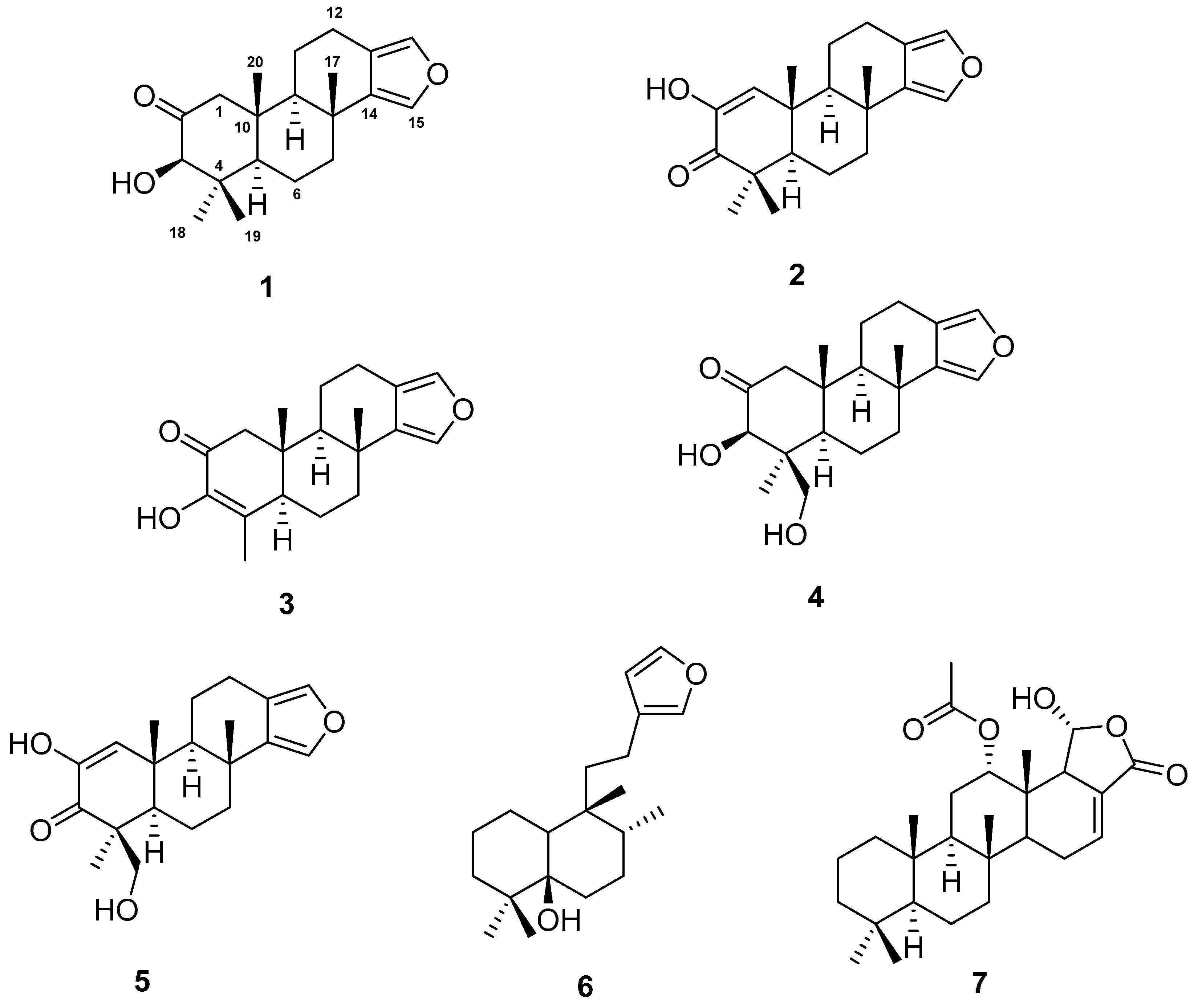
1. Introduction
Specimens belonging to the genus
Spongia have been subjected to numerous chemical investigations yielding a wide variety of C21 and other linear furanoterpenes, spongian diterpenes, scalarane sesterterpenoids, sesquiterpene quinones, sterols (including secosterols), and macrolides [
1], many of which have shown biological activities including antibacterial [
2,
3], antiviral [
4], antitumoral [
5,
6], and anti-inflammatory functions [
7].
In our continuing investigations of diterpenes from marine organisms [
8,
9], and in particular from marine sponges [
10], we have focused our attention on the sponge
Spongia tubulifera, collected in the Mexican Caribbean, because of the cytotoxic activity found in its methanolic extract. To the best of our knowledge, the only previous reports of
S. tubulifera were a comparative study of the fatty acids composition of specimens of this sponge collected at Ahogado Reef near La Parguera, Puerto Rico [
11] and the assays of the antimicrobial activity against
Staphylococcus aureus and
Candida albicans of the organic extracts of specimens collected at Urabá Gulf reefs in the Colombian Caribbean [
12].
