et al. Bioactive compounds from aquatic and terrestrial sources. J. Nat. Prod. 1990, 53, 771–792. [Google Scholar] [CrossRef]
- Izquierdo, M.A.; Bowman, A.; García, M.; Jodrell, D.; Martinez, M.; Pardo, B.; Gómez, J.; López–Martin, J.A.; Jimeno, J.; Germá, J.R.; et al. Phase I clinical and pharmacokinetic study of plitidepsin as a 1–hour weekly intravenous infusion in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 3105–3112. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Álvarez, S.; Pardal, E.; Sánchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martín, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Devel. Ther. 2017, 11, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Cibeira, M.T.; Richardson, P.G.; Prosper, F.; Oriol, A.; de la Rubia, J.; Lahuerta, J.J.; García–Sanz, R.; Extremera, S.; Szyldergemajn, S.; et al. Phase II clinical and pharmacokinetic study of plitidepsin 3-hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin. Cancer Res. 2010, 16, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Spicka, I.; Ocio, E.M.; Oakervee, H.E.; Greil, R.; Banh, R.H.; Catley, L.; Huang, S.-Y.; Rozario, J.M.; Dimopoulos, M.A.; Rodríguez, J.; et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Blood 2017, 130, 1886. [Google Scholar]
- Toulmonde, M.; Le Cesne, A.; Piperno-Neumann, S.; Penel, N.; Chevreau, C.; Duffaud, F.; Bellera, C.; Italiano, A. Aplidin in patients with advanced dedifferentiated liposarcomas: A French Sarcoma Group Single-Arm phase II study. Ann. Oncol. 2015, 26, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Guillem, V.; Garcia, M.; Rivera, F.; Tabernero, J.; Cullell, M.; Lopez–Martin, J.A.; Pollard, P.; Dumez, H.; del Muro, X.G.; et al. Phase II randomized study of Plitidepsin (Aplidin), alone or in association with l-carnitine, in patients with unresectable advanced renal cell carcinoma. Mar. Drugs 2009, 7, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Danu, A.; Willekens, C.; Ribrag, V. Plitidepsin: An orphan drug. Expert Opin. Orphan Drugs 2013, 1, 569–580. [Google Scholar] [CrossRef]
- Elez, M.E.; Tabernero, J.; Geary, D.; Macarulla, T.; Kang, S.P.; Kahatt, C.; Pita, A.S.; Teruel, C.F.; Siguero, M.; Cullell-Young, M.; et al. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2205–2214. [Google Scholar] [CrossRef]
- Poveda, A.; del Campo, J.M.; Ray-Coquard, I.; Alexandre, J.; Provansal, M.; Guerra Alía, E.M.; Casado, A.; Gonzalez-Martin, A.; Fernández, C.; Rodriguez, I.; et al. Phase II randomized study of PM01183 versus topotecan in patients with platinum-resistant/refractory advanced ovarian cancer. Ann. Oncol. 2017, 28, 1280–1287. [Google Scholar] [CrossRef]
- Gaillard, S.; Ghamande, S.A.; Pardo, B.; Lorusso, D.; Vergote, I.; Papai, Z.; O’Malley, D.; Kristeleit, R.S.; Redondo, A.; Timcheva, C.; et al. CORAIL trial: Randomized phase III study of lurbinectedin (PM01183) versus pegylated liposomal doxorubicin (PLD) or topotecan (T) in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 2016, 34, TPS5597. [Google Scholar] [CrossRef]
- Farago, A.F.; Paz-Ares, L.G.; Ciuleanu, T.-E.; Fülop, A.; Navarro, A.; Bonanno, L.; Lopez-Vilariño, J.A.; Nuñez, R.; Kahatt, C.M.; Kos, G.; et al. ATLANTIS: Global, randomized phase III study of lurbinectedin (L) with doxorubicin (DOX) vs. CAV or topotecan (T) in small–cell lung cancer after platinum therapy. J. Clin. Oncol. 2018, 36, TPS8587. [Google Scholar] [CrossRef]
- Gomes, N.G.M.; Lefranc, F.; Kijjoa, A.; Kiss, R. Can some marine–derived fungal metabolites become actual anticancer agents? Mar. Drugs 2015, 13, 3950–3991. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S.T. NPI–2358 is a tubulin–depolymerizing agent: In–vitro evidence for activity as a tumor vascular–disrupting agent. Anticancer Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef]
- Blayney, D.W.; Bazhenova, L.; Lloyd, G.K.; Huang, L.; Mohanlal, R. Plinabulin, a novel small molecule that ameliorates chemotherapy-induced neutropenia, is administered on the same day of chemotherapy and has anticancer efficacy. Blood 2016, 128, 2508. [Google Scholar]
