Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products


Abstract
:1. Introduction
2. Results
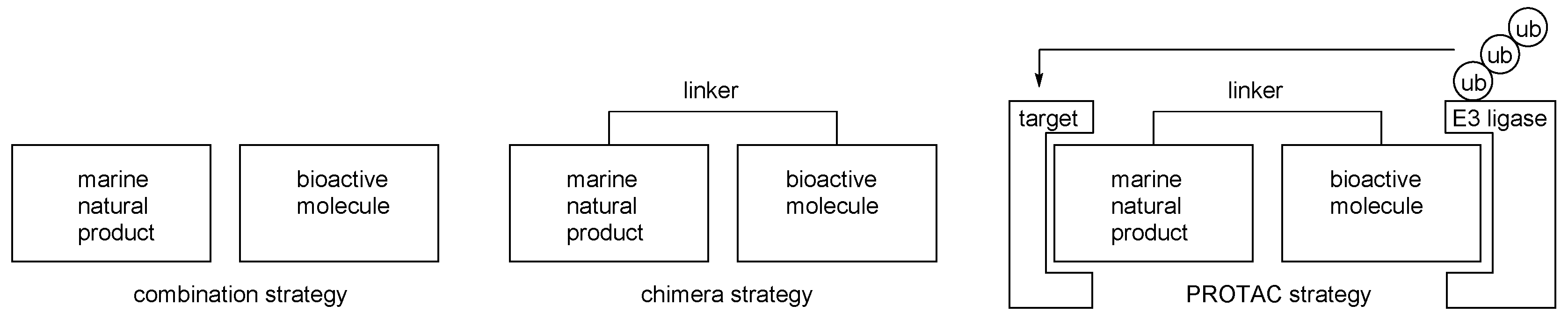
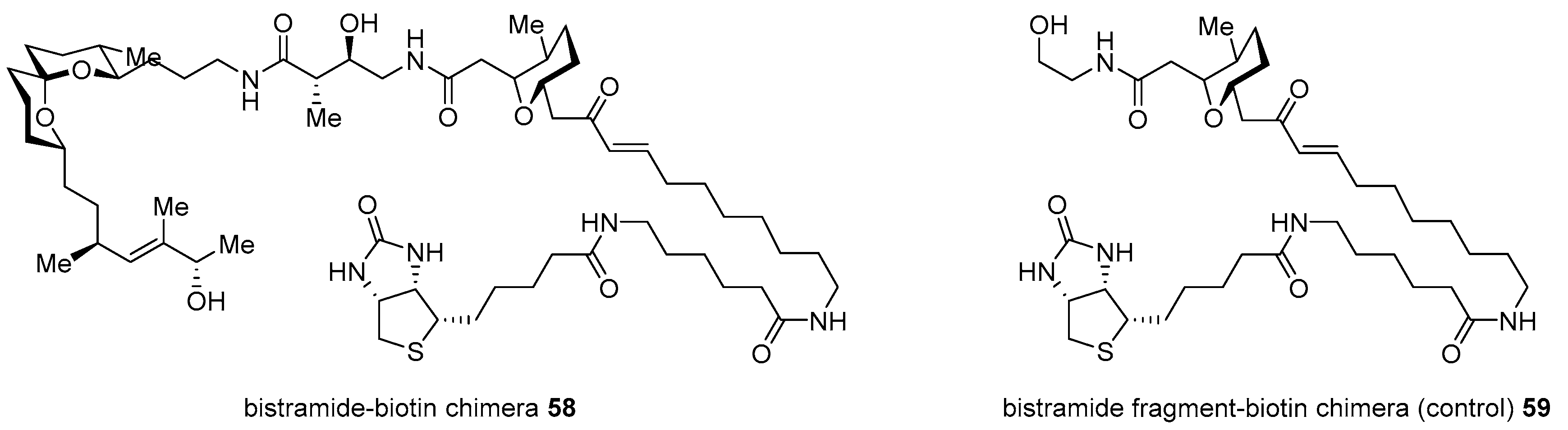
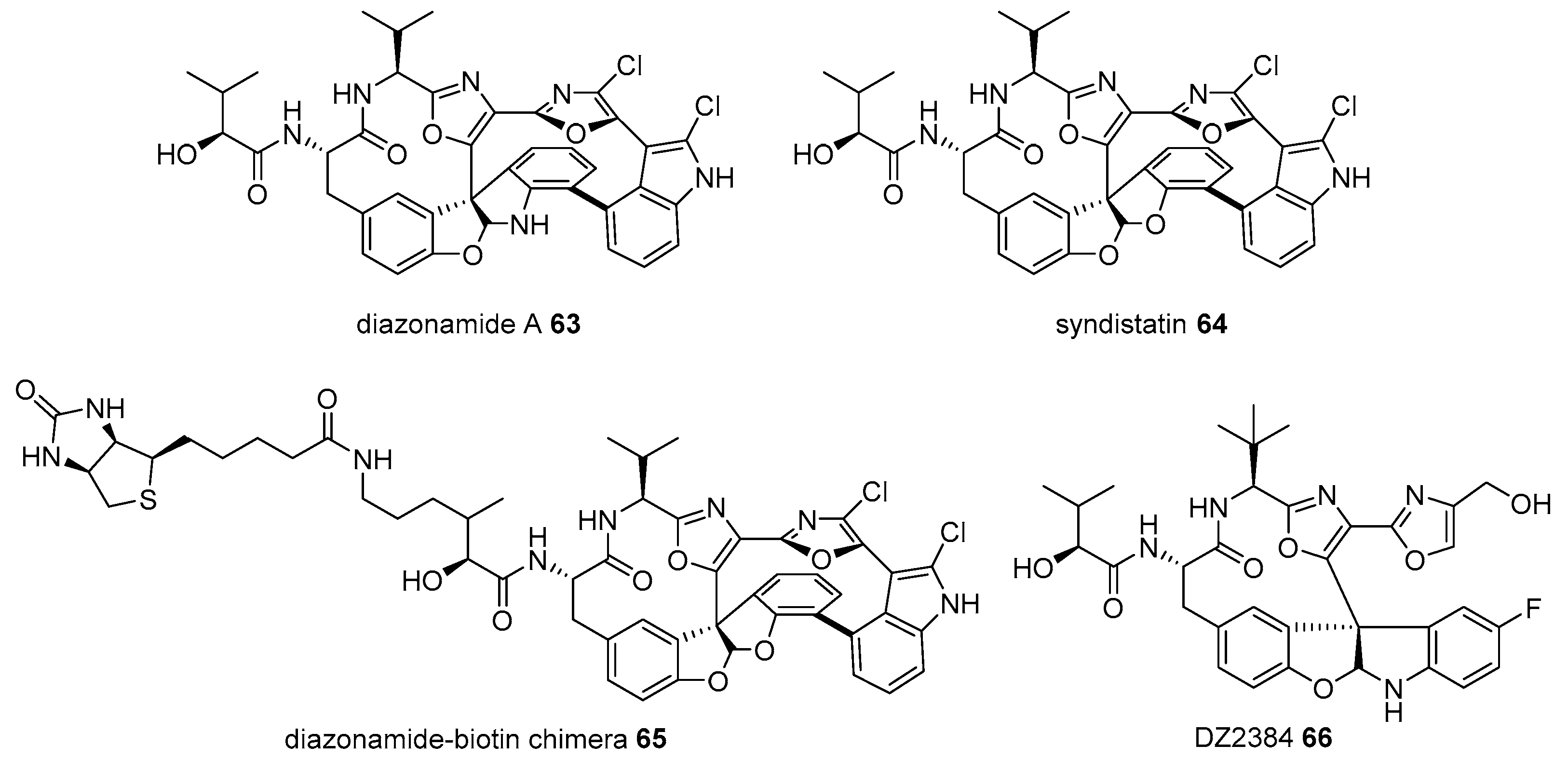
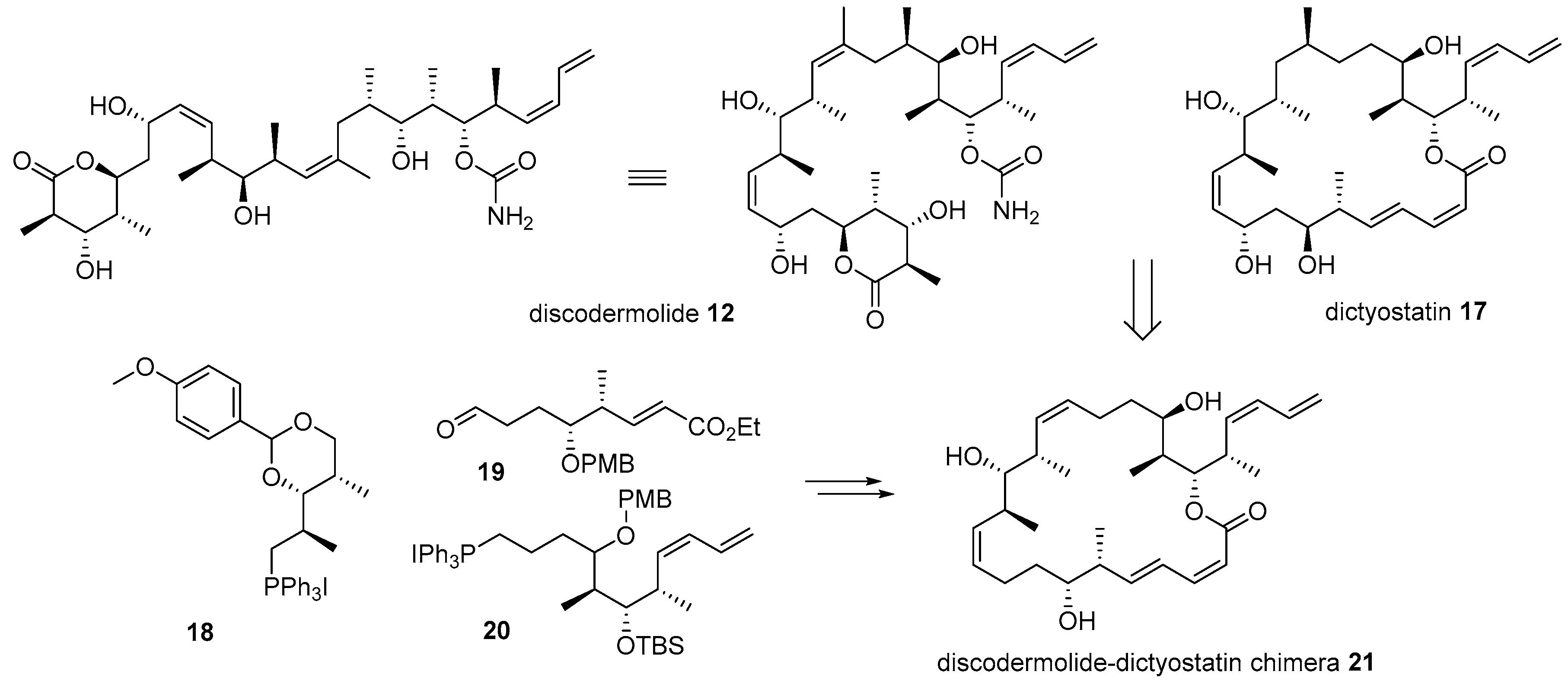
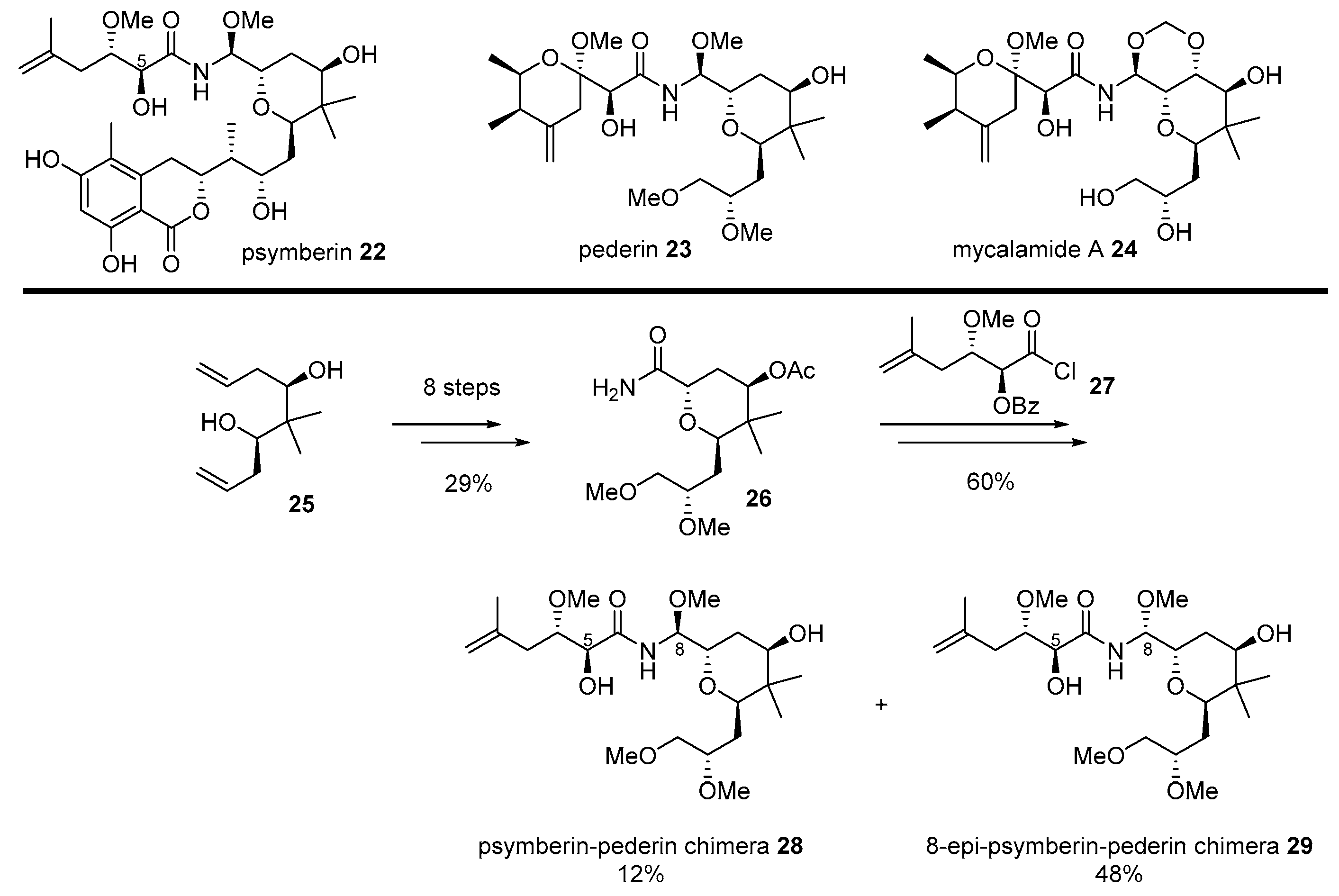
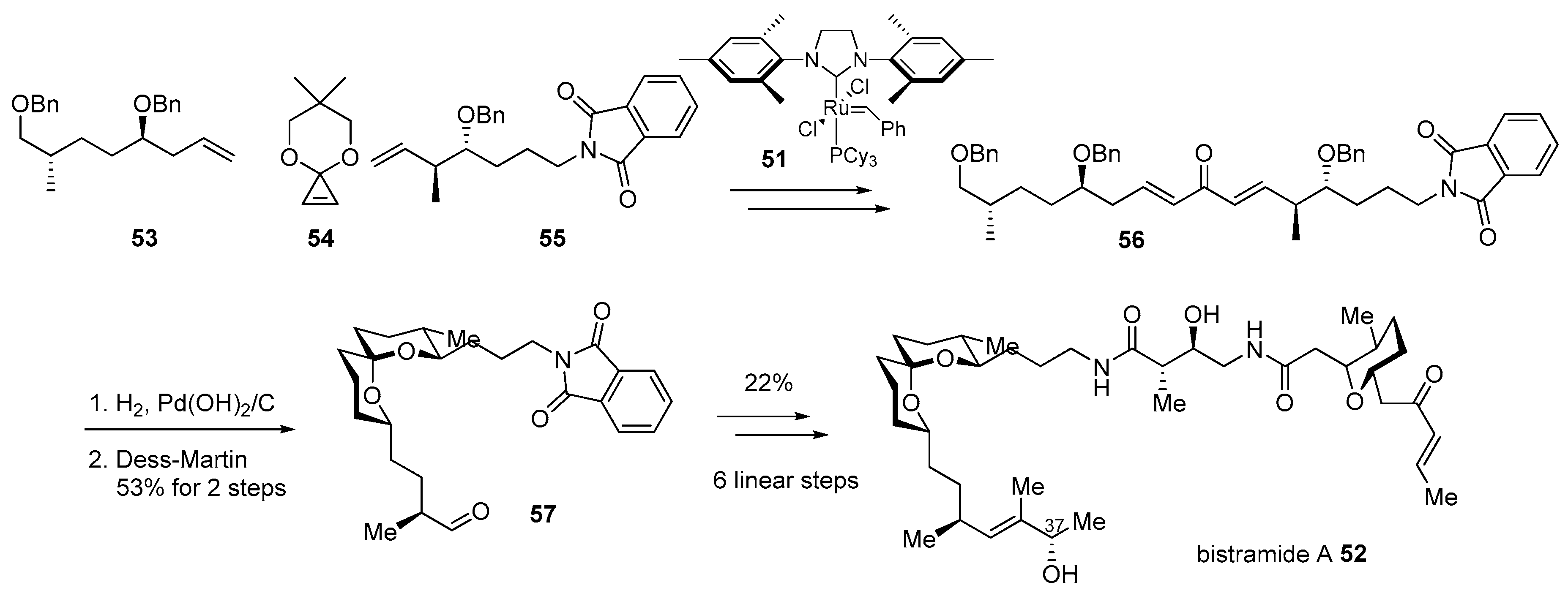
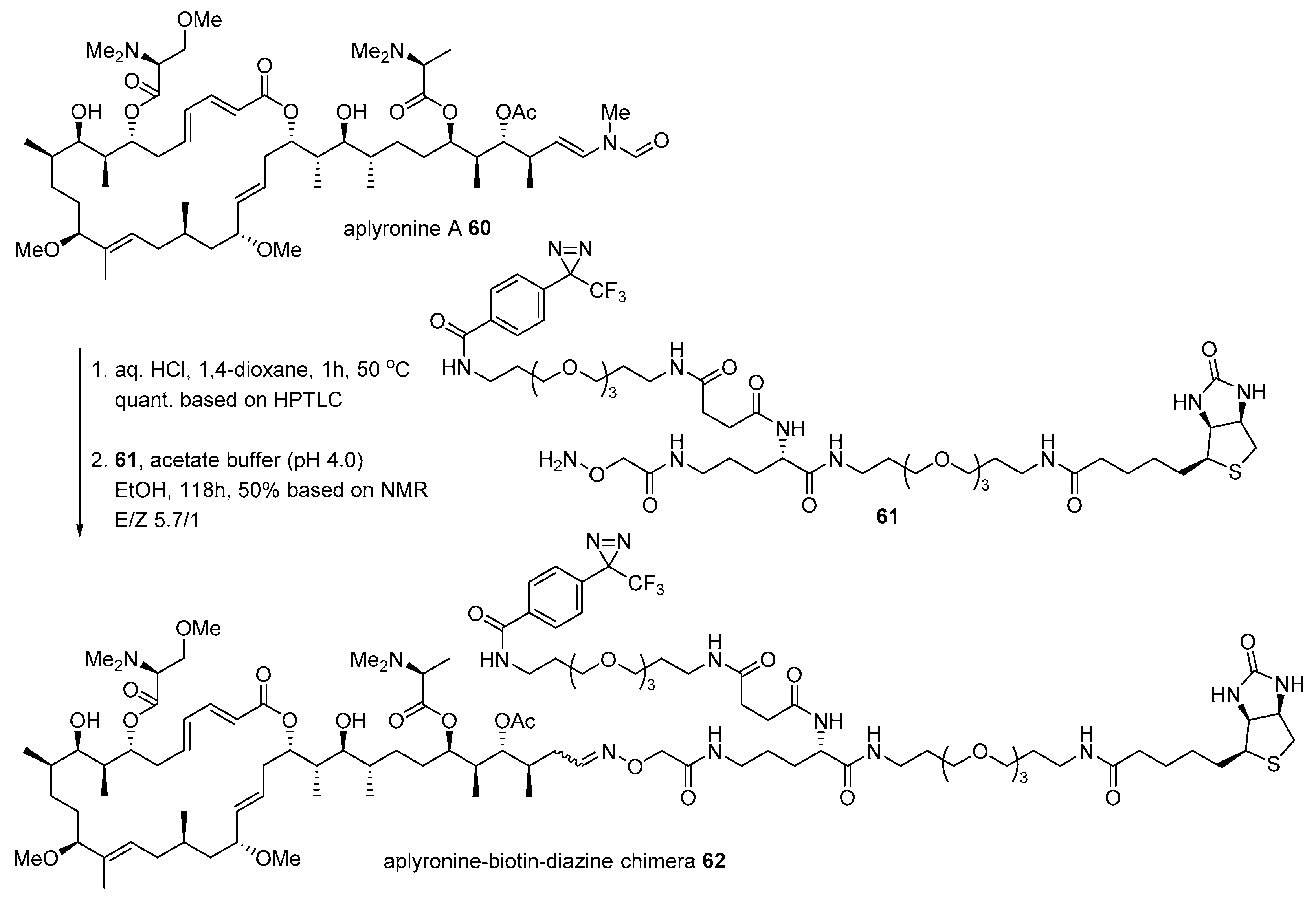
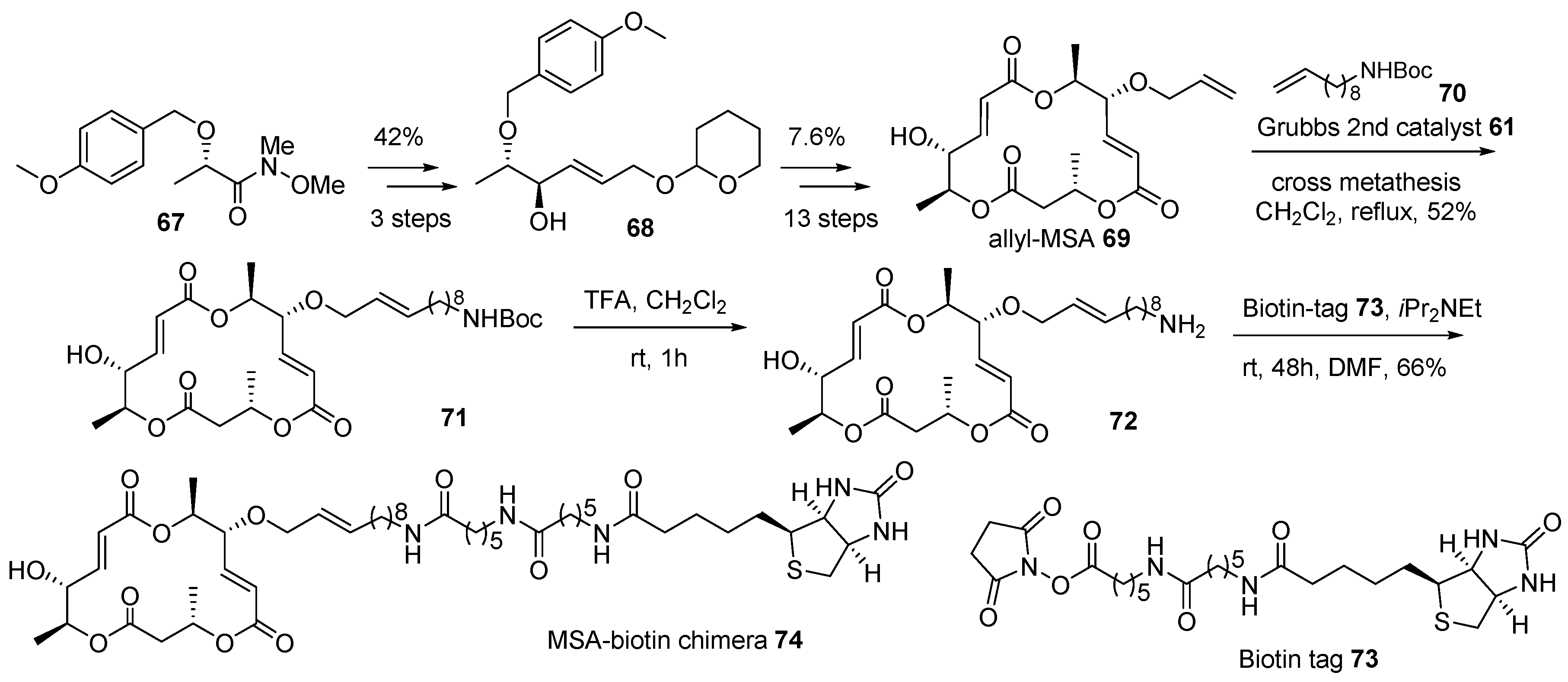
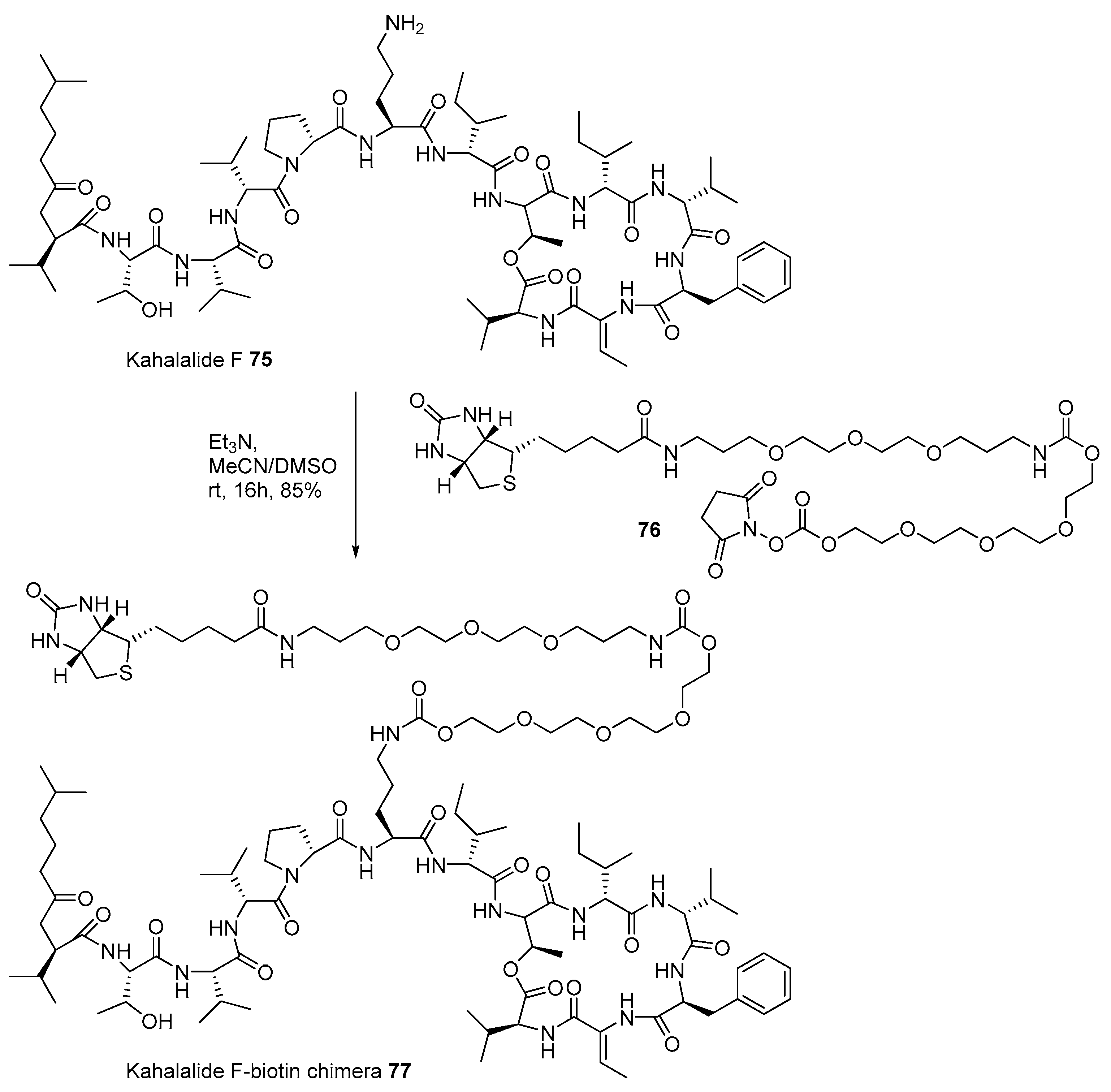
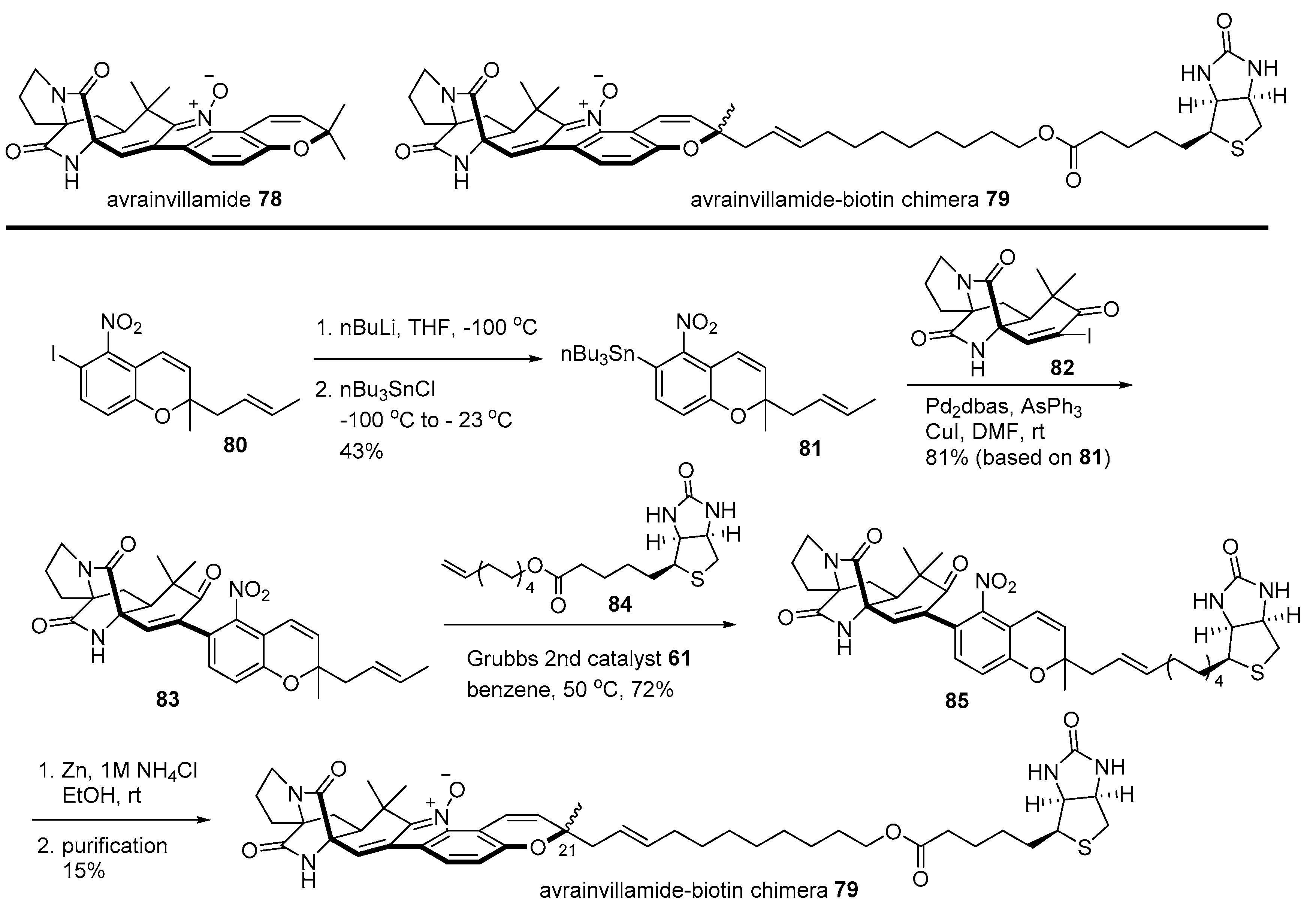
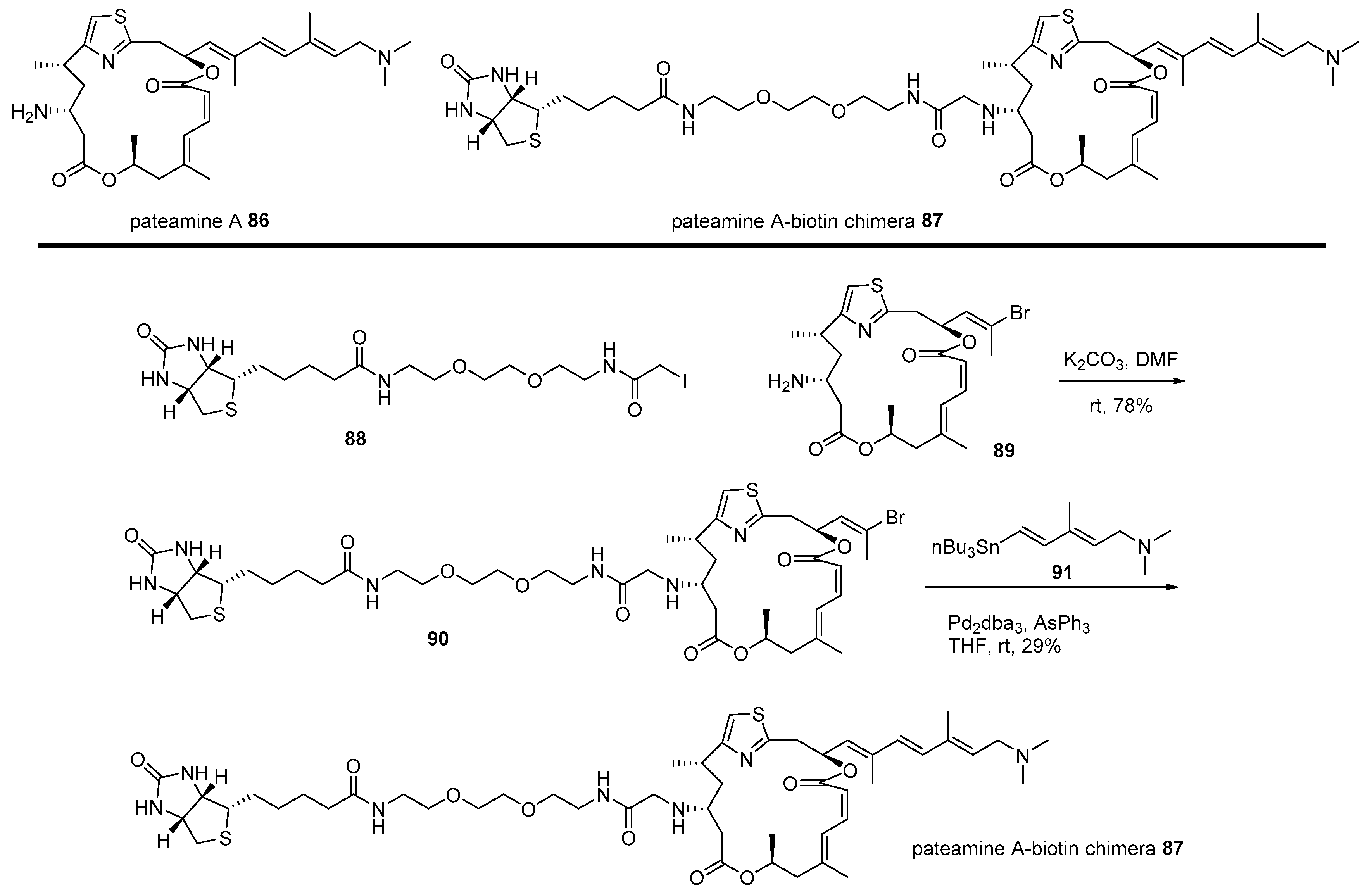
2.1. Conjugation with Other Active Molecules
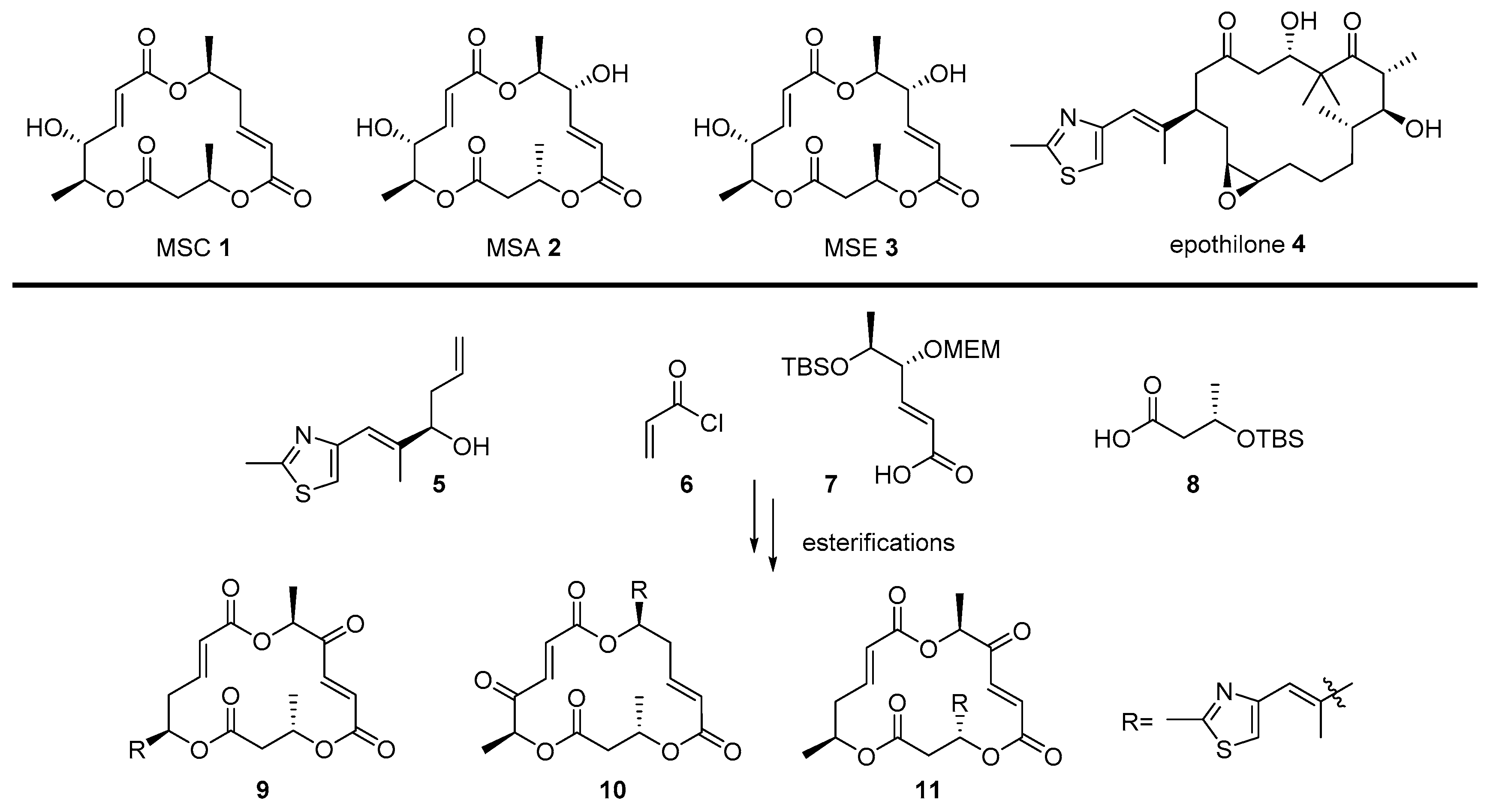
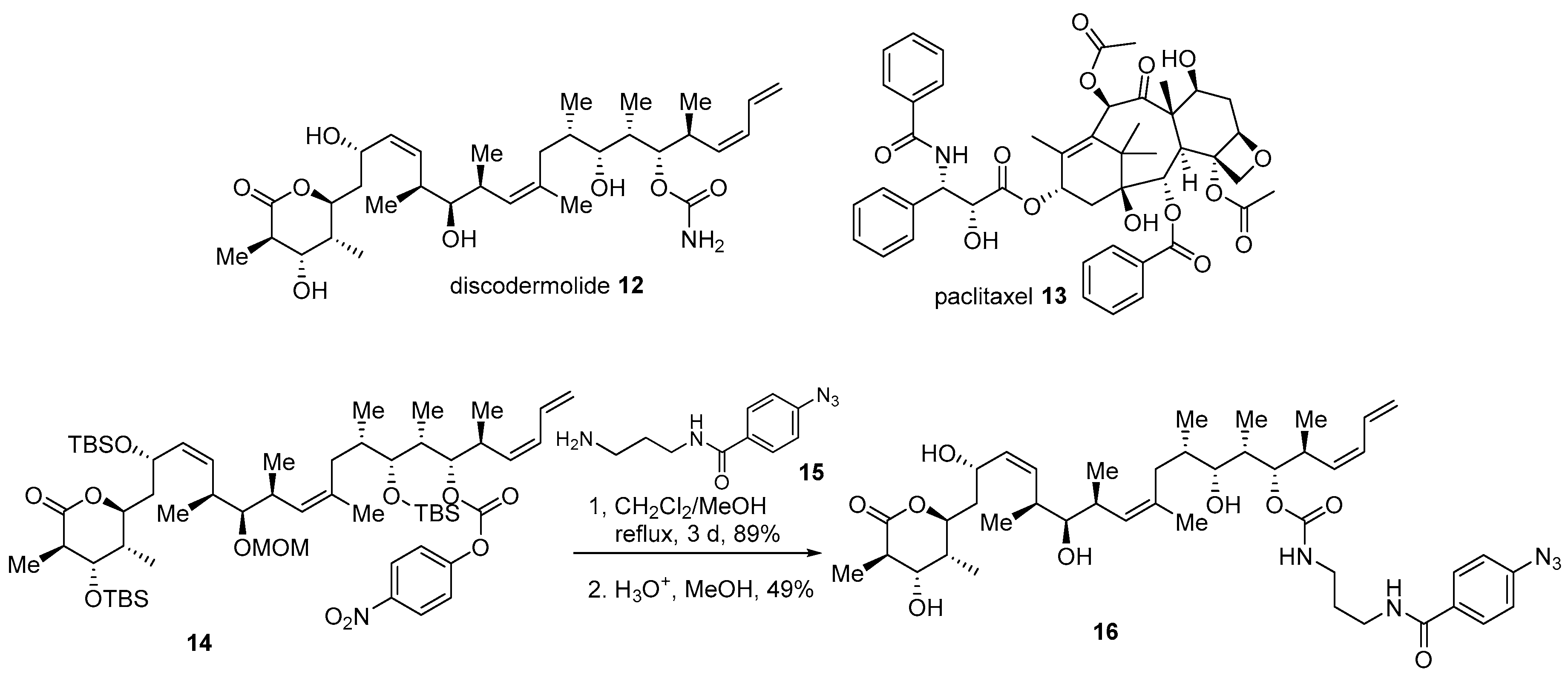
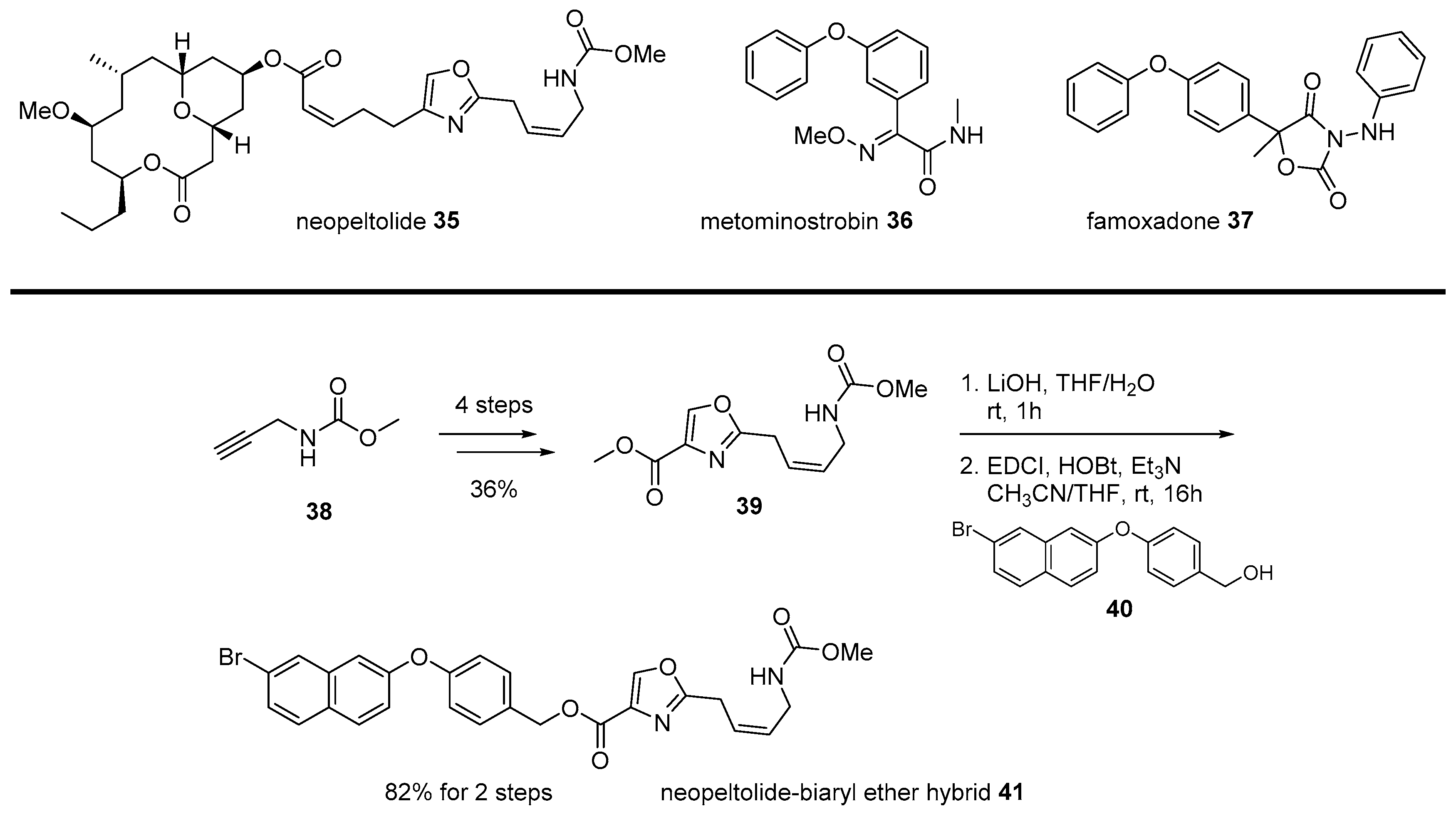
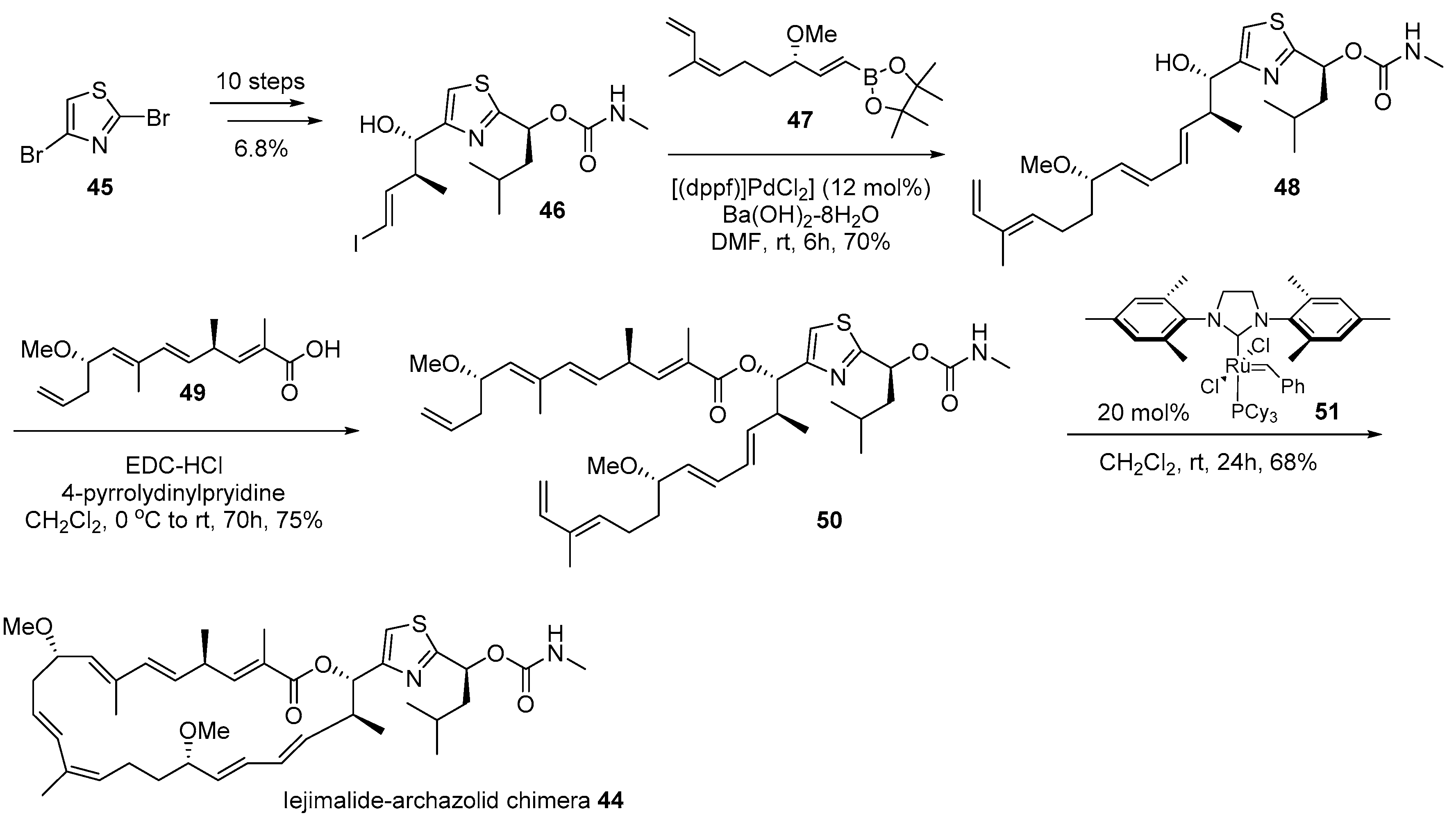
2.2. Conjugation with Other Functional Compounds
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, J.M.; Goncalves, B.D.; Gomez, M.V.; Vieira, L.B.; Ribeiro, F.M. Animal toxins as therapeutic tools to treat neurodegenerative diseases. Front. Pharmacol. 2018, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, P.; D’Auria, M.V.; Muller, C.D.; Tammela, P.; Vuorela, H.; Yli-Kauhaluoma, J. Exploring marine resources for bioactive compounds. Planta Med. 2014, 80, 1234–1246. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lou, H.-X. Strategies to diversify natural products for drug discovery. Med. Res. Rev. 2018, 38, 1255–1294. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
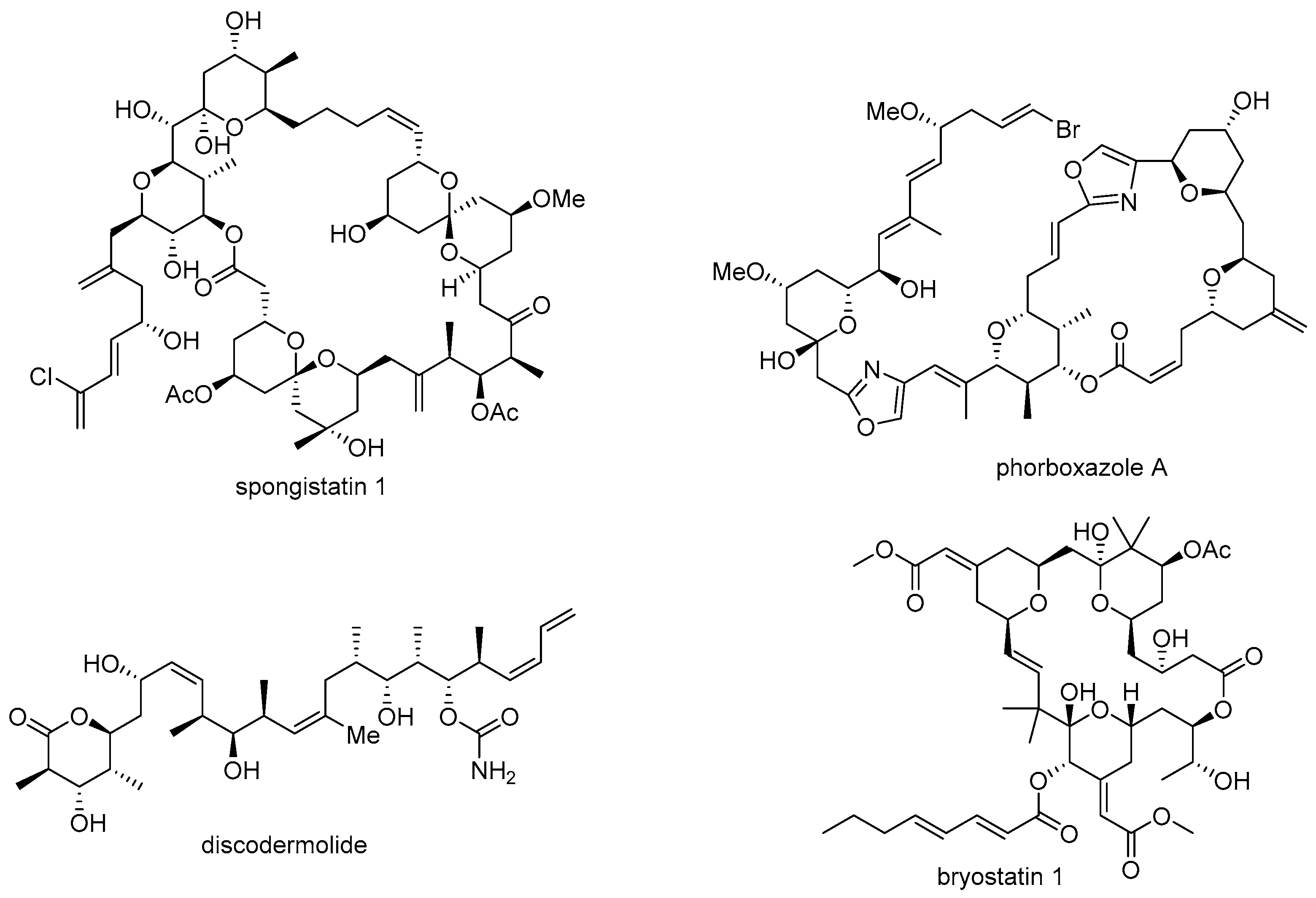
- Pettit, G.R.; Chicacz, Z.A.; Gao, F.; Herald, C.L.; Boyd, M.R.; Schmidt, J.M.; Hooper, J.N. Antineoplastic agents. 257. Isolation and structure of spongistatin 1. J. Org. Chem. 1993, 58, 1302–1304. [Google Scholar] [CrossRef]
- Smith, A.B.; Razler, T.M.; Meis, R.M.; Pettit, G.R. Synthesis and biological evaluation of phorboxazole congeners leading to the discovery and preparative-scale synthesis of (+)-chlorophorboxazole a possessing picomolar human solid tumor cell growth inhibitory activity. J. Org. Chem. 2008, 73, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.P.; Gunasekera, M.; Longley, R.E.; Schulte, G.K. Discodermolide: A new bioactive polyhydroxylated lactone from the marine sponge Discodermia dissoluta. J. Org. Chem. 1990, 55, 4912–4915. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Schaufelberger, D.E.; Koleck, M.P.; Beutler, J.A.; Vatakis, A.M.; Alvarado, A.B.; Andrews, P.; Marzo, L.; Muschik, G.; Roach, J.; Ross, J.T. The large-scale isolation of bryostatin 1 from Bugula neritina following current good manufacturing practices. J. Nat. Prod. 1991, 54, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B., III; Tomioka, T.; Risatti, C.A.; Sperry, J.B.; Sfouggatakis, C. Gram-scale synthesis of (+)-spongistatin 1: Development of an improved, scalable synthesis of the F-ring subunit, fragment union, and final elaboration. Org. Lett. 2008, 10, 4359–4362. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Beauchamp, T.J.; LaMarche, M.J.; Kaufman, M.D.; Qiu, Y.; Arimoto, H.; Jones, D.R.; Kobayashi, K. Evolution of a gram-scale synthesis of (+)-discodermolide. J. Am. Chem. Soc. 2000, 122, 8654–8664. [Google Scholar] [CrossRef]
