Echinochrome A Promotes Ex Vivo Expansion of Peripheral Blood-Derived CD34+ Cells, Potentially through Downregulation of ROS Production and Activation of the Src-Lyn-p110δ Pathway

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
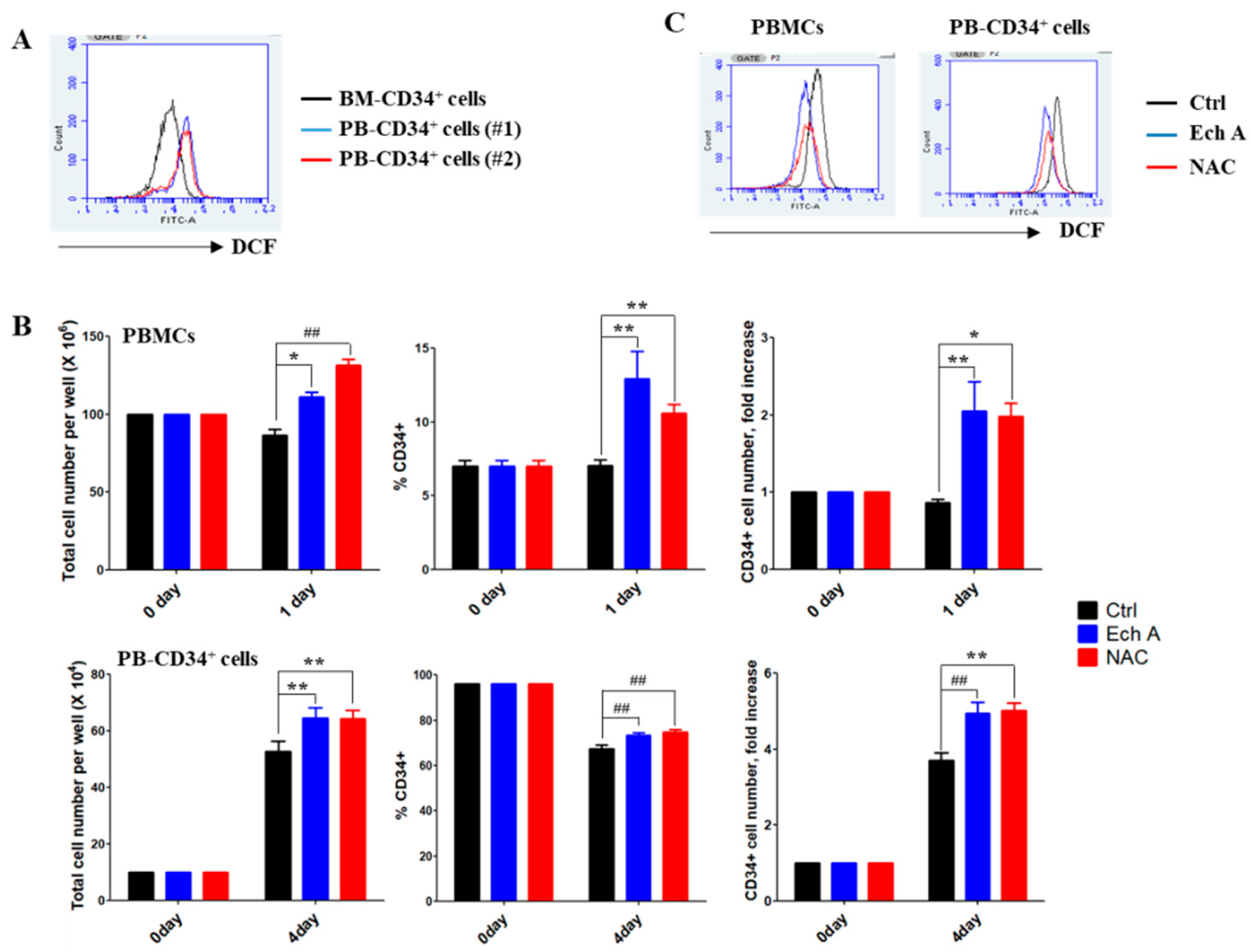
2.1. Ech A Suppresses ROS Production and Promotes Expansion of PBMC-Derived CD34+ Cells
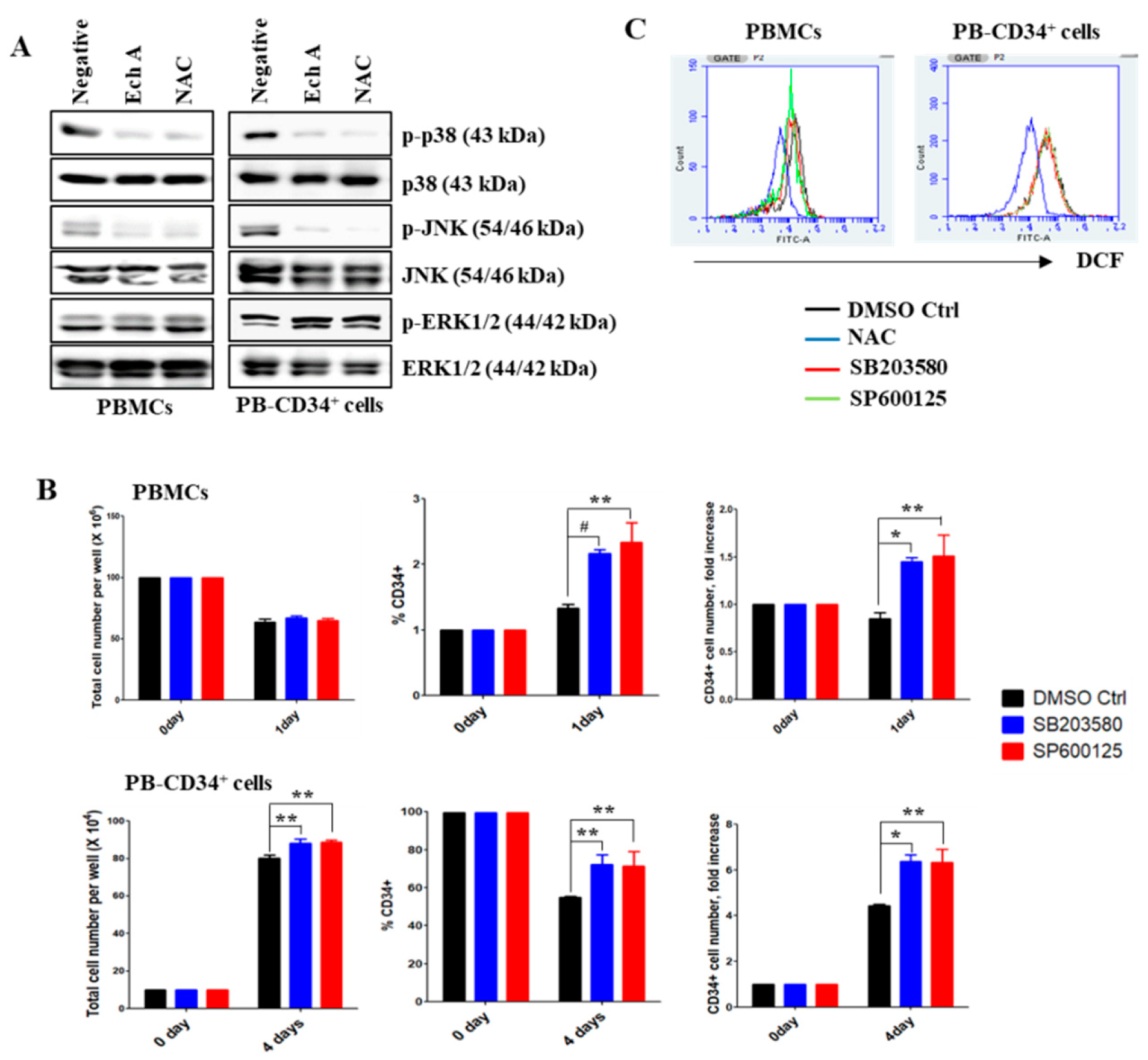
2.2. Ech A Inhibits the Activation of p38-MAPK and JNK in PB-CD34+ Cells
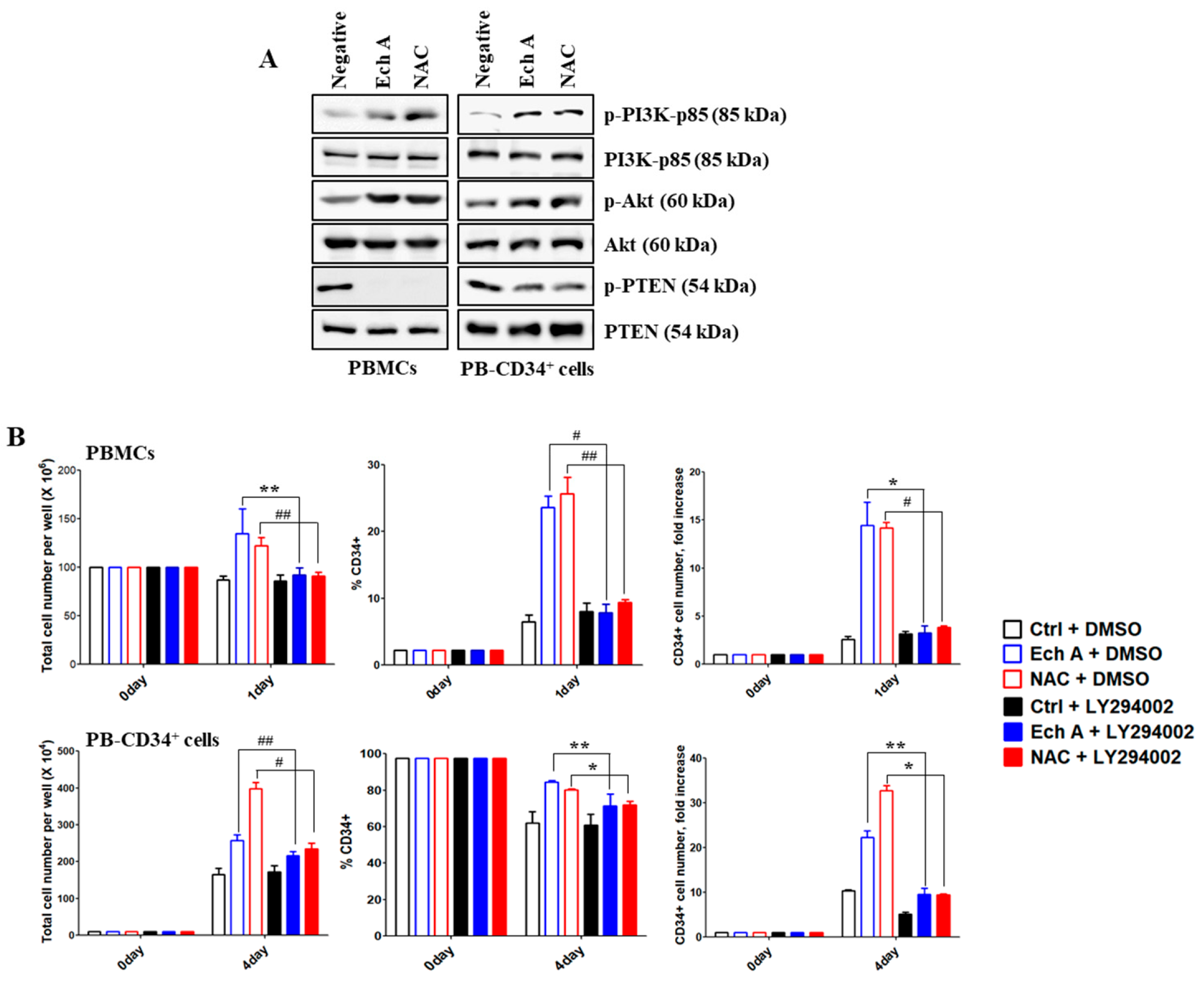
2.3. Ech A Regulates PB-CD34+ Cell Expansion via PI3K/Akt Pathway
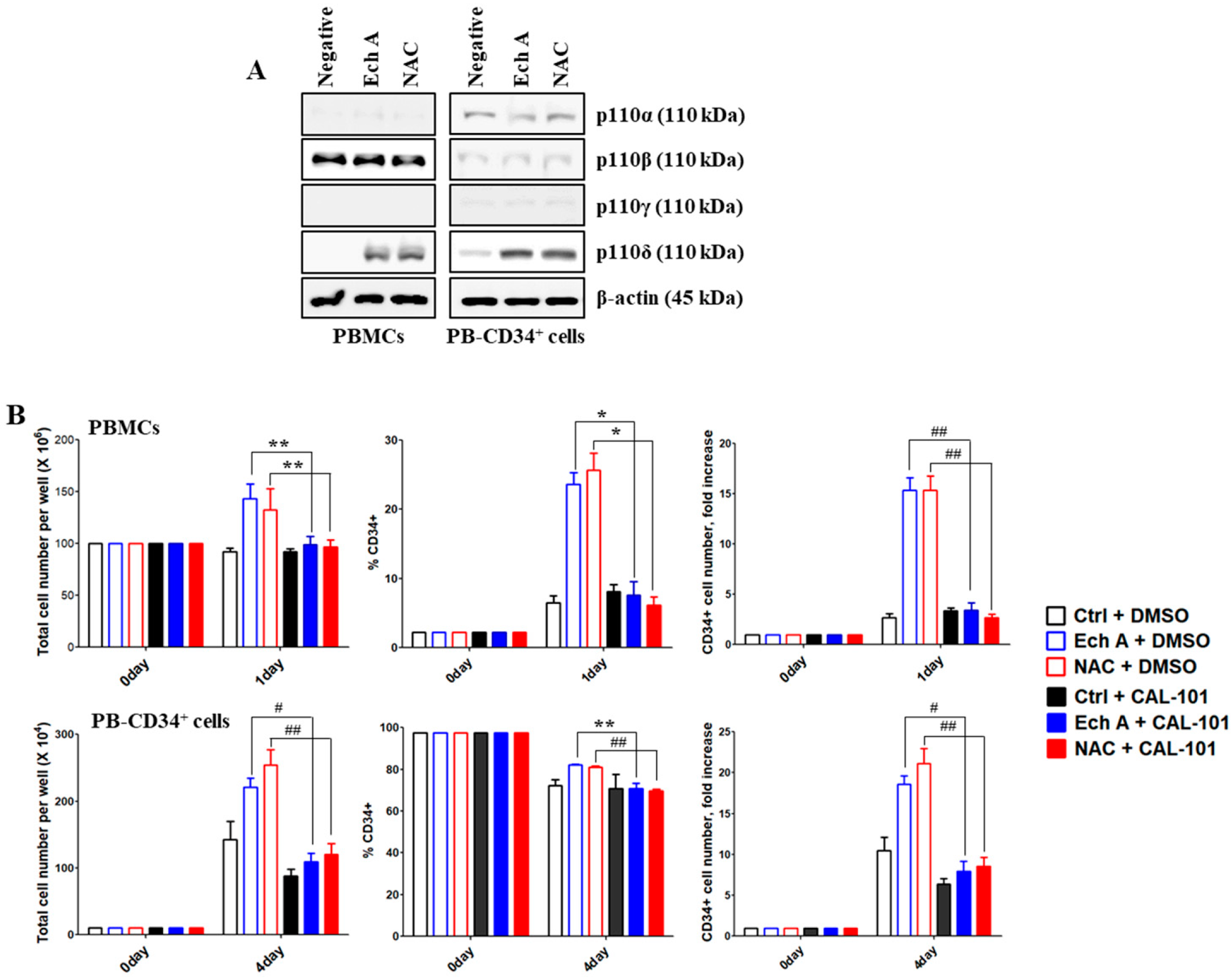
2.4. The PI3K p110δ Isoform Is Required for Ech A-Induced CD34+ Cell Expansion
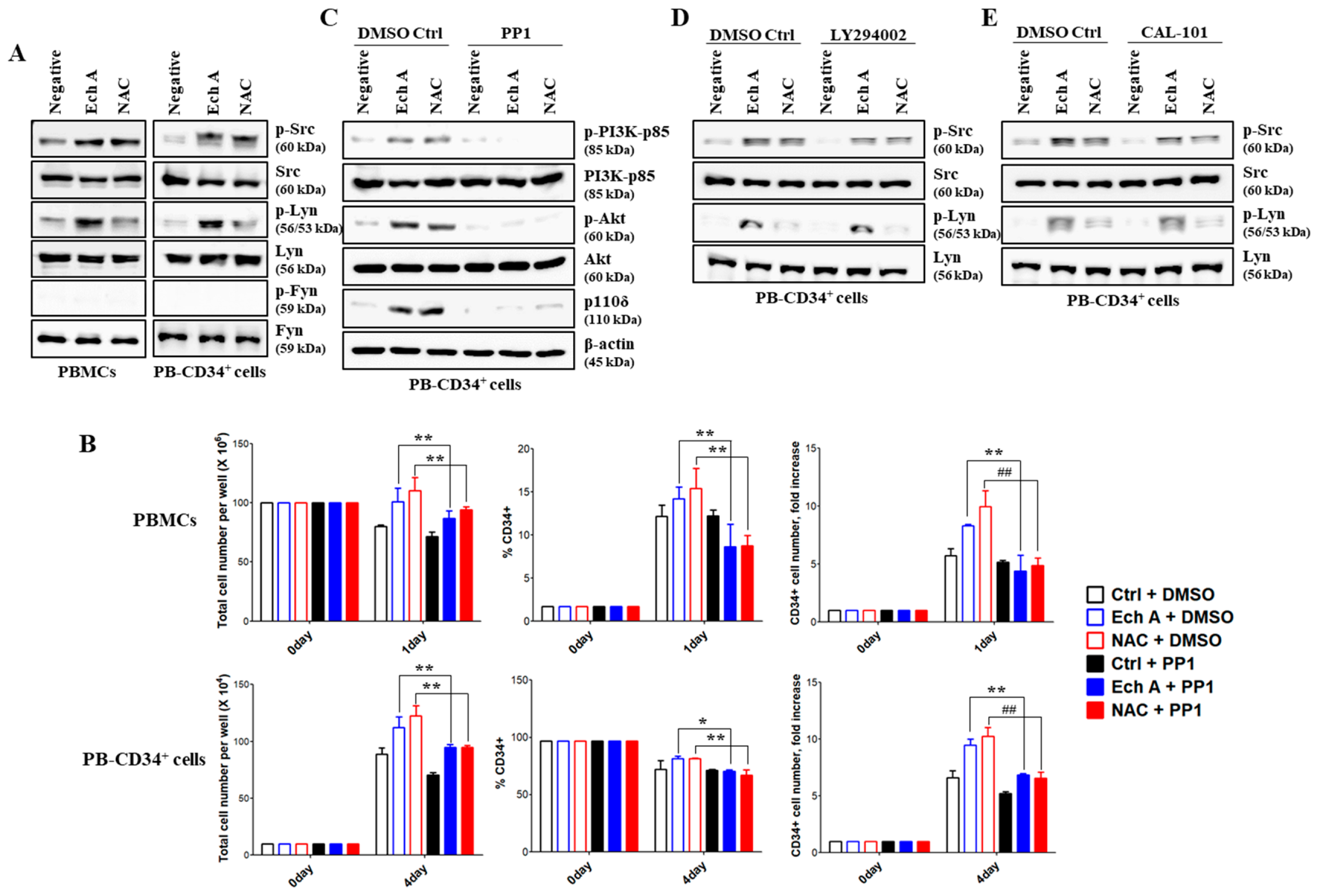
2.5. Src/Lyn Is a Major Upstream Signal for Ech A-Induced p110δ-Mediated CD34+ Cell Expansion
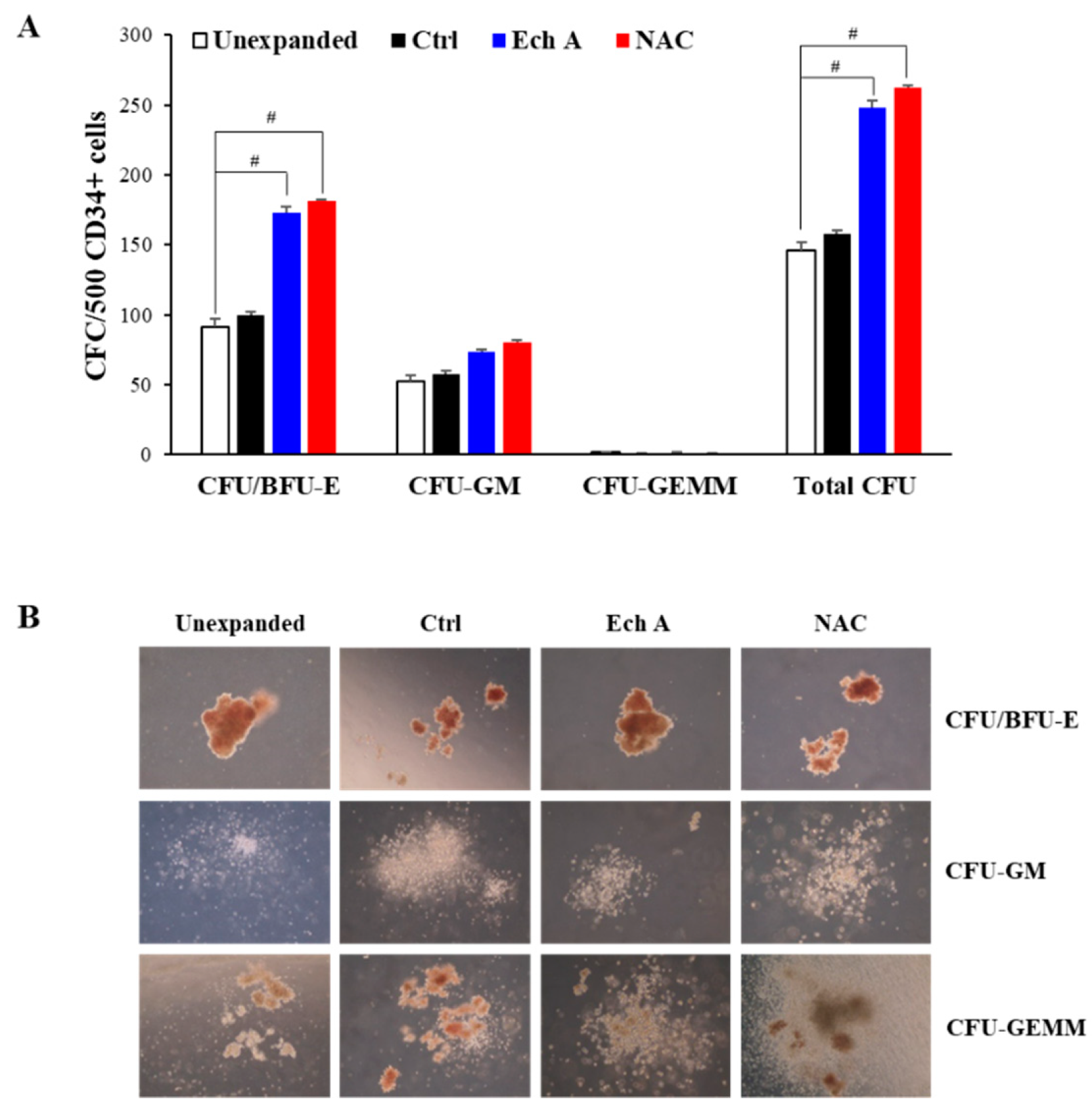
2.6. Ex Vivo Expanded CD34+ Cells Maintain Their Colony-Forming Capacity
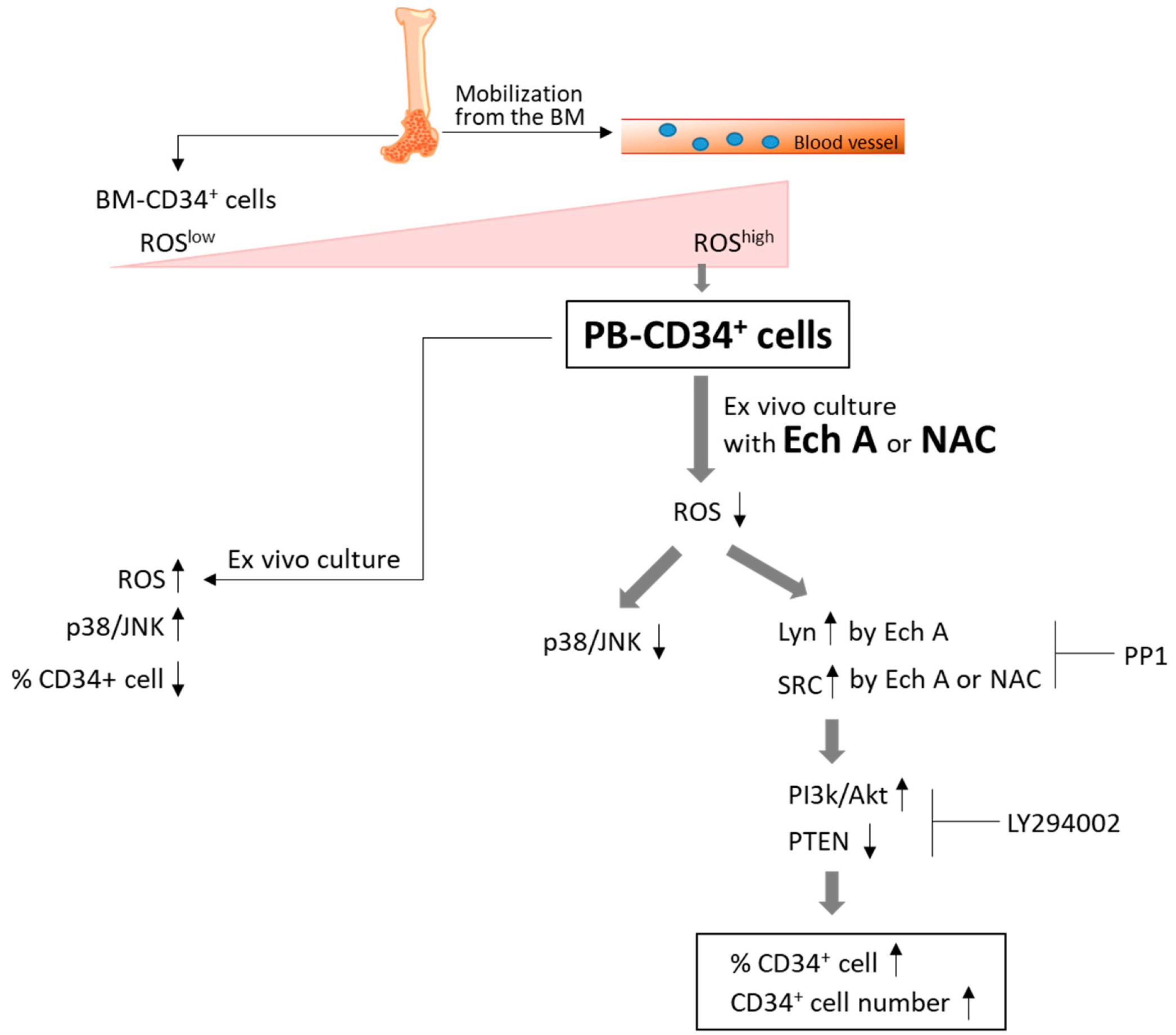
3. Discussion
4. Materials and Methods
4.1. Isolation of PBMCs and Purification of PB-CD34+ Cells
4.2. Chemicals and Reagents
4.3. Immunophenotypic Analysis by Flow Cytometry
4.4. Determination of Intracellular ROS Production
4.5. Immunoblotting
4.6. Colony-Forming Cell (CFC) Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gratwohl, A.; Baldomero, H.; Schwendener, A.; Rocha, V.; Apperley, J.; Frauendorfer, K.; Niederwieser, D. The EBMT activity survey 2007 with focus on allogeneic HSCT for AML and novel cellular therapies. Bone Marrow Transpl. 