In this paper, we elucidate the structures of two new spongian furanoditerpenes, 3β-hydroxyspongia-13(16),14-dien-2-one (1) and 19-dehydroxyspongian diterpene 17 (2), along with five known terpenes, 3–7, and we evaluate their cytotoxic activity against a panel of five human tumour cell lines.
2. Results and Discussion
Specimens of the sponge
S. tubulifera, collected by hand and scuba diving off the coast of the Mexican Caribbean, were extracted several times with CH
3OH/CH
2Cl
2 to give an extract which showed cytotoxic activity. The organic extract was subsequently partitioned between H
2O/CH
2Cl
2, and the CH
2Cl
2 portion was further fractionated into hexane, CH
2Cl
2, and aqueous methanolic fractions. The hexane fraction was submitted to silica gel flash chromatography using a gradient mixture of hexane and EtOAc to yield enriched terpene fractions that were then submitted repeatedly to reversed-phase HPLC separation (H
2O/CH
3OH mixtures) to yield
1–
3 and
6. The CH
2Cl
2 fraction was fractionated by solid phase extraction (SPE) with a RP-18 column using a stepped gradient from H
2O, CH
3OH, and CH
2Cl
2 to yield enriched terpene fractions that were separated by RP-HPLC using H
2O/CH
3OH mixtures to afford
4,
5, and
7 (
Figure 1).
Compound 1 was obtained as a colorless white powder. The molecular formula of 1 was determined on the basis of the M+. peak at m/z 316.2014, observed in its HREIM spectrum (calculated for C20H28O3, 316.2038, 7 degrees of unsaturation) and from its 13C NMR spectrum. Its IR spectrum displayed signals at 3500 and 1745 cm−1, suggesting the presence of a hydroxyl group and a ketone carbonyl functionality, respectively.
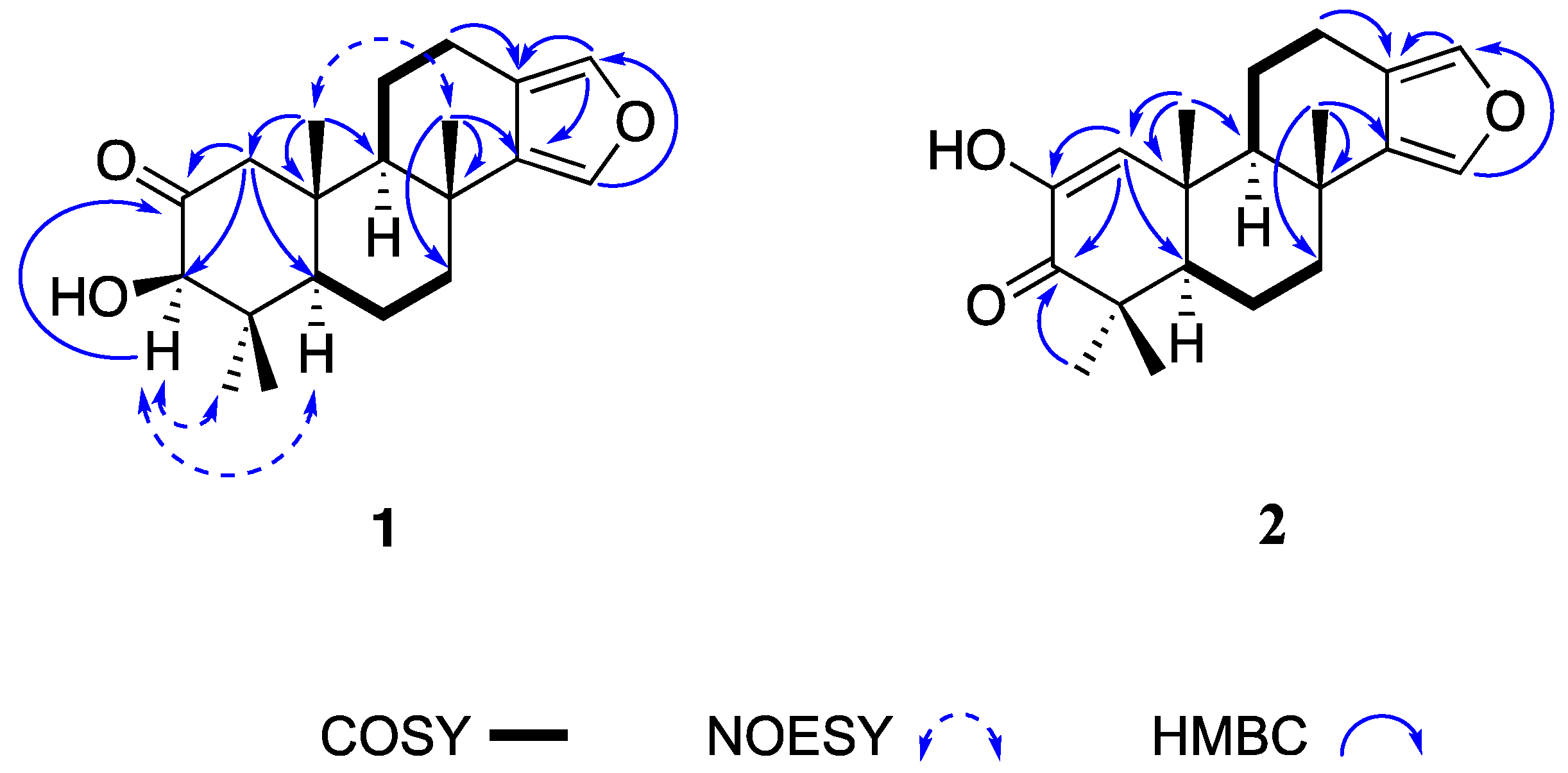
The
13C NMR spectrum of
1 shows 20 signals (
Table 1,
Supplementary Material, Table S1) that, in combination with the