- Mickel, S.J.; Niederer, D.; Daeffler, R.; Osmani, A.; Kuesters, E.; Schmid, E.; Schaer, K.; Gamboni, R.; Chen, W.; Loeser, E. Large-scale synthesis of the anti-cancer marine natural product (+)-Discodermolide. Part 5: Linkage of fragments C1-6 and C7-24 and finale. Org. Process. Res. Dev. 2004, 8, 122–130. [Google Scholar] [CrossRef]
- Choudhary, A.; Naughton, L.; Montánchez, I.; Dobson, A.; Rai, D. Current status and future prospects of marine natural products (MNPs) as antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Lear, M.J.; Hirai, K.; Ogawa, K.; Yamashita, S.; Hirama, M. A convergent total synthesis of the kedarcidin chromophore: 20-years in the making. J. Antibiot. 2019, 72, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Morris-Natschke, S.L.; Lee, K.-H. Strategies for the optimization of natural leads to anticancer drugs or drug candidates. Med. Res. Rev. 2016, 36, 32–91. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Drugs and drug candidates from marine sources: An assessment of the current “state of play”. Planta Med. 2016, 82, 775–789. [Google Scholar] [CrossRef] [PubMed]
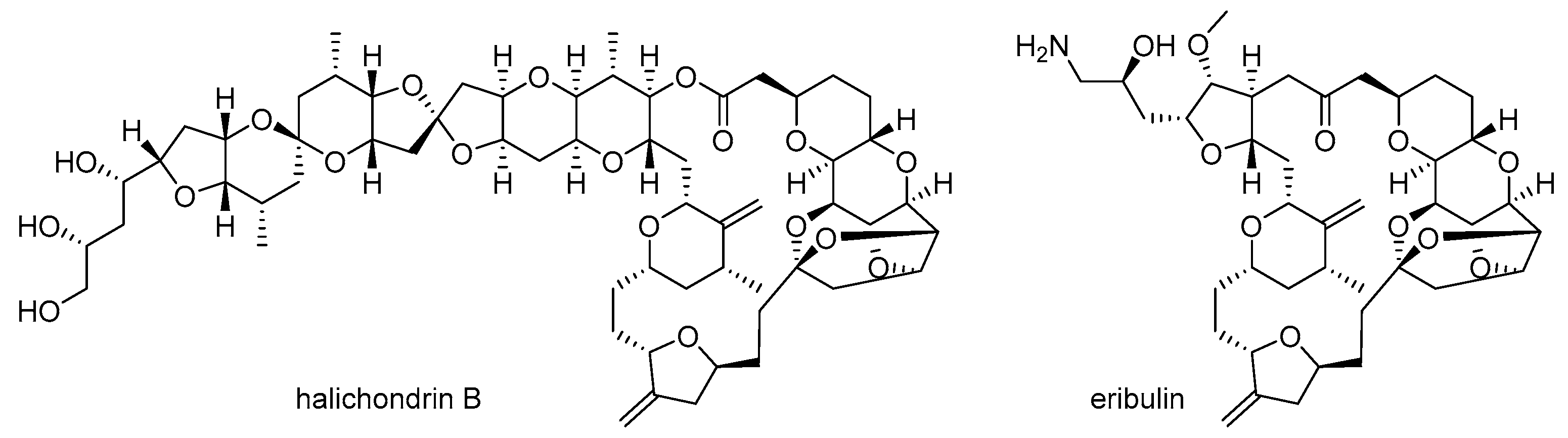
- Hirata, Y.; Uemura, D. Halichondrins-antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- McBride, A.; Butler, S.K. Eribulin mesylate: A novel halichondrin B analogue for the treatment of metastatic breast cancer. Am. J. Health Syst. Pharm. 2012, 69, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Aicher, T.D.; Buszek, K.R.; Fang, F.G.; Forsyth, C.J.; Jung, S.H.; Kishi, Y.; Matelich, M.C.; Scola, P.M.; Spero, D.M.; Yoon, S.K. Total synthesis of halichondrin B and norhalichondrin B. J. Am. Chem. Soc. 1992, 114, 3162–3164. [Google Scholar] [CrossRef]
- Lindequist, U. Marine-derived pharmaceuticals—Challenges and opportunities. Biomol. Ther. 2016, 24, 561. [Google Scholar] [CrossRef] [PubMed]
- Bebbington, M.W. Natural product analogues: Towards a blueprint for analogue-focused synthesis. Chem. Soc. Rev. 2017, 46, 5059–5109. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.E. Design and synthesis of analogues of natural products. Org. Biomol. Chem. 2015, 13, 5302–5343. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chem. Int. Edit. 2013, 52, 2744–2792. [Google Scholar] [CrossRef]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Edit. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Mehta, G.; Singh, V. Hybrid systems through natural product leads: An approach towards new molecular entities. Chem. Soc. Rev. 2002, 31, 324–334. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent progress in the development of synthetic hybrids of natural or unnatural bioactive compounds for medicinal chemistry. Mini Rev. Med. Chem. 2010, 10, 773–793. [Google Scholar] [CrossRef]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101. [Google Scholar] [CrossRef]
- Salami, J.; Crews, C.M. Waste disposal-An attractive strategy for cancer therapy. Science 2017, 355, 1163–1167. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Olaáh, J.; Ovaádi, J.; Sippl, W.; Jung, M. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2017, 61, 482–491. [Google Scholar] [CrossRef]
- Leslie, B.J.; Hergenrother, P.J. Identification of the cellular targets of bioactive small organic molecules using affinity reagents. Chem. Soc. Rev. 2008, 37, 1347–1360. [Google Scholar] [CrossRef]