2009, 43, 257–291. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, C.M. Hematopoietic stem cells for transplantation. Nat. Immunol. 2002, 3, 314–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, A.; Lee, S.; Zhu, A.; Pasquini, M. Current use and trends in hematopoietic cell transplantation in the united states. Biol. Blood Marrow Transpl. 2017, 23, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Kallinikou, K.; Anjos-Afonso, F.; Blundell, M.P.; Ings, S.J.; Watts, M.J.; Thrasher, A.J.; Limch, D.C.; Bonnet, D.; Yong, K.L. Engraftment defect of cytokine-cultured adult human mobilized CD34(+) cells is related to reduced adhesion to bone marrow niche elements. Br. J. Haematol. 2012, 158, 778–787. [Google Scholar] [CrossRef]
- Kobayashi, C.I.; Suda, T. Regulation of reactive oxygen species in stem cells and cancer stem cell. J. Cell Physiol. 2012, 227, 421–430. [Google Scholar] [CrossRef]
- Kim, S.M.; Hwang, K.A.; Choi, K.C. Potential roles of reactive oxygen species derived from chemical substances involved in cancer development in the female reproductive system. BMB Rep. 2018, 51, 557–562. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3058–3063. [Google Scholar] [CrossRef]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef]
- Ergen, A.V.; Goodell, M.A. Mechanisms of hematopoietic stem cell aging. Exp. Gerontol. 2010, 45, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Tasdogan, A.; Kumar, S.; Allies, G.; Bausinger, J.; Beckel, F.; Hofemeister, H.; Mulaw, M.; Madan, V.; Scharffetter-Kochanek, K.; Feuring-Buske, M.; et al. DNA damage-induced HSPC malfunction depends on ROS accumulation downstream of IFN-1 signaling and bid mobilization. Cell Stem Cell 2016, 19, 752–767. [Google Scholar] [PubMed]
- Pervaiz, S.; Taneja, R.; Ghaffari, S. Oxidative stress regulation of stem and progenitor cells. Antioxid. Redox Signal. 2009, 11, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Cai, H.; Li, Q.; Du, Z.; Tan, W. The effects of ROS-mediating oxygen tension on human CD34(+)CD38(−) cells induced into mature dendritic cells. J. Biotechnol. 2012, 158, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Jang, H.K.; Choi, Y.J.; Eum, W.S.; Park, J.; Choi, S.Y.; Kwon, H.Y. PEP-1-paraoxonase 1 fusion protein prevents cytokine-induced cell destruction and impaired insulin secretion in rat insulinoma cells. BMB Rep. 2018, 51, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.Y.; Zyriyantey, A.H.; Taib, I.S.; Tan, H.Y.; Muhd, K.A.W.H.; Chow, P.W. Effects of n-acetyl-cysteine supplementation on ex-vivo clonogenicity and oxdative profile of lineage-committed hematopoietic stem/progenitor cells. J. Teknologi. 2018, 80, 1–8. [Google Scholar]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yu, D. Targeting Src family kinases in anti-cancer therapies: Turning promise into triumph. Trends Pharmacol. Sci. 2012, 33, 122–128. [Google Scholar] [CrossRef]