1H NMR and HSQC spectra, indicated the presence of a spongian furanditerpene bearing four tertiary methyl groups (δ
H/δ
C 1.23, s/26.0; 1.21, s/29.4; 0.73, s/16.5; and 0.88, s/17.3), a 3,4-disubstituted furan ring (δ
H/δ
C 7.12, s/135.3; 7.07, s/137.1; 119.4; and 136.8), one ketone carbonyl group (δ
C 211.1), a hydroxyl group at δ
H 3.48, and an oxymethine sp
3 carbon (δ
H/δ
C 3.90, d/ 83.1). Comparison of the NMR data of
1 with those of reported for other spongian furanoditerpenes, along with the HMBC correlations shown in
Figure 2, indicated that
1 has a similar structure to 3α-hydroxyspongia-13(16),14-dien-2-one isolated from an unidentified
Spongia collected in Australia [
13]. The differences of the proton and carbon chemical shifts at C-3, e.g., δ
H/δ
C 3.90 (d,
J = 1.5 Hz)/ 83.1 in
1 instead of δ
H/δ
C 4.36 (d,
J = 1.5 Hz)/ 80.1 in 3α-hydroxyspongia-13(16),14-dien-2-one, suggested that they differed only in the stereochemistry at C-3, and thus,
1 must be its 3β isomer. The NOESY correlations from H-3 at δ
H 3.90 to H-5 at δ
H 1.62 and H-18 at δ
H 1.21 indicated that these protons were in the same face of the molecule, confirming the β-orientation of the hydroxyl group at C-3. The relative configuration of the remaining stereogenic centers in
1 was also confirmed by its NOESY correlations (
Figure 2). These data indicated that
1 is a new spongian furanoditerpene derivative with a 3β-hydroxyspongia-13(16),14-dien-2-one structure.
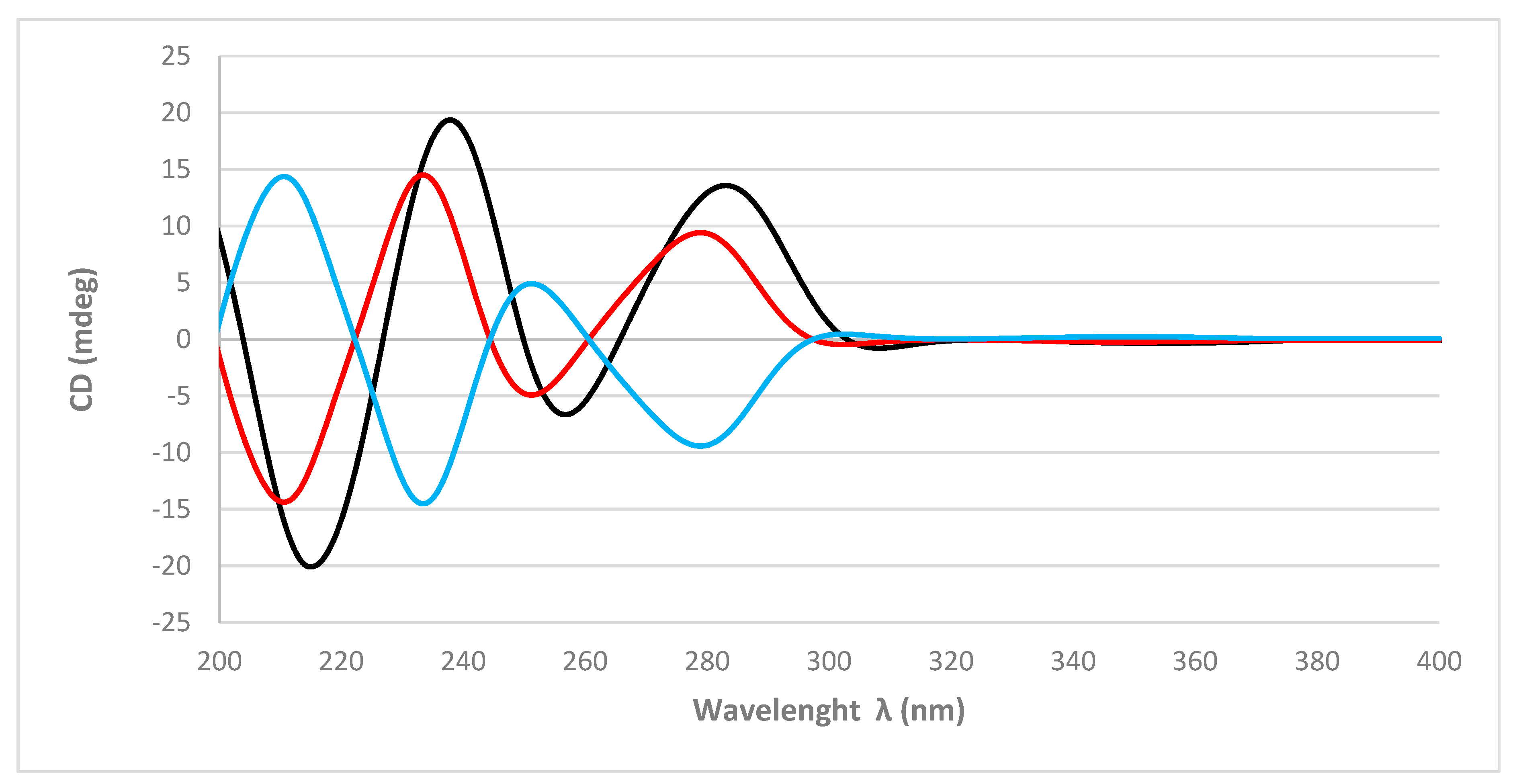
The absolute configurations of the stereogenic carbons of
1 were determined by comparison of the experimental and simulated electronic circular dichroism (ECD) spectra generated by time-dependent density functional theory (TDDFT) calculations. Overall, the two possible enantiomers for
1, (3
R,5
R,8
R,9
R,10
R)-
1 and (3
S,5
S,8
S,9
S,10
S)-