- Paek, S. Synthetic advances in macrosphelides: Natural anticancer agents. Molecules 2014, 19, 15982–16000. [Google Scholar] [CrossRef]
- Yamada, T.; Iritani, M.; Doi, M.; Minoura, K.; Ito, T.; Numata, A. Absolute stereostructures of cell-adhesion inhibitors, macrosphelides C, E–G and I, produced by a Periconia species separated from an Aplysia sea hare. J. Chem. Soc. Perkin Trans. 1 2001, 3046–3053. [Google Scholar] [CrossRef]
- Hayashi, M.; Kim, Y.; Hiraoka, H.; Natori, M.; Takamatsu, S.; Kawakubo, T.; Masuma, R.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule. J. Antibiot. 1995, 48, 1435–1439. [Google Scholar] [CrossRef]
- Sunazuka, T.; Hirose, T.; Harigaya, Y.; Takamatsu, S.; Hayashi, M.; Komiyama, K.; Ōmura, S.; Sprengeler, P.A.; Smith, A.B. Relative and absolute stereochemistries and total synthesis of (+)-macrosphelides A and B, potent, orally bioavailable inhibitors of cell—Cell adhesion. J. Am. Chem. Soc. 1997, 119, 10247–10248. [Google Scholar] [CrossRef]
- Paek, S. Development of advanced macrosphelides: Potent anticancer agents. Molecules 2015, 20, 4430–4449. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kawaguchi, T.; Ishihara, K.; Ahmed, K.; Zhao, Q.; Kondo, T.; Nemoto, H. Synthesis of macrosphelides with a thiazole side chain: New antitumor candidates having apoptosis-inducing property. Org. Lett. 2006, 8, 4609–4612. [Google Scholar] [CrossRef]
- Meng, D.; Bertinato, P.; Balog, A.; Su, D.; Kamenecka, T.; Sorensen, E.J.; Danishefsky, S.J. Total syntheses of epothilones A and B. J. Am. Chem. Soc. 1997, 119, 10073–10092. [Google Scholar] [CrossRef]
- Xia, S.; Kenesky, C.S.; Rucker, P.V.; Smith, A.B.; Orr, G.A.; Horwitz, S.B. A photoaffinity analogue of discodermolide specifically labels a peptide in β-tubulin. Biochemistry 2006, 45, 11762–11775. [Google Scholar] [CrossRef]
- Smith, A.B., III; Sugasawa, K.; Atasoylu, O.; Yang, C.H.; Horwitz, S.B. Design and synthesis of (+)-discodermolide–paclitaxel hybrids leading to enhanced biological activity. J. Med. Chem. 2011, 54, 6319–6327. [Google Scholar] [CrossRef]
- Shin, Y.; Choy, N.; Balachandran, R.; Madiraju, C.; Day, B.W.; Curran, D.P. Discodermolide/dictyostatin hybrids: Synthesis and biological evaluation. Org. Lett. 2002, 4, 4443–4446. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Boyd, M.R.; Schmidt, J.M. Isolation and structure of the cancer cell growth inhibitor dictyostatin 1. J. Chem. Soc. Chem. Commun. 1994, 1111–1112. [Google Scholar] [CrossRef]
- Isbrucker, R.A.; Cummins, J.; Pomponi, S.A.; Longley, R.E.; Wright, A.E. Tubulin polymerizing activity of dictyostatin-1, a polyketide of marine sponge origin. Biochem. Pharmacol. 2003, 66, 75–82. [Google Scholar] [CrossRef]
- Zanato, C.; Pignataro, L.; Hao, Z.; Gennari, C. A practical synthesis of the C1–C9 fragment of dictyostatin. Synthesis 2008, 2008, 2158–2162. [Google Scholar]
- Cichewicz, R.H.; Valeriote, F.A.; Crews, P. Psymberin, a potent sponge-derived cytotoxin from Psammocinia distantly related to the pederin family. Org. Lett. 2004, 6, 1951–1954. [Google Scholar] [CrossRef]
- Clark, W.D.; Corbett, T.; Valeriote, F.; Crews, P. Cyclocinamide A. An unusual cytotoxic halogenated hexapeptide from the marine sponge Psammocinia. J. Am. Chem. Soc. 1997, 119, 9285–9286. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tanaka, J.I.; Katori, T.; Matsuura, M.; Kitagawa, I. Structure of swinholide A, a potent cytotoxic macrolide from the Okinawan marine sponge Tehonella Swinhoei. Tetrahedron Lett. 1989, 30, 2963–2966. [Google Scholar] [CrossRef]