- Lowell, C.A. Src-family kinases: Rheostats of immune cell signaling. Mol. Immunol. 2004, 41, 631–643. [Google Scholar] [CrossRef]
- Yamanashi, Y.; Mori, S.; Yoshida, M.; Kishinoto, T.; Yamamoto, T.; Toyoshima, K. Selective expression of a protein-tyrosine kinase, p56lyn, in hematopoietic cells and association with production of human T-cell lymphotropic virus type I. Proc. Natl Acad. Sci. USA 1989, 86, 6538–6542. [Google Scholar] [CrossRef]
- Hibbs, M.L.; Tarlinton, D.M.; Armes, J.; Grail, D.; Hodgson, G.; Maglitto, R.; Stacker, S.A.; Dunn, A.R. Multiple defects in the immune system of Lun-deficient mice, culminating in autoimmune disease. Cell 1995, 83, 301–311. [Google Scholar] [CrossRef]
- Scapini, P.; Pereira, S.; Zhang, H.; Lowell, C.A. Multiple roles of Lyn kinase in myeloid cell signaling and function. Immunol. Rev. 2009, 228, 23–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilbrook, P.A.; Ingley, E.; Williams, J.H.; Hibbs, M.L.; Klinken, S.P. Lyn tyrosine kinase is essential for erythropoietin-induced differentiation of J2E erythroid cells. EMBO J. 1997, 16, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Linnekin, D.; DeBerry, C.S.; Mou, S. Lyn associates with the juxtamembrane region of c-kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J. Biol. Chem. 1997, 272, 27450–27455. [Google Scholar] [CrossRef] [PubMed]
- Ingley, E.; Sarna, M.K.; Beaumont, J.G.; Tilbrook, P.A.; Tsai, S.; Takemoto, Y.; Williams, J.H.; Klinken, S.P. HS1 interacts with Lyn and is critical for erythropoietin-induced differentiation of erythroid cells. J. Biol. Chem. 2000, 275, 7887–7893. [Google Scholar] [CrossRef] [PubMed]