1, were initially submitted to a conformational search with the Maestro Suite (Schrödinger). Four conformers were found within a 10.0 kcal/mol energy threshold from global minimum. All these conformers were geometrically optimized by a density functional theory (DFT) method at the HSEH1PBE/cc-pVDZ functions (see computational details in the experimental section). The resulting ECD spectra were combined by Boltzmann weighting to give a composite spectrum for each enantiomer. Comparison of the experimental and calculated ECD spectra for
1 showed excellent agreement with the (3
R,5
R,8
R,9
R,10
R)-
1 enantiomer (
Figure 3). Thus, the absolute configurations of C-3, C-5, C-8, C-9, and C-10 were determined as 3
R, 5
R, 8
R, 9
R, and 10
R, respectively.
The molecular formula of
2, isolated as a colorless white powder, was established as C
20H
26O
3 based on the [M + Na]
+ at
m/
z 337.1803 in its (+)-HRESIM spectrum (calculated for C
20H
26O
3Na, 337.1780, 8 degrees of unsaturation) and on NMR data (
Table 1,
Supplementary Material, Table S2). The IR spectrum of
2 shows absorptions from the hydroxyl (3505 cm
−1) and a conjugated ketone carbonyl (1650 cm
−1) groups.
The 20 carbon signals observed in the
13C NMR spectrum of
2 along with the presence of two α-furan proton signals (δ
H 7.09 and 7.06) and four tertiary methyl groups (δ
H 1.28, 1.23, 1.22, and 1.16) in its
1H NMR spectrum were indicative of a spongian furanoditerpene structure. The planar structure of
2 was established by a combination of 1D and 2D NMR spectroscopy. Comparison of the NMR data of
2 with those of
1 (see
Table 1) revealed that they shared the same framework at the B, C, and D rings but differed in the A-ring. Signals in the
13C NMR spectrum of
2 for the conjugated ketone carbonyl group at δ
C 201.2 (C-3) and two
sp2 carbons, the non-protonated carbon at δ
C 144.3 (C-2) and the methine carbon at δ
C 128.3 (C-1), were consistent with the presence of a conjugated α,β-unsaturated ketone moiety.