- Mita, M.M.; Spear, M.A.; Yee, L.K.; Mita, A.C.; Heath, E.I.; Papadopoulos, K.P.; Federico, K.C.; Reich, S.D.; Romero, O.; Malburg, L.; et al. Phase 1 first–in–human trial of the vascular disrupting agent plinabulin(NPI-2358) in patients with solid tumors or lymphomas. Clin. Cancer Res. 2010, 16, 5892–5899. [Google Scholar] [CrossRef] [PubMed]
- Mohanlal, R.W.; Lloyd, K.; Huang, L.; Bazhenova, L. Plinabulin as a novel small molecule clinical stage immune-oncology agent for NSCLC. J. Clin. Oncol. 2017, 35, 139. [Google Scholar]
- Mohanlal, R.W.; Lloyd, K.; Huang, L. Plinabulin, a novel small molecule clinical stage IO agent with anti-cancer activity, to prevent chemo–induced neutropenia and immune related AEs. J. Clin. Oncol. 2018, 36, 126. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Plinabulin. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=plinabulin&cntry=&state=&city=&dist=&Search=Search (accessed on 17 March 2019).
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew. Chem. Int. Ed. Engl. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Di, K.; Lloyd, G.K.; Abraham, V.; MacLaren, A.; Burrows, F.J.; Desjardins, A.; Trikha, M.; Bota, D.A. Marizomib activity as a single agent in malignant gliomas: ability to cross the blood-brain barrier. Neuro Oncol. 2016, 18, 840–848. [Google Scholar] [CrossRef]
- Harrison, S.J.; Mainwaring, P.; Price, T.; Millward, M.J.; Padrik, P.; Underhill, C.R.; Cannell, P.K.; Reich, S.D.; Trikha, M.; Spencer, A. Phase I clinical trial of marizomib (NPI-0052) in patients with advanced malignancies including multiple myeloma: study NPI-0052-102 final results. Clin. Cancer Res. 2016, 22, 4559–4566. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Diao, A. Marizomib, a potent second generation proteasome inhibitor from natural origin. Anticancer Agents Med. Chem. 2015, 15, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Kesari, S.; Piccioni, D.E.; Aregawi, D.G.; Roth, P.; Stupp, R.; Desjardins, A.; Reich, S.D.; Elias, I.; Li, M.; et al. A phase 1, multicenter, open–label study of marizomib (MRZ) with temozolomide (TMZ) and radiotherapy (RT) in newly diagnosed WHO grade IV malignant glioma (glioblastoma, ndGBM): dose–escalation results. J. Clin. Oncol. 2018, 36, e14083. [Google Scholar] [CrossRef]
- Pera, B.; Barasoain, I.; Pantazopoulou, A.; Canales, A.; Matesanz, R.; Rodriguez-Salarichs, J.; García-Fernandez, L.F.; Moneo, V.; Jiménez-Barbero, J.; Galmarini, C.M.; et al. New interfacial microtubule inhibitors of marine origin, PM050489/PM060184, with potent antitumor activity and a distinct mechanism. ACS Chem. Biol. 2013, 8, 2084–2094. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.J.; Coello, L.; Fernández, R.; Reyes, F.; Rodríguez, A.; Murcia, C.; Garranzo, M.; Mateo, C.; Sánchez–Sancho, F.; Bueno, S.; et al. Isolation and first total synthesis of PM050489 and PM060184, two new marine anticancer compounds. J. Am. Chem. Soc. 2013, 135, 10164–10171. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Martin, M.; Bouchet, B.P.; Guillen-Navarro, M.J.; Martínez-Diez, M.; Martinez-Leal, J.F.; Akhmanova, A.; Aviles, P. Plocabulin, a novel tubulin-binding agent, inhibits angiogenesis by modulation of microtubule dynamics in endothelial cells. BMC Cancer 2018, 18, 164. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Current status of marine-derived compounds as warheads in anti-tumor drug candidates. Mar. Drugs 2017, 15, 99. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Flinn, I.; Advani, R.H.; Diefenbach, C.S.; Kolibaba, K.; Press, O.W.; Sehn, L.H.; Chen, A.I.; Salles, G.; Tilly, H.; et al. Updated results of a phase II randomized study (ROMULUS) of polatuzumab vedotin or pinatuzumab vedotin plus rituximab in patients with relapsed/refractory non-Hodgkin lymphoma. Blood 2014, 124, 4457. [Google Scholar]
- ClinicalTrials.gov. Polatuzumab vedotin. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=polatuzumab+vedotin&cntry=&state=&city=&dist=&Search=Search (accessed on 29 March 2019).