- Chin, H.; Arai, A.; Wakao, H.; Kamiyama, R.; Miyasaka, N.; Miura, O. Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood 1998, 91, 3734–3745. [Google Scholar] [PubMed]
- Dos Santos, C.; Demur, C.; Bardet, V.; Prade-Houdellier, N.; Payrastre, B.; Recher, C. A critical role for Lyn in acute myeloid leukemia. Blood 2008, 111, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Ptasznik, A.; Nakata, Y.; Kalota, A.; Emerson, S.G.; Gewirtz, A.M. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat. Med. 2004, 10, 1187–1189. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D. PI3K catalytic isoform alteration promotes the LIMK1-related metastasis through the PAK1 or ROCK1/2 activation in cigarette smoke-exposed ovarian cancer cells. Anticancer Res. 2017, 37, 1805–1818. [Google Scholar]
- Park, G.B.; Kim, D. Insulin-like growth factor-1 activates different catalytic subunits p110 of PI3K in a cell-type-dependent manner to induce lipogenesis-dependent epithelial-mesenchymal transition through the regulation of ADAM10 and ADAM17. Mol. Cell Biochem. 2018, 439, 199–211. [Google Scholar] [CrossRef]
- Anderson, H.A.; Mathieson, J.W.; Thomson, R.H. Distribution of spinochrome pigments in echinoids. Comp. Biochem. Physiol. 1969, 28, 333–345. [Google Scholar] [CrossRef]