The key
1H-
13C long range correlations between the olefinic proton at δ
H 6.54 (H-1) and the olefinic carbon at δ
C 144.3 (C-2), the ketone carbonyl carbon at δ
C 201.2 (C-3) and the carbon at δ
C 54.5 (C-5), along with the HMBC correlation from the methyl singlet at δ
H 1.22 (H-20) to the olefinic carbon at δ
C 128.3 (C-1) placed the α,β-unsaturated ketone in the A-ring (
Figure 2). The exchangeable proton signal at δ
H 5.93 was indicative of an enolized α-diketone moiety in the A-ring. The NMR data for this part of the molecule (see
Table 1) are in agreement with those observed for other diterpenes containing the same A-ring in the tetracyclic framework such as spongian diterpene 17 (
5), previously reported from the nudibranch
Doriprismatica (=
Glossodoris)
atromarginata [
14]. The diagnostic HMBC correlations displayed by the α-furan proton signals and the methyl groups Me-17 and Me-18 displayed in
Figure 2 confirm
2 as a new spongian furanoditerpene that was named 19-dehydroxy-spongian diterpene 17.
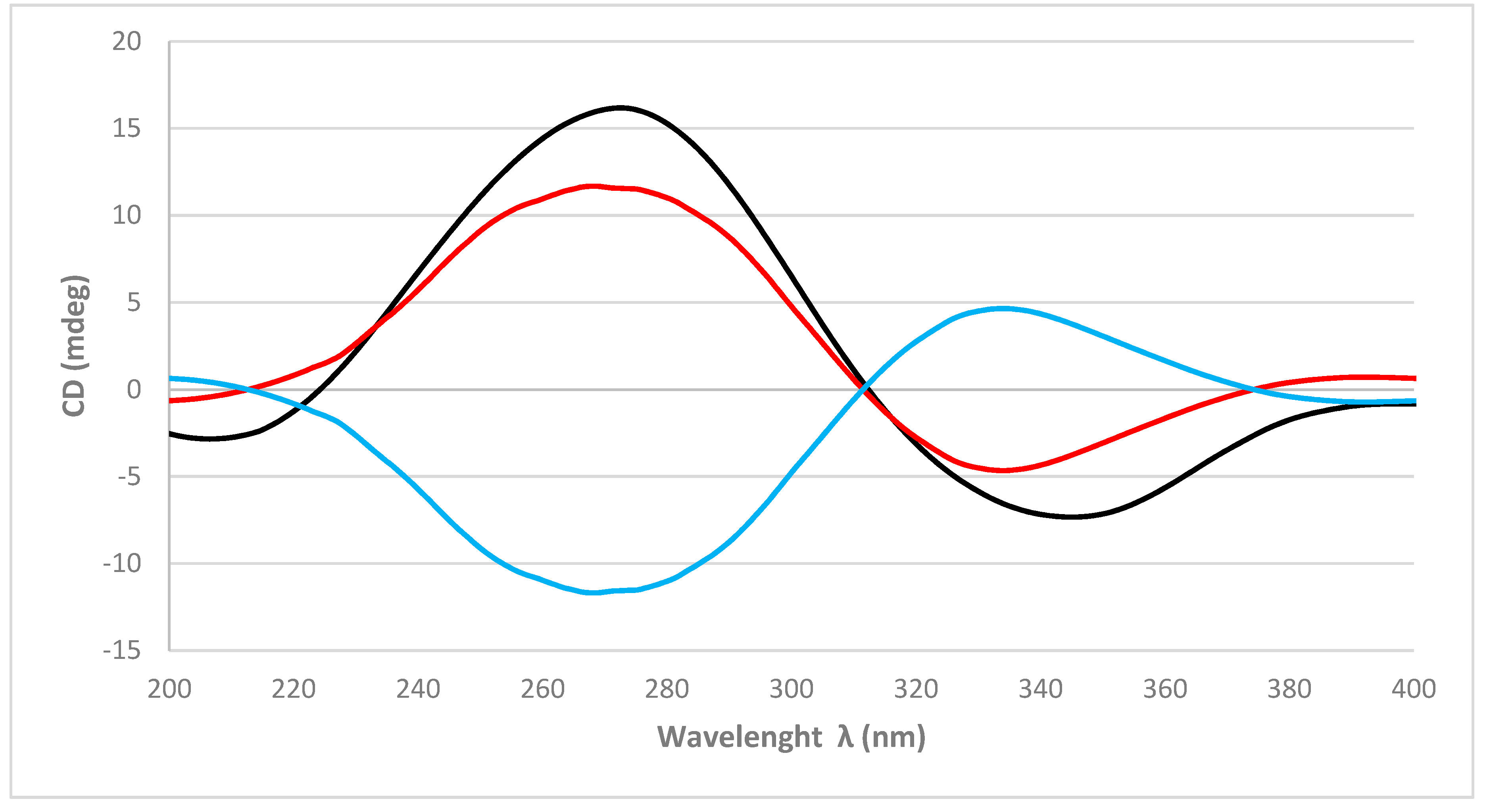
As in
1, the absolute configurations of the stereogenic carbons of
2 were determined by comparison of the experimental to those generated by TDDFT on the two possible enantiomers. The two possible enantiomers for
2, (5
R,8
R,9
R,10
R)-
2 and (5
S,8
S,9
S,10
S)-
2, were initially submitted to a conformation search with the Maestro Suite (Schrödinger). Thus, 4 conformers were found within a 10.0 Kcal/mol energy threshold from a global minimum. All these conformers were geometrically optimized by density functional theory method at the HSEH1PBE/cc-pVDZ function (see computational details in experimental section). As shown in
Figure 4, the calculated ECD spectra for the (5
R,8
R,9
R,10
R)-
2 and its experimental data were almost identical. Thus, the absolute configurations of C-5, C-8, C-9, and C-10 of
2 were determined as 5
R, 8
R, 9
R, and 10
R, respectively.