- European Union Clinical Trials Register. Polatuzumab Vedotin. Available online: https://www.clinicaltrialsregister.eu/ctr–search/search?query=polatuzumab+vedotin (accessed on 29 March 2019).
- European Medicines Agency. Polatuzumab vedotin. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Annex_to_CHMP_highlights/2017/06/WC500230050.pdf (accessed on 29 March 2019).
- Petrylak, D.P.; Perez, R.P.; Zhang, J.; Smith, D.C.; Ruether, J.D.; Sridhar, S.S.; Sangha, R.S.; Lang, J.M.; Heath, E.I.; Merchan, J.R.; et al. A phase I study of enfortumab vedotin (ASG-22CE.; ASG-22ME): Updated analysis of patients with metastatic urothelial cancer. J. Clin. Oncol. 2017, 35, 106. [Google Scholar] [CrossRef]
- De Goeij, B.E.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Yardley, D.; Melisko, M.; Forero, A.; Daniel, B.R.; Montero, A.J.; Guthrie, T.H.; Canfield, V.A.; Oakman, C.A.; Chew, H.K.; Ferrario, C.; et al. METRIC: A randomized international study of the antibody-drug conjugate glembatumumab vedotin (GV or CDX-011) in patients (pts) with metastatic gpNMB-overexpressing triple-negative breast cancer (TNBC). J. Clin. Oncol. 2015, 33, TPS1110. [Google Scholar] [CrossRef]
- Rose, A.A.N.; Biondini, M.; Curiel, R.; Siegel, P.M. Targeting GPNMB with glembatumumab vedotin: Current developments and future opportunities for the treatment of cancer. Pharmacol. Ther. 2017, 179, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Mitten, M.J.; Norvell, S.; Falls, H.D.; DeVries, P.J.; Cheng, D.; Meulbroek, J.A.; Buchanan, F.G.; et al. ABT-414, an antibody–drug conjugate targeting a tumor-selective EGFR epitope. Mol. Cancer Ther. 2016, 15, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Motzer, R.; Molina, A.M.; Choueiri, T.K.; Heath, E.I.; Kollmannsberger, C.K.; Redman, B.G.; Sangha, R.S.; Ernst, D.S.; Pili, R.; et al. Phase I studies of anti-ENPP3 antibody drug conjugates (ADCs) in advanced refractory renal cell carcinomas (RRCC). J. Clin. Oncol. 2015, 33, 2503. [Google Scholar] [CrossRef]
- Tai, Y.T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef] [Green Version]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Van Den Bent, M.J.; French, P.; Eoli, M.; Sepúlveda, J.M.; Walenkamp, A.M.E.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Weller, M.; de Heer, I.; et al. Updated results of the INTELLANCE 2/EORTC trial 1410 randomized phase II study on Depatux-M alone, Depatux-M in combination with temozolomide (TMZ) and either TMZ or lomustine (LOM) in recurrent EGFR amplified glioblastoma (NCT02343406). J. Clin. Oncol. 2018, 36, 2023. [Google Scholar] [CrossRef]
- AbbVie. FDA rare pedriatic disease designation for investigational ABT–414 for the treatment of a type of pediatric brain tumor known as Diffuse Intrinsic Pontine Glioma (DIPG). Available online: https://news.abbvie.com/news/abbvie–receives–us–fda–rare–pediatric–disease–designation–for–investigational–abt–414–for–treatment–type–pediatric–brain–tumor–known–as–diffuse–intrinsic–pontine–glioma–dipg.htm (accessed on 29 March 2019).