- Faulkner, D.J. Variabilin, an antibiotic from the sponge, Ircinia variabilis. Tetrahedron Lett. 1973, 39, 3821–3822. [Google Scholar] [CrossRef]
- Pettit, G.R.; Xu, J.; Chapuis, J.; Pettit, R.K.; Tackett, L.P.; Doubek, D.L.; Hooper, J.N.; Schmidt, J.M. Antineoplastic Agents. 520. Isolation and Structure of Irciniastatins A and B from the Indo-Pacific Marine Sponge Ircinia r amosa. J. Med. Chem. 2004, 47, 1149–1152. [Google Scholar] [CrossRef]
- Jiang, X.; García-Fortanet, J.; De Brabander, J.K. Synthesis and complete stereochemical assignment of psymberin/irciniastatin A. J. Am. Chem. Soc. 2005, 127, 11254–11255. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.; Pannell, L.K. Mycalamide A, an antiviral compound from a New Zealand sponge of the genus Mycale. J. Am. Chem. Soc. 1988, 110, 4850–4851. [Google Scholar] [CrossRef]
- Jiang, X.; Williams, N.; De Brabander, J.K. Synthesis of psymberin analogues: Probing a functional correlation with the pederin/mycalamide family of natural products. Org. Lett. 2007, 9, 227–230. [Google Scholar] [CrossRef]
- Bartik, K.; Braekman, J.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Topsentins, new toxic bis-indole alkaloids from the marine sponge Topsentia genitrix. Can. J. Chem. 1987, 65, 2118–2121. [Google Scholar] [CrossRef]
- Tsujii, S.; Rinehart, K.L.; Gunasekera, S.P.; Kashman, Y.; Cross, S.S.; Lui, M.S.; Pomponi, S.A.; Diaz, M.C. Topsentin, bromotopsentin, and dihydrodeoxybromotopsentin: Antiviral and antitumor bis (indolyl) imidazoles from Caribbean deep-sea sponges of the family Halichondriidae. Structural and synthetic studies. J. Org. Chem. 1988, 53, 5446–5453. [Google Scholar] [CrossRef]
- Gu, X.; Wan, X.; Jiang, B. Syntheses and biological activities of bis (3-indolyl) thiazoles, analogues of marine bis (indole) alkaloid nortopsentins. Bioorg. Med. Chem. Lett. 1999, 9, 569–572. [Google Scholar] [CrossRef]
- Hogan, I.; Sainsbury, M. The synthesis of dendrodoine, 5-[3-(N, N-dimethylamino-1, 2, 4-thiadiazolyl]-3-indolylmethanone, a metabolite of the marine tunicate dendroda grossular. Tetrahedron 1984, 40, 681–682. [Google Scholar] [CrossRef]
- Juneja, M.; Vanam, U.; Paranthaman, S.; Bharathan, A.; Keerthi, V.S.; Reena, J.K.; Rajaram, R.; Rajasekharan, K.N.; Karunagaran, D. 4-amino-2-arylamino-5-indoloyl/cinnamoythiazoles, analogs of topsentin-class of marine alkaloids, induce apoptosis in hela cells. Eur. J. Med. Chem. 2013, 63, 474–483. [Google Scholar] [CrossRef]
- Wright, A.E.; Botelho, J.C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef]
- Custar, D.W.; Zabawa, T.P.; Scheidt, K.A. Total synthesis and structural revision of the marine macrolide neopeltolide. J. Am. Chem. Soc. 2008, 130, 804–805. [Google Scholar] [CrossRef]
- Custar, D.W.; Zabawa, T.P.; Hines, J.; Crews, C.M.; Scheidt, K.A. Total synthesis and structure—Activity investigation of the marine natural product neopeltolide. J. Am. Chem. Soc. 2009, 131, 12406–12414. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Janjic, J.; Suzuki, M.; Sabharwal, S.S.; Schumacker, P.T.; Kron, S.J.; Kozmin, S.A. Synthesis enables identification of the cellular target of leucascandrolide A and neopeltolide. Nat. Chem. Biol. 2008, 4, 418. [Google Scholar] [CrossRef]