- Pozharitskaya, O.N.; Shikov, A.N.; Laakso, I.; Seppänen-Laakso, T.; Makarenko, I.E.; Faustova, N.M.; Makarova, M.N.; Makarov, V.G. Bioactivity and chemical characterization of gonads of green sea urchin Strongylocentrotus droebachiensis from Barents Sea. J. Funct. Foods 2015, 17, 227–234. [Google Scholar] [CrossRef]
- Fedoreyev, S.A.; Krylova, N.V.; Mishchenko, N.P.; Vasileva, E.A.; Pislyagin, E.A.; Lunikhina, O.V.; Lavrov, V.F.; Svitich, O.A.; Ebralidze, L.K.; Leonova, G.N. Antiviral and antioxidant properties of Echinochrome A. Mar. Drugs 2018, 16, 509. [Google Scholar] [CrossRef] [PubMed]
- Ultkina, N.K.; Pokhilo, N.D. Free radical scavenging activities of naturally occurring and synthetic analogues of sea urchin naphthazarin pigments. Nat. Prod. Commun. 2012, 7, 901–904. [Google Scholar]
- Lebed’ko, O.A.; Ryzhavskii, B.Y.; Demidova, O.V. Effect of antioxidant Echinochrome A on bleomycin-induced pulmonary fibrosis. Bull. Exp. Biol. Med. 2015, 159, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.V.; Ivanova, M.V.; Levitsky, D.O. Iron chelators and free radical scavengers in naturally occurring polyhydroxylated 1,4-naphthoquinones. Hemoglobin 2008, 32, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Abdul Hamid, Z.; Lin Lin, W.H.; Abdalla, B.J.; Bee Yuen, O.; Latif, E.S.; Mohamed, J.; Rajab, N.F.; Paik Wah, C.; Wak Harto, M.K.A.; Budin, S.B. The role of Hibiscus sabdariffa L. (Roselle) in maintenance of ex vivo murine bone marrow-derived hematopoietic stem cells. Sci. World J. 2014, 1–10. [Google Scholar] [CrossRef]
- Liu, A.M.; Qu, W.W.; Liu, X.; Qu, C.K. Chromosomal instability in in vitro cultured mouse hematopoietic cells associated with oxidative stress. Am. J. Blood Res. 2012, 2, 71–76. [Google Scholar]