- ClinicalTrials.gov. Depatuxizumab mafodotin. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=depatuxizumab+mafodotin&cntry=&state=&city=&dist=&Search=Search (accessed on 29 March 2019).
- European Union Clinical Trials Register. Depatuxizumab Mafodotin. Available online: https://www.clinicaltrialsregister.eu/ctr–search/search?query=depatuxizumab+mafodotin (accessed on 29 March 2019).
- Maderna, A.; Doroski, M.; Subramanyam, C.; Porte, A.; Leverett, C.A.; Vetelino, B.C.; Chen, Z.; Risley, H.; Parris, K.; Pandit, J.; et al. Discovery of cytotoxic dolastatin 10 analogues with N-terminal modifications. J. Med. Chem. 2014, 57, 10527–10543. [Google Scholar] [CrossRef]
- Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K.W. DNA sequence- and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compounds from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry 1996, 35, 13303–13309. [Google Scholar] [CrossRef]
- Zewail-Foote, M.; Hurley, L.H. Ecteinascidin 743: A minor groove alkylator that bends DNA toward the major groove. J. Med. Chem. 1999, 42, 2493–2497. [Google Scholar] [CrossRef]
- Erba, E.; Bergamaschi, D.; Bassano, L.; Damia, G.; Ronzoni, S.; Faircloth, G.T.; D’Incalci, M. Ecteinascidin–743 (ET–743), a natural marine compound, with a unique mechanism of action. Eur. J. Cancer 2001, 37, 97–105. [Google Scholar] [CrossRef]
- Herrero, A.B.; Martin-Castellanos, C.; Marco, E.; Gago, F.; Moreno, S. Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res. 2006, 66, 8155–8162. [Google Scholar] [CrossRef]
- Marco, E.; David-Cordonnier, M.H.; Bailly, C.; Cuevas, C.; Gago, F. Further insight into the DNA recognition mechanism of trabectedin from the differential affinity of its demethylated analogue ecteinascidin ET729 for the triplet DNA binding site CGA. J. Med. Chem. 2006, 49, 6925–6929. [Google Scholar] [CrossRef]
- Le Cesne, A. 13 Years of trabectedin, 5 years of Yondelis®: What have we learnt? Expert Rev. Anticancer Ther. 2013, 13, 11–19. [Google Scholar] [CrossRef]
- Di Giandomenico, S.; Frapolli, R.; Bello, E.; Uboldi, S.; Licandro, S.A.; Marchini, S.; Beltrame, L.; Brich, S.; Mauro, V.; Tamborini, E.; et al. Mode of action of trabectedin in myxoid liposarcomas. Oncogene 2014, 33, 5201–5210. [Google Scholar] [CrossRef]
- Grosso, F.; Jones, R.L.; Demetri, G.D. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: A retrospective study. Lancet Oncol. 2007, 8, 595–602. [Google Scholar] [CrossRef]
- Damia, G.; D’Incalci, M. Targeting DNA repair as a promising approach in cancer therapy. Eur. J. Cancer 2007, 43, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.G.; Battistella, A.; Rocca, C.J.; Matuo, R.; Henriques, J.A.; Larsen, A.K.; Escargueil, A.E. Ataxia telangiectasia mutated- and Rad3-related kinase drives both the early and the late DNA-damage response to the monofunctional antitumour alkylator S23906. Biochem. J. 2011, 437, 63–73. [Google Scholar] [CrossRef]