Spectral data (
1H and
13C NMR, MS,
) of
3 and
4 were identical with those reported for 19-
nor-3-hydroxyspongia-3,13(16),14-trien-2-one (epispongiadiol) [
15] and 3β-19-dihydroxyspongia-13(16),14-dien-2-one [
16], respectively, isolated from an unidentified
Spongia; while the NMR/spectroscopic data for
5 matched with those reported for spongian diterpene 17, isolated from the nudibranch
Doriprismatica (=
Glossodoris)
atromarginata [
14]; the NMR/spectroscopic data for
6 were identical with those reported for ambliol C, isolated from the sponge
Dysidea amblia [
17], and the NMR/spectroscopic data for
7 matched with those reported for scalarin from the sponge
Cacospongia scalaris by Fattorusso et al. [
18] and later on from
Spongia nitens by Cimino et al. [
19].
The isolated compounds were submitted to biological activity assays. MTT ((3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)) assays were performed on human lung carcinoma A549 ATCC® CCL-185TM, human skin melanoma A2058 ATCC® CRL-11147TM, hepatocyte carcinoma HepG2 ATCC® HB-8065TM, breast adenocarcinoma MCF7 ATCC® HTB-22TM, and pancreas carcinoma MiaPaca-2 ATCC® CRL-1420TM with doxorubicin as a positive control [
20]. Compounds
1 and
4 showed a weak cytotoxic activity, while
6 exhibited the highest cytotoxic activity with IC
50 values from 28.3 to 11.7 µM (
Table 2). Previous biological studies of
4 indicated cytotoxic activity against the human tumor cell lines A549 (human lung carcinoma cells), HT-29 (human colorectal carcinoma cells), and P388 (leukemia cells lines) [
21] and antiviral activity against VSV (vesicular stomalitis virus) and HSV-1 (herpes simplex virus type 1) [
4]. On the other hand, it was reported that
6 induced
Artemia sp. to death in a test of settlement and metamorphosis inhibition of larvae or juveniles [
22]. Additionally,
1–
7 did not show any significant antibacterial activity against
Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella pneumoniae, Staphylococcus aureus, or antiviral activity against human adenoviruses (HAdV5 and HAdV5-GFP).
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured on a JASCO DIP-1000 polarimeter, with a Na (589 nm) lamp and filter. IR spectra were measured on a FTIR Bruker Vector 22 spectrometer. 1H, 13C, and 2D NMR spectra were recorded on a Bruker Avance 500 spectrometer at 500 and 125 MHz, respectively, using CDCl3. Low resolution electrospray mass spectrometry (LRESIMS) and high resolution electrospray mass spectrometry (HRESIMS) experiments were performed on the Applied Biosystems QSTAR Elite system. LREIMS and HREIMS were performed on the Mass Spectrometer Thermo MAT95XP. HPLC separations were performed on the Agilent 1100 liquid chromatography system equipped with a solvent degasser, quaternary pump, and diode array detector (Agilent Technologies, Waldbronn, Germany) using a semipreparative reversed phase column Luna C18, 5 µ, 100 Å, 250 × 10 mm. Precoated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) were used for TLC analysis, and the spots were visualized under a UV light (254 nm) or by heating the plate pretreated with H2SO4/H2O/AcOH (1:4:20).