- Zhu, X.L.; Z’hang, R.; Wu, Q.; Song, Y.; Wang, Y.; Yang, J.; Yang, G. Natural Product Neopeltolide as A Cytochrome bc1 Complex Inhibitor: Mechanism of Action and Structural Modification. J. Agric. Food Chem. 2019, 67, 2774–2781. [Google Scholar] [CrossRef]
- Vintonyak, V.V.; Kunze, B.; Sasse, F.; Maier, M.E. Total synthesis and biological activity of neopeltolide and analogues. Chem. Eur. J. 2008, 14, 11132–11140. [Google Scholar] [CrossRef]
- Xiong, L.; Li, H.; Jiang, L.; Ge, J.; Yang, W.; Zhu, X.L.; Yang, G. Structure-based discovery of potential fungicides as succinate ubiquinone oxidoreductase inhibitors. J. Agric. Food Chem. 2017, 65, 1021–1029. [Google Scholar] [CrossRef]
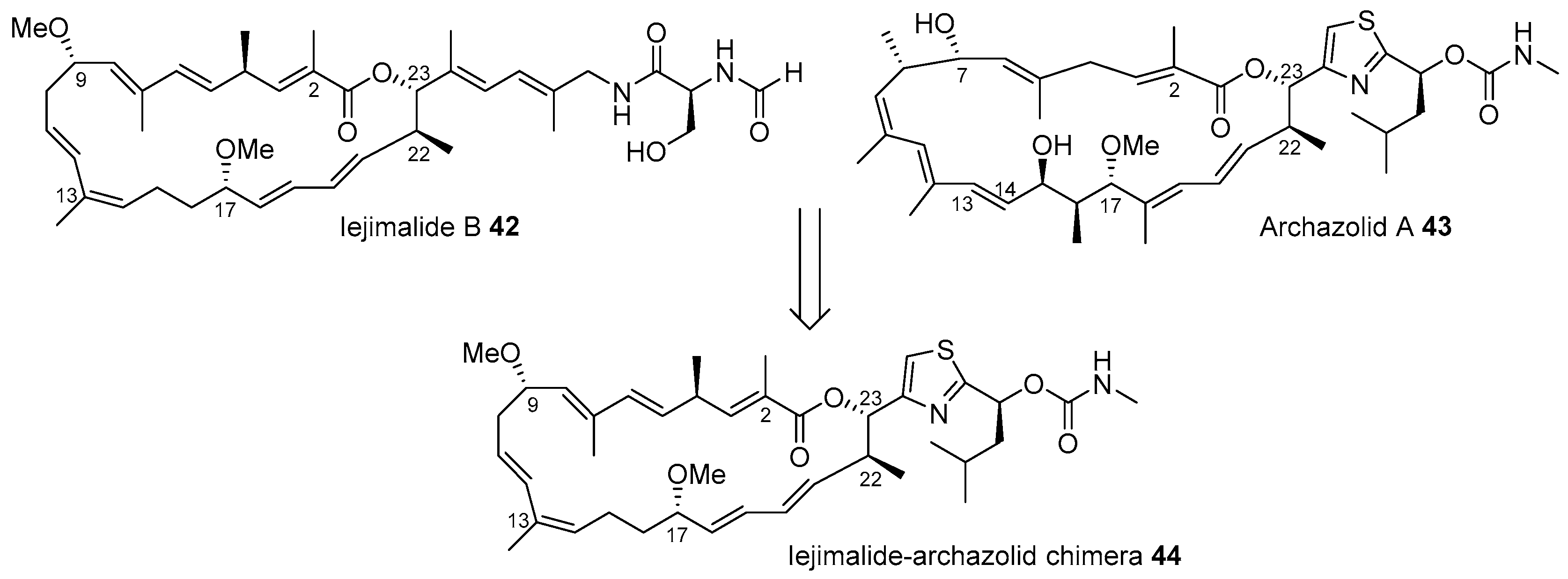
- Moulin, E.; Nevado, C.; Gagnepain, J.; Kelter, G.; Fiebig, H.; Fürstner, A. Synthesis and evaluation of an Iejimalide-archazolid chimera. Tetrahedron 2010, 66, 6421–6428. [Google Scholar] [CrossRef]
- Kobayashi, J.; Cheng, J.; Ohta, T.; Nakamura, H.; Nozoe, S.; Hirata, Y.; Ohizumi, Y.; Sasaki, T. Iejimalides A and B, novel 24-membered macrolides with potent antileukemic activity from the Okinawan tunicate Eudistoma cf. rigida. J. Org. Chem. 1988, 53, 6147–6150. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Ishibashi, M.; Sasaki, T.; Kobayashi, J. Iejimalides C and D, new antineoplastic 24-membered macrolide sulfates from the Okinawan marine tunicate Eudistoma cf. rigida. Tetrahedron Lett. 1991, 32, 797–798. [Google Scholar] [CrossRef]
- Fürstner, A.; Nevado, C.; Tremblay, M.; Chevrier, C.; Teplý, F.; Aïssa, C.; Waser, M. Total synthesis of iejimalide B. Angew. Chem. Int. Edit. 2006, 45, 5837–5842. [Google Scholar] [CrossRef]
- Fürstner, A.; Nevado, C.; Waser, M.; Tremblay, M.; Chevrier, C.; Teplý, F.; Aïssa, C.; Moulin, E.; Müller, O. Total Synthesis of Iejimalide A–D and Assessment of the Remarkable Actin-Depolymerizing Capacity of These Polyene Macrolides. J. Am. Chem. Soc. 2007, 129, 9150–9161. [Google Scholar] [CrossRef]
- Sasse, F.; Steinmetz, H.; Hoefle, G.; Reichenbach, H. Archazolids, New Cytotoxic Macrolactones from Archangium gephyra (Myxobacteria). J. Antibiot. 2003, 56, 520–525. [Google Scholar] [CrossRef]
- Corey, E.J.; Helal, C.J. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: A new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem. Int. Edit. 1998, 37, 1986–2012. [Google Scholar] [CrossRef]
- Marshall, J.A. Chiral allylic and allenic metal reagents for organic synthesis. J. Org. Chem. 2007, 72, 8153–8166. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Ogba, O.; Warner, N.; O’Leary, D.; Grubbs, R. Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, O.H.; Nordlund, H.R.; Hytönen, V.P.; Kulomaa, M.S. Brave new (strept) avidins in biotechnology. Trends Biotechnol. 2007, 25, 269–277. [Google Scholar] [CrossRef]
- Dundas, C.M.; Demonte, D.; Park, S. Streptavidin–biotin technology: Improvements and innovations in chemical and biological applications. Appl. Microbiol. Biotechnol. 2013, 97, 9343–9353. [Google Scholar] [CrossRef]