- Tavecchio, M.; Simone, M.; Erba, E.; Chiolo, I.; Liberi, G.; Foiani, M.; D’Incalci, M.; Damia, G. Role of homologous recombination in trabectedin-induced DNA damage. Eur. J. Cancer 2008, 44, 609–618. [Google Scholar] [CrossRef]
- Damia, G.; Silvestri, S.; Carrassa, L.; Filiberti, L.; Faircloth, G.T.; Liberi, G.; Foiani, M.; D’Incalci, M. Unique pattern of ET-743 activity in different cellular systems with defined deficiencies in DNA-repair pathways. Int. J. Cancer 2001, 92, 583–588. [Google Scholar] [CrossRef]
- Schöffski, P.; Taron, M.; Jimeno, J.; Grosso, F.; Sanfilipio, R.; Casali, P.G.; Le Cesne, A.; Jones, R.L.; Blay, J.Y.; Poveda, A.; et al. Predictive impact of DNA repair functionality on clinical outcome of advanced sarcoma patients treated with trabectedin: A retrospective multicentric study. Eur. J. Cancer 2011, 47, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Frapolli, R.; Zangarini, M.; Bello, E.; Porcu, L.; Galmarini, C.M.; García-Fernández, L.F.; Cuevas, C.; Allavena, P.; Erba, E.; et al. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Int. J. Cancer 2013, 133, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, A.; Takebayashi, Y.; Ren, X.Q.; Miyashita, H.; Mori, S.; Akiyama, S. Overcoming multidrug resistance in P-glycoprotein/MDR1-overexpressing cell lines by ecteinascidin 743. Mol. Cancer Ther. 2002, 1, 1327–1334. [Google Scholar] [PubMed]
- Galmarini, C.M.; D’Incalci, M.; Allavena, P. Trabectedin and plitidepsin: Drugs from the sea that strike the tumor microenvironment. Mar. Drugs 2014, 12, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Germano, G.; Belgiovine, C.; D’Incalci, M.; Mantovani, A. Trabectedin: A drug from the sea that strikes tumor-associated macrophages. Oncoimmunology 2013, 2, e24614. [Google Scholar] [CrossRef] [PubMed]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumor-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef]
- Dabydeen, D.A.; Burnett, J.C.; Bai, R.; Verdier-Pinard, P.; Hickford, S.J.; Pettit, G.R.; Blunt, J.W.; Munro, M.H.; Gussio, R.; Hamel, E. Comparison of the activities of the truncated halichondrin B analog NSC 707389 (E7389) with those of the parent compound and a proposed binding site on tubulin. Mol. Pharmacol. 2006, 70, 1866–1875. [Google Scholar] [CrossRef]
- Smith, J.A.; Wilson, L.; Azarenko, O.; Zhu, X.; Lewis, B.M.; Littlefield, B.A.; Jordan, M.A. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry 2010, 49, 1331–1337. [Google Scholar] [CrossRef]
- Kuznetsov, G.; Towle, M.J.; Cheng, H.; Kawamura, T.; TenDyke, K.; Liu, D.; Kishi, Y.; Yu, M.J.; Littlefield, B.A. Induction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin B macrocyclic ketone analog E7389. Cancer Res. 2004, 64, 5760–5766. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ozawa, Y.; Kimura, T.; Sato, Y.; Kuznetsov, G.; Xu, S.; Uesugi, M.; Agoulnik, S.; Taylor, N.; Funahashi, Y.; et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial–mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Agoulnik, S.I.; Kawano, S.; Taylor, N.; Oestreicher, J.; Matsui, J.; Chow, J.; Oda, Y.; Funahashi, Y. Eribulin mesylate exerts specific gene expression changes in pericytes and shortens pericyte-driven capillary network in vitro. Vasc. Cell 2014, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Díez, M.; Guillén-Navarro, M.J.; Pera, B.; Bouchet, B.P.; Martínez-Leal, J.F.; Barasoain, I.; Cuevas, C.; Andreu, J.M.; García-Fernández, L.F.; Díaz, J.F.; et al. PM060184, a new tubulin binding agent with potent antitumor activity including P-glycoprotein over-expressing tumors. Biochem. Pharmacol. 2014, 88, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.H.; Steinmetz, M.O. A new tubulin–binding site and pharmacophore for microtubule–destabilizing anticancer drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Huber, R.; Potts, B.C. Crystal structures of salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. [Google Scholar] [CrossRef]
- Macherla, V.R.; Mitchell, S.S.; Manam, R.R.; Reed, K.A.; Chao, T.H.; Nicholson, B.; Deyanat-Yazdi, G.; Mai, B.; Jensen, P.R.; Fenical, W.F.; et al. Structure-activity relationship studies of salinosporamide A (NPI-0052), a novel marine derived proteasome inhibitor. J. Med. Chem. 2005, 48, 3684–3687. [Google Scholar] [CrossRef]
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720. [Google Scholar] [CrossRef]
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef]
- Niewerth, C.; Franke, N.E.; Janesen, G.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Degenhardt, J.; Anderl, J.; Schimmer, A.D.; Zweegman, S.; et al. Higher ratio immune vs. constitutive proteasome level as novel indicator of sensitivity of pediatric acute leukemia cells to proteasome inhibitors. Haematologica 2013, 98, 1896–1904. [Google Scholar] [CrossRef] [PubMed]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C., Jr.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Lebovic, D.; Chen, A. Phase I study of the anti-CD22 antibody-drug conjugate pinatuzumab vedotin with/without rituximab in patients with relapsed/refractory B-cell non-Hodgkin’s lymphoma. Clin. Cancer Res. 2017, 23, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Breij, E.C.; de Goeij, B.E.; Verploegen, S. An antibody-drug conjugate that targets tissue factor exhibits potent therapeutic activity against a broad range of solid tumors. Cancer Res. 2014, 74, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- de Goeij, B.E.; Satijn, D.; Freitag, C.M. High turnover of tissue factor enables efficient intracellular delivery of antibody-drug conjugates. Mol. Cancer Ther. 2015, 14, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Sussman, D.; Smith, L.M.; Anderson, M.E.; Duniho, S.; Hunter, J.H.; Kostner, H.; Miyamoto, J.B.; Nesterova, A.; Westendorf, L.; Van Epps, H.A.; et al. SGN-LIV1A: A novel antibody-drug conjugate targeting LIV-1 for the treatment