3.2. Animal Material
The sponge Spongia tubulifera was collected by hand and traditional scuba diving off the coast of the Mexican Caribbean (18°48′22.17″N / 87°39′32.61″W) at depths ranging from 10 to 15 m in March 2017. Samples were frozen immediately after collection. A voucher specimen 17YUE11ST was deposited in the Phylum Porifera Gerardo Green National Collection of the Instituto de Ciencias del Mar y Limnología (ICMyL) at the National Autonomous University of México (UNAM) in Ciudad de Mexico.
3.3. Extraction and Isolation
Sliced bodies of S. tubulifera (wet weight, 157.7 g; dry weight, 40.2 g) were exhaustively extracted with CH3OH-CH2Cl2 (1:1, 3 × 1.5 L) at 25 °C for 24 h each extraction. The combined extracts were concentrated under reduced pressure to give 12.0 g of a crude residue that was first partitioned between CH2Cl2/H2O (1:1 v/v). The resulting aqueous portion was extracted with n-butanol (200 mL) to yield the n-butanol fraction (1.44 g). The organic phase was concentrated under reduced pressure and partitioned between 10% aqueous CH3OH (400 mL) and hexane (2 × 400 mL) to give, after removing the solvent under reduced pressure, 526 mg of the hexane fraction. The H2O content (% v/v) of the methanolic fraction was adjusted to 50% aqueous CH3OH, and the mixture was extracted with CH2Cl2 (100 mL) to afford, after removing the solvent under reduced pressure, 1.51 g of the CH2Cl2 fraction and 2.38 g of the remaining aqueous methanolic fraction. The hexane fraction (526 mg) was subjected to a flash chromatography column on silica gel using a stepped gradient from hexane to EtOAc to give 14 fractions (FHC1-C14). Separation of the fraction FHC2, eluted with hexane/EtOAc (9:1, 93.9 mg), by RP-HPLC with a mobile phase consisting of an isocratic at 100% CH3OH at a flow rate of 2.0 mL/min afforded 6 (13.5 mg; tR = 8.4 min). Separation of the fraction FHC3, eluted with hexane/EtOAc (9:1, 20.0 mg), by RP-HPLC (isocratic 100% CH3OH, flow rate 2.0 mL/min) gave 2 (2.6 mg; tR = 9.7 min) and 6 (2.6 mg; tR = 8.9 min). Separation of the fraction FHC4, eluted with hexane/EtOAc (8:2, 9.0 mg), by RP-HPLC with a mobile phase consisting of 5 min gradient from 90% to 95% of CH3OH in H2O, followed by a 10 min isocratic at 95% of CH3OH in H2O and, finally, a 5 min gradient from 95% to 100% of CH3OH in H2O at a flow rate of 2.0 mL/min yielded 2 (2.0 mg; tR = 12.6 min). Separation of the fraction FHC5, eluted with hexane/EtOAc (8:2, 21.0 mg), by RP-HPLC with a mobile phase consisting of 5 min gradient from 90% to 95% of CH3OH in H2O, followed by a 15 min isocratic at 95% of CH3OH in H2O and, finally, a 10 min gradient from 95% to 100% of CH3OH in H2O at a flow rate of 2.0 mL/min afforded 2 (1.7 mg; tR = 13.0 min) and 3 (1.5 mg; tR = 12.0 min). Separation of the fraction FHC7, eluted with hexane/EtOAc (8:2, 27.2 mg), by RP-HPLC (isocratic 100% CH3OH, flow rate 2.0 mL/min) yielded 1 (3.0 mg; tR = 16.8 min). Separation of the fraction FHC8, eluted with hexane/EtOAc (8:2, 20.9 mg), by RP-HPLC with a mobile phase consisting of 5 min gradient from 90% to 95% of CH3OH in H2O followed by a 15 min isocratic at 95% of CH3OH in H2O and, finally, a 1 min gradient from 95% to 100% of CH3OH in H2O at a flow rate of 2.0 mL/min afforded 1 (1.8 mg; tR = 11.0 min). The dicloromethane fraction (1.51 g) was subjected to solid phase extraction (SPE) with RP-18 column (Merck KGaA) using a stepped gradient from H2O to CH3OH and then CH2Cl2, to give 6 fractions: H2O (100%), H2O/CH3OH (2:1, 1:1, and 1:2), CH3OH (100%), and CH2Cl2 100%. The fraction eluted with H2O/CH3OH (1:2) was submitted to RP-HPLC separation using a mobile phase consisting of 20 min gradient from 50% to 100% of CH3OH in H2O followed by a 10 min isocratic at 100% of CH3OH at a flow rate of 2.0 mL/min to afford 4 (9.2 mg; tR = 10.0 min). Separation of the fraction eluted with CH3OH (100%) by RP-HPLC using a mobile phase consisting of 30 min gradient from 80% to 100% of CH3OH in H2O at a flow rate of 2.0 mL/min afforded 7 (5.0 mg; tR = 26.7 min) and 5 (3.1 mg; tR = 29.7 min).