- Kim, J.; Cantor, A.B.; Orkin, S.H.; Wang, J. Use of in vivo biotinylation to study protein–protein and protein–DNA interactions in mouse embryonic stem cells. Nat. Protoc. 2009, 4, 506–517. [Google Scholar] [CrossRef]
- Manz, B.; Heubner, A.; Kohler, I.; Grill, H.; Pollow, K. Synthesis of Biotin-Labelled Dexamethasone Derivatives: Novel Hormone-Affinity Probes. Eur. J. Biochem. 1983, 131, 333–338. [Google Scholar] [CrossRef]
- Wright, M.; Sieber, S. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33, 681–708. [Google Scholar] [CrossRef] [Green Version]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of direct protein targets of small molecules. ACS Chem. Biol. 2010, 6, 34–46. [Google Scholar] [CrossRef]
- Sato, S.; Murata, A.; Shirakawa, T.; Uesugi, M. Biochemical target isolation for novices: Affinity-based strategies. Chem. Biol. 2010, 17, 616–623. [Google Scholar] [CrossRef]
- Gouiffes, D.; Moreau, S.; Helbecque, N.; Bernier, J.; Henichart, J.; Barbin, Y.; Laurent, D.; Verbist, J. Proton nuclear magnetic study of bistramide A, a new cytotoxic drug isolated from Lissoclinum bistratum Sluiter. Tetrahedron 1988, 44, 451–459. [Google Scholar] [CrossRef]
- Degnan, B.M.; Hawkins, C.J.; Lavin, M.F.; McCaffrey, E.J.; Parry, D.L.; Watters, D.J. Novel cytotoxic compounds from the ascidian Lissoclinum bistratum. J. Med. Chem. 1989, 32, 1354–1359. [Google Scholar] [CrossRef]
- Biard, J.; Roussakis, C.; Kornprobst, J.; Gouiffes-Barbin, D.; Verbist, J.; Cotelle, P.; Foster, M.P.; Ireland, C.M.; Debitus, C. Bistramides A, B, C, D, and K: A new class of bioactive cyclic polyethers from Lissoclinum bistratum. J. Nat. Prod. 1994, 57, 1336–1345. [Google Scholar] [CrossRef]
- Riou, D.; Roussakis, C.; Biard, J.F.; Verbist, J.F. Comparative study of the antitumor activity of bistramides A, D and K against a non-small cell broncho-pulmonary carcinoma. Anticancer Res. 1993, 13, 2331–2334. [Google Scholar]
- Wipf, P.; Uto, Y.; Yoshimura, S. Total synthesis of a stereoisomer of bistramide C and assignment of configuration of the natural product. Chem. Eur. J. 2002, 8, 1670–1681. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Liu, D.; Kozmin, S.A. Synthesis of bistramide A. J. Am. Chem. Soc. 2004, 126, 9546–9547. [Google Scholar] [CrossRef]
- Gagne, M.R.; Grubbs, R.H.; Feldman, J.; Ziller, J.W. Catalytic activity of a well-defined binuclear ruthenium alkylidene complex. Organometallics 1992, 11, 3933–3935. [Google Scholar] [CrossRef]
- Foster, M.P.; Mayne, C.L.; Dunkel, R.; Pugmire, R.J.; Grant, D.M.; Kornprobst, J.M.; Verbist, J.F.; Biard, J.F.; Ireland, C.M. Revised structure of bistramide A (bistratene A): Application of a new program for the automated analysis of 2D INADEQUATE spectra. J. Am. Chem. Soc. 1992, 114, 1110–1111. [Google Scholar] [CrossRef]
- Johnson, W.E.B.; Watters, D.J.; Suniara, R.K.; Brown, G.; Bunce, C.M. Bistratene A induces a microtubule-dependent block in cytokinesis and altered stathmin expression in HL60 cells. Biochem. Biophys. Res. Commun. 1999, 260, 80–88. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Bai, R.; Baryza, J.L.; Verma, V.A.; Hamel, E.; Wender, P.A.; Kozmin, S.A. Actin is the primary cellular receptor of bistramide A. Nat. Chem. Biol. 2005, 1, 383–388. [Google Scholar] [CrossRef]