of metastatic breast cancer. Mol. Cancer Ther. 2014, 13, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.S.; Guevara, C.I.; Jin, L.; Mbong, N.; Verlinsky, A.; Hsu, S.J.; Aviña, H.; Karki, S.; Abad, J.D.; Yang, P.; et al. AGS67E, an anti-CD37 monomethyl auristatin E antibody-drug conjugate as a potential therapeutic for B/T-cell malignancies and AML: A new role for CD37 in AML. Mol. Cancer Ther. 2015, 14, 1650–1660. [Google Scholar] [CrossRef]
- Morrison, K.; Challita-Eid, P.M.; Raitano, A.; An, Z.; Yang, P.; Abad, J.D.; Liu, W.; Lortie, D.R.; Snyder, J.T.; Capo, L.; et al. Development of ASG-15ME, a novel antibody-drug conjugate targeting SLITRK6, a new urothelial cancer biomarker. Mol. Cancer Ther. 2016, 15, 1301–1310. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R.; Palladino, M.A.; Lam, K.S.; Lloyd, G.K.; Potts, B.C. Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg. Med. Chem. 2009, 17, 2175–2180. [Google Scholar] [CrossRef] [Green Version]
- Corey, E.J.; Gin, D.Y.; Kania, R.S. Enantioselective total synthesis of ecteinascidin 743. J. Am. Chem. Soc. 1996, 118, 9202–9203. [Google Scholar] [CrossRef]
- Cuevas, C.C.; Perez, M.; Martin, M.J.; Chicharro, J.L.; Fernández–Rivas, C.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; de La Calle, F.; et al. Synthesis of ecteinascidin ET–743 and phthalascidin Pt-650 from cyanosafracin B. Org. Lett. 2000, 2, 2545–2548. [Google Scholar] [CrossRef] [PubMed]


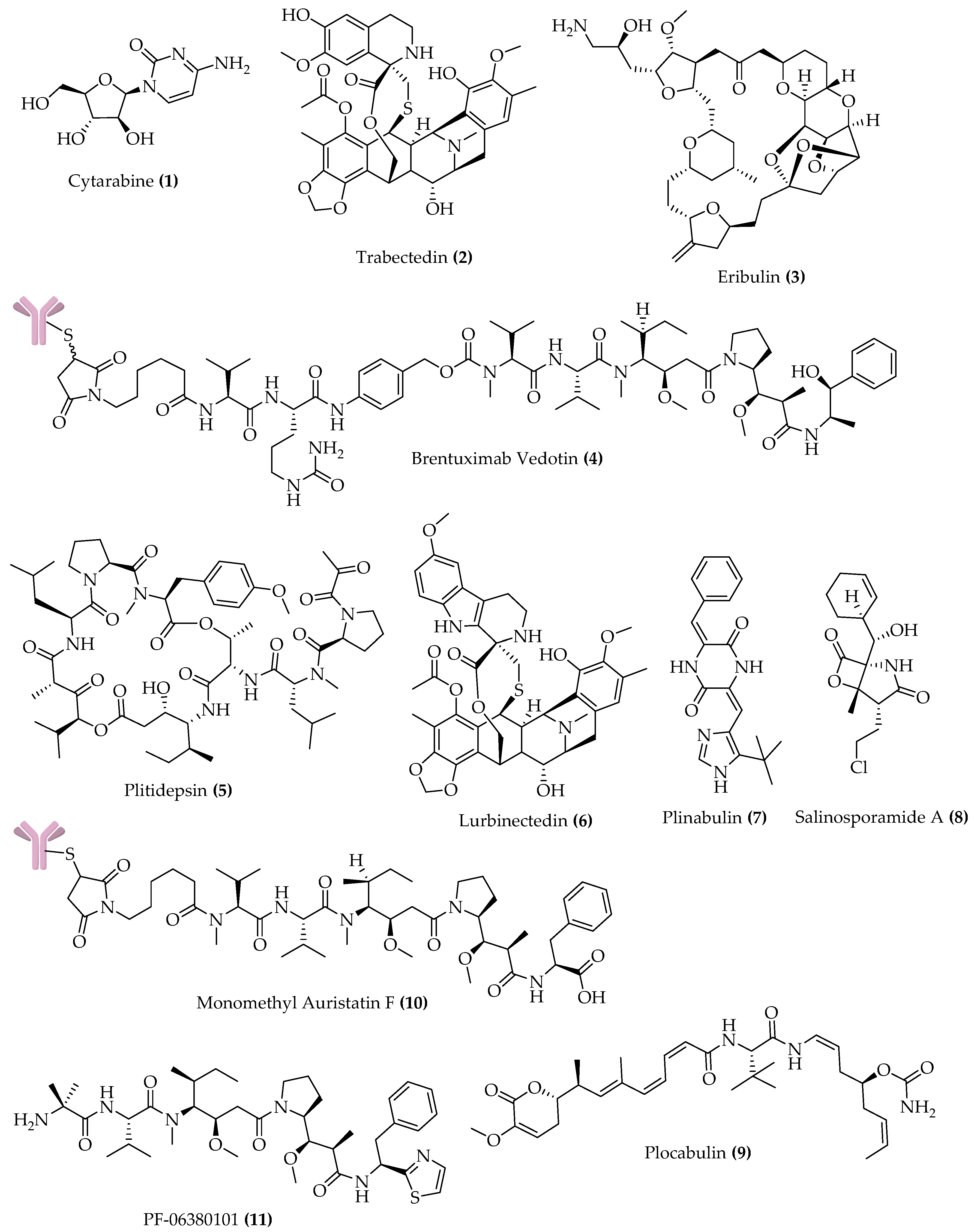{kind=link}
| Compound Name (Trademark) | Lead Compound (Source) | Chemical Class | Molecular Target | Cancer Conditions |
|---|---|---|---|---|
| APPROVED | ||||
| Cytarabine (Cytosar-U®; DepoCyt®) | Spongothymidine (Sponge) | Nucleoside | DNA polymerase | Leukemia; lymphomatous meningitis |
| Trabectedin (Yondelis®) | Trabectedin (Tunicate) | Alkaloid | DNA minor groove | Soft tissue sarcoma; ovarian cancer |
| Eribulin mesylate (Halaven®) | Halichondrin B (Sponge) | Macrolide | Microtubules | Metastatic breast cancer; advanced liposarcoma |
| Brentuximab vedotin (Adcetris®) | Dolastatin 10 (Mollusk/cyanobacterium) | ADC a (MMAE) b | CD30 and microtubules | sALCL c; Hodgkin lymphoma |
| PHASE 3 | ||||
| Plitidepsin (Aplidin®) | Plitidepsin (Tunicate) | Depsipeptide | Rac1 and JNK activation | Relapsed/refractory multiple myeloma |
| Lurbinectedin (Zepsyre®) | Trabectedin | Alkaloid | DNA minor groove | Ovarian cancer; SCLC d |
| Plinabulin | Halimide (Fungus) | Diketopiperazine | Microtubules | NSCLC e; CIN f |
| Salinosporamide A Marizomib | Salinosporamide A (Bacterium) | γ-lactam-β-lactone | 20S proteasome | Newly diagnosed glioblastoma |
| Polatuzumab vedotin DCDS-4501A | Dolastatin 10 | ADC (MMAE) | CD79b and microtubules | DLBCL g |
| Depatuxizumab vedotin ABT-414 | Dolastatin 10 | ADC (MMAF h) | EGFR and microtubules | Newly diagnosed glioblastoma |
| PHASE 2 | ||||
| PM060184 Plocabulin | PM060184 (Sponge) | Polyketide | Microtubules | Advanced colorectal cancer |