- Rausch, O.; Marshall, C.J. Cooperation of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways during granulocyte colony-stimulating factor-induced hemopoietic cell proliferation. J. Biol. Chem. 1999, 274, 4096–4105. [Google Scholar] [CrossRef]
- Katsoulidis, E.; Li, Y.; Yoon, P.; Sassano, A.; Altman, J.; Kannan-Thulasiraman, P.; Balasubramanian, L.; Parmar, S.; Varga, J.; Tallman, M.S.; et al. Role of the p38 mitogen-activated protein kinase pathway in cytokine-mediated hematopoietic suppression in myelodysplastic syndromes. Cancer Res. 2005, 65, 9029–9037. [Google Scholar] [CrossRef]
- Verma, A.; Deb, D.K.; Sassano, A.; Kambhampati, S.; Wickrema, A.; Uddin, S.; Mohindru, M.; Van Besien, K.; Platanias, L.C. Cutting edge: Activation of the p38 mitogen-activated protein kinase signaling pathway mediates cytokine-induced hemopoietic suppression in aplastic anemia. J. Immunol. 2002, 168, 5984–5988. [Google Scholar] [CrossRef]
- Miyamato, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. FOXO3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; Mauch, P.; Vergilio, J.A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Du, Z.; Cai, H.; Ye, Z.; Fan, J.; Tan, W.S. Low oxygen tension favored expansion and hematopoietic reconstitution of CD34(+) CD38(−) cells expanded from human cord blood-derived CD34(+) cells. Biotechonol. J. 2016, 11, 945–953. [Google Scholar] [CrossRef]