- Sun, H.; Low, K.E.; Woo, S.; Noble, R.L.; Graham, R.J.; Connaughton, S.S.; Gee, M.A.; Lee, L.G. Real-time protein kinase assay. Anal. Chem. 2005, 77, 2043–2049. [Google Scholar] [CrossRef]
- Yamada, K.; Ojika, M.; Ishigaki, T.; Yoshida, Y.; Ekimoto, H.; Arakawa, M. Aplyronine A, a potent antitumor substance and the congeners aplyronines B and C isolated from the sea hare Aplysia kurodai. J. Am. Chem. Soc. 1993, 115, 11020–11021. [Google Scholar] [CrossRef]
- Kita, M.; Hirayama, Y.; Yoneda, K.; Yamagishi, K.; Chinen, T.; Usui, T.; Sumiya, E.; Uesugi, M.; Kigoshi, H. Inhibition of microtubule assembly by a complex of actin and antitumor macrolide aplyronine A. J. Am. Chem. Soc. 2013, 135, 18089–18095. [Google Scholar] [CrossRef]
- Paterson, I.; Fink, S.J.; Lee, L.Y.; Atkinson, S.J.; Blakey, S.B. Total synthesis of aplyronine C. Org. Lett. 2013, 15, 3118–3121. [Google Scholar] [CrossRef]
- Lindquist, N.; Fenical, W.; Van Duyne, G.D.; Clardy, J. Isolation and structure determination of diazonamides A and B, unusual cytotoxic metabolites from the marine ascidian Diazona chinensis. J. Am. Chem. Soc. 1991, 113, 2303–2304. [Google Scholar] [CrossRef]
- Li, J.; Jeong, S.; Esser, L.; Harran, P.G. Total synthesis of nominal Diazonamides—Part 1: Convergent preparation of the structure proposed for (−)-Diazonamide A. Angew. Chem. Int. Edit. 2001, 40, 4765–4769. [Google Scholar] [CrossRef]
- Li, J.; Burgett, A.W.; Esser, L.; Amezcua, C.; Harran, P.G. Total synthesis of nominal diazonamides—Part 2: On the true structure and origin of natural isolates. Angew. Chem. Int. Edit. 2001, 40, 4770–4773. [Google Scholar] [CrossRef]
- Nicolaou, K.; Bella, M.; Chen, D.Y.; Huang, X.; Ling, T.; Snyder, S.A. Total synthesis of diazonamide A. Angew. Chem. Int. Edit. 2002, 41, 3495–3499. [Google Scholar] [CrossRef]
- Wang, G.; Shang, L.; Burgett, A.W.; Harran, P.G.; Wang, X. Diazonamide toxins reveal an unexpected function for ornithine δ-amino transferase in mitotic cell division. Proc. Natl. Acad. Sci. USA 2007, 104, 2068–2073. [Google Scholar] [CrossRef]
- Seiler, N. Ornithine aminotransferase, a potential target for the treatment of hyperammonemias. Curr. Drug Targets 2000, 1, 119–154. [Google Scholar] [CrossRef]
- Wieczorek, M.; Tcherkezian, J.; Bernier, C.; Prota, A.E.; Chaaban, S.; Rolland, Y.; Godbout, C.; Hancock, M.A.; Arezzo, J.C.; Ocal, O. The synthetic diazonamide DZ-2384 has distinct effects on microtubule curvature and dynamics without neurotoxicity. Sci. Transl. Med. 2016, 8, 365ra159. [Google Scholar] [CrossRef]
- Yun, H.; Sim, J.; An, H.; Lee, J.; Lee, H.S.; Shin, Y.K.; Paek, S.; Suh, Y. Design and synthesis of a macrosphelide A-biotin chimera. Org. Biomol. Chem. 2014, 12, 7127–7135. [Google Scholar] [CrossRef]
- Paek, S.; Seo, S.; Kim, S.; Jung, J.; Lee, Y.; Jung, J.; Suh, Y. Concise syntheses of (+)-macrosphelides A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar] [CrossRef]
- Hamann, M.T.; Scheuer, P.J. Kahalalide F: A bioactive depsipeptide from the sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Faircloth, G.; Marchante, M.d.C.C. Kahalalide F and ES285: Potent anticancer agents from marine molluscs. In Molluscs; Springer: Berlin/Heidelberg, Germany, 2006; pp. 363–379. [Google Scholar]
- Piggott, A.M.; Karuso, P. Rapid identification of a protein binding partner for the marine natural product kahalalide F by using reverse chemical proteomics. ChemBioChem 2008, 9, 524–530. [Google Scholar] [CrossRef]