| Enfortumab vedotin ASG-22ME | Dolastatin 10 | ADC (MMAE) | Nectin-4 and microtubules | Carcinoma, transitional cell; urinary bladder, urologic, renal pelvis, ureteral and urethral neoplasms; urothelial cancer; |
| Glembatumumab vedotin CDX-011 | Dolastatin 10 | ADC (MMAE) | gpNMB and microtubules | Metastatic gpNMB over-expressing triple negative breast cancer; recurrent osteosarcoma; recurrent uveal melanoma; stage IV uveal melanoma AJCC v7; melanoma; squamous cell carcinoma of the lung |
| AGS-16C3F | Dolastatin 10 | ADC (MMAF) | ENPP3 and microtubules | Metastatic renal cell carcinoma |
| GSK2857916 | Dolastatin 10 | ADC (MMAF) | BCMA | Multiple myeloma |
| Tisotumab vedotin (HuMax®-TF-ADC) | Dolastatin 10 | ADC (MMAE) | Tissue factor and microtubules | NSCLC; Ovary, cervical, endometrium, bladder, prostate and esophagus cancer; squamous cell carcinoma of the head and neck |
| Ladiratuzumab vedotin SGN-LIV1A | Dolastatin 10 | ADC (MMAE) | LIV-1 and microtubules | Breast cancer |
| Telisotuzumab vedotin ABBV-399 | Dolastatin 10 | ADC (MMAE) | c-Met | Recurrent and stage IV squamous cell lung carcinoma; NSCLC |
| PHASE 1 | ||||
| ABBV-085 | Dolastatin 10 | ADC (MMAE) | LRRC15 | Advanced solid tumors; undifferentiated pleomorphic sarcoma; squamous cell carcinoma of the head and neck; breast carcinoma |
| AGS-67E | Dolastatin 10 | ADC (MMAE) | CD37 and microtubules | Refractory/relapsed lymphoid malignancy |
| ASG-15ME | Dolastatin 10 | ADC (MMAE) | SLITRK6 and microtubules | Metastatic urothelial cancer |
| PF-06647020 | Dolastatin 10 | ADC (PF-06380101) | PTK7 and microtubules | Advanced solid tumors; triple negative and metastatic breast cancer |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, R.B.; Evdokimov, N.M.; Lefranc, F.; Valentão, P.; Kornienko, A.; Pereira, D.M.; Andrade, P.B.; Gomes, N.G.M. Marine-Derived Anticancer Agents: Clinical Benefits, Innovative Mechanisms, and New Targets. Mar. Drugs 2019, 17, 329. https://doi.org/10.3390/md17060329
Pereira RB, Evdokimov NM, Lefranc F, Valentão P, Kornienko A, Pereira DM, Andrade PB, Gomes NGM. Marine-Derived Anticancer Agents: Clinical Benefits, Innovative Mechanisms, and New Targets. Marine Drugs. 2019; 17(6):329. https://doi.org/10.3390/md17060329
Chicago/Turabian StylePereira, Renato B., Nikolai M. Evdokimov, Florence Lefranc, Patrícia Valentão, Alexander Kornienko, David M. Pereira, Paula B. Andrade, and Nelson G. M. Gomes. 2019. "Marine-Derived Anticancer Agents: Clinical Benefits, Innovative Mechanisms, and New Targets" Marine Drugs 17, no. 6: 329. https://doi.org/10.3390/md17060329
APA StylePereira, R. B., Evdokimov, N. M., Lefranc, F., Valentão, P., Kornienko, A., Pereira, D. M., Andrade, P. B., & Gomes, N. G. M. (2019). Marine-Derived Anticancer Agents: Clinical Benefits, Innovative Mechanisms, and New Targets. Marine Drugs, 17(6), 329. https://doi.org/10.3390/md17060329