- Shao, L.; Li, H.; Pazhanisamy, S.K.; Meng, A.; Wang, Y.; Zhou, D. Reactive oxygen species and hematopoietic stem cell senescence. Int. J. Hematol. 2011, 94, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geest, C.R.; Coffer, P.J. MAPK signaling pathways in the regulation of hematopoiesis. J. Luekoc. Biol. 2009, 86, 237–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinge, A.; Xu, J.; Javier, J.; Mose, E.; Kumar, S.; Kapur, R.; Srour, E.F.; Malik, P.; Aronow, B.J.; Filippi, M.D. p190-B RhoGAP and intracellular cytokine signals balance hematopoietic stem and progenitor cell self-renewal and differentiation. Nat. Commun. 2017, 8, 14382. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P.; Montgomery, C.; Geske, R.; Bradley, A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 1991, 64, 693–702. [Google Scholar] [CrossRef]
- Harder, K.W.; Parsons, L.M.; Armes, J.; Evans, N.; Kountouri, N.; Clark, R.; Quilici, C.; Grail, D.; Hodgson, G.S.; Dunn, A.R.; et al. Gain-and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity 2001, 15, 603–615. [Google Scholar] [CrossRef]
- Debuire, B.; Henry, C.; Bernissa, M.; Bierte, G.; Claverie, J.M.; Saule, S.; Martin, P.; Stehelin, D. Sequencing the erbA gene of avian erythroblastosis virus reveals a new type of oncogene. Science 1984, 224, 1456–1459. [Google Scholar] [CrossRef]
- Satterthwaite, A.B.; Lowell, C.A.; Khan, W.N.; Sideras, P.; Alt, F.W.; Witte, O.N. Independent and opposing roles for Btk and lyn in B and myeloid signaling pathways