- Wulff, J.E.; Siegrist, R.; Myers, A.G. The natural product avrainvillamide binds to the oncoprotein nucleophosmin. J. Am. Chem. Soc. 2007, 129, 14444–14451. [Google Scholar] [CrossRef]
- Sugie, Y.; Hirai, H.; Inagaki, T.; Ishiguro, M.; Kim, Y.; Kojima, Y.; Sakakibara, T.; Sakemi, S.; Sugiura, A.; Suzuki, Y. A new antibiotic CJ-17, 665 from Aspergillus ochraceus. J. Antibiot. 2001, 54, 911–916. [Google Scholar] [CrossRef]
- Herzon, S.B.; Myers, A.G. Enantioselective synthesis of stephacidin B. J. Am. Chem. Soc. 2005, 127, 5342–5344. [Google Scholar] [CrossRef]
- Wulff, J.E.; Herzon, S.B.; Siegrist, R.; Myers, A.G. Evidence for the rapid conversion of stephacidin B into the electrophilic monomer avrainvillamide in cell culture. J. Am. Chem. Soc. 2007, 129, 4898–4899. [Google Scholar] [CrossRef]
- Chan, W.Y.; Liu, Q.R.; Borjigin, J.; Busch, H.; Rennert, O.M.; Tease, L.A.; Chan, P.K. Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry 1989, 28, 1033–1039. [Google Scholar] [CrossRef]
- Myers, A.G.; Herzon, S.B.; Wulff, J.E.; Siegrist, R.; Svenda, J.; Zajac, M.A. Synthesis of Avrainvillamide, Stephacidin B, and Analogues Thereof. U.S. Patent 20110166170A1, 1 July 2011. [Google Scholar]
- Northcote, P.T.; Blunt, J.W.; Munro, M.H. Pateamine: A potent cytotoxin from the New Zealand marine sponge, Mycale sp. Tetrahedron Lett. 1991, 32, 6411–6414. [Google Scholar] [CrossRef]
- Romo, D.; Rzasa, R.M.; Shea, H.A.; Park, K.; Langenhan, J.M.; Sun, L.; Akhiezer, A.; Liu, J.O. Total synthesis and immunosuppressive activity of (−)-pateamine A and related compounds: Implementation of a β-lactam-based macrocyclization. J. Am. Chem. Soc. 1998, 120, 12237–12254. [Google Scholar] [CrossRef]
- Low, W.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Merrick, W.C.; Romo, D.; Liu, J.O. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol. Cell 2005, 20, 709–722. [Google Scholar] [CrossRef]
- Low, W.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Rzasa, R.M.; Shea, H.A.; Li, S.; Park, K.; Ma, G. Isolation and identification of eukaryotic initiation factor 4A as a molecular target for the marine natural product Pateamine A. Meth. Enzymol. 2007, 431, 303–324. [Google Scholar]
- Low, W.; Dang, Y.; Bhat, S.; Romo, D.; Liu, J.O. Substrate-dependent targeting of eukaryotic translation initiation factor 4A by pateamine A: Negation of domain-linker regulation of activity. Chem. Biol. 2007, 14, 715–727. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, M.W.; Song, B.R.; Chung, H.J.; Paek, S.-M. Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Mar. Drugs 2019, 17, 500. https://doi.org/10.3390/md17090500
Ha MW, Song BR, Chung HJ, Paek S-M. Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Marine Drugs. 2019; 17(9):500. https://doi.org/10.3390/md17090500
Chicago/Turabian StyleHa, Min Woo, Bo Reum Song, Hye Jin Chung, and Seung-Mann Paek. 2019. "Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products" Marine Drugs 17, no. 9: 500. https://doi.org/10.3390/md17090500
APA StyleHa, M. W., Song, B. R., Chung, H. J., & Paek, S. -M. (2019). Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Marine Drugs, 17(9), 500. https://doi.org/10.3390/md17090500