3.4. Computational Calculations
Conformational searches were performed by using the corresponding module implemented in the Maestro Quantum mechanical software. An OPLS 2005 force field with chloroform as the solvent was used, and torsional enhanced sampling with 1000 or 10,000 steps was fixed using an energy window of 10 kcal/mol. Molecular geometry optimizations were performed at the DFT theoretical level using the Gaussian 09W package firstly with a B3LYP/6-31G(d) combination and then with HSEH1PBE/cc-pVDZ auto for energy and frequency calculations. After removing redundant conformers, theoretical Boltzmann energy population-weighted ECD was calculated by using two combinations: PBEPBE/6-311++(3d,2p) or CAM-B3LYP/6-311++(3d,2p), both with 24 states. Graphical theoretical ECD curves were obtained using the open software SpecDis V.1.71 (Berlin, Germany, 2017,
https:/specdis-software.jimdo.com) [
23].
3.5. Metabolite Characterization
(3R, 5R, 8R, 9R, 10R) 3β-Hydroxyspongia-13(16),14-dien-2-one (1): Colorless white powder;
− 10.5 (c 0.1, CHCl
3); IR (ATR neat) υ
max 3500, 2920, 2810, 1745, 1430, 1371, 1229, 1120, 1050, 1038, 955, 882 cm
−1;
1H NMR (500 MHz) and
13C NMR (125 MHz) see
Table 1; HREIMS
m/
z 316.2014 [M]
+ (calcd. for C
20H
28O
3, 316.2038).
(5R, 8R, 9R, 10R) 19-Dehydroxy-spongian diterpene 17 (2): Colorless white powder;
+ 18.7 (c 0.1, CHCl
3); IR (ATR neat) υ
max 3505, 2950, 2855, 2325, 1650, 1430, 1370, 1230, 1120, 1050, 1038, 955, 880, cm
−1;
1H NMR (500 MHz) and
13C NMR (125 MHz) see
Table 1; (+)-HRESIMS
m/
z 337.1803 [M + Na]
+ (calcd. for C
20H
26O
3Na, 337.1780).
19-nor-3-Hydroxyspongia-3,13(16),14-trien-2-one (3): Colorless white powder; + 2.7 (c 0.1, CHCl3); (+)-HREIMS m/z 300.1719 [M]+ (calcd. for C19H24O3, 300.1725).
3β, 19-Dihydroxyspongia-13(16),14-dien-2-one (epispongiadiol) (4): Yellow powder; + 18.2 (c 0.1, CHCl3); (+)-HRESIMS m/z 355.1890 [M + Na]+ (calcd. for C20H28O4Na, 355.1885).
Spongian diterpene 17 (5): Yellow powder; − 20.4 (c 0.1, CHCl3); (+)-HRESIMS m/z 353.1723 [M + Na]+ (calcd. for C20H26O4Na, 353.1723).
Ambliol C (6) Yellow powder; − 33.3 (c 0.1, CHCl3); (+)-HREIMS m/z 304.2379 [M]+ (calcd. for C20H32O2, 304.2397).
Scalarin (7): Yellow powder; + 41.2 (c 0.1, CHCl3); (+)-HRESIMS m/z 467.2767 [M + Na]+ (calcd. for C27H40O5Na, 467.2773).
3.6. Cytotoxic Assays
Colorimetric MTT ((3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)) assays were carried out to assess the cell viability of the samples against a panel of five different cancer cell lines (i.e., human lung carcinoma A549 ATCC® CCL-185TM, human skin melanoma A2058 ATCC® CRL-11147TM, hepatocyte carcinoma HepG2 ATCC® HB-8065TM, breast adenocarcinoma MCF7 ATCC® HTB-22TM, and pancreas carcinoma MiaPaca-2 ATCC® CRL-1420TM). All cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). A549 cells were grown in Ham’s F12K medium with 2 mM glutamine, 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. A2058 and HepG2 were grown in ATCC formulated Eagle’s M essential medium (MEM) with 10% FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, and 100 μM MEM nonessential amino acids. MCF-7 cells were grown in the previous medium supplemented with 0.01 mg/mL of bovine insulin. MiaPaca-2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS, 100U/mL penicillin, and 100 μg/mL streptomycin. The bioassays were performed as reported by Audoin et al. [
20]. The cytotoxic activity was assessed after 72 h of treatment of the compound at the concentrations 0.098, 0.195, 0.391, 0.781, 1.563, 3.125, 6.250, 12.5, 25, 50, and 100 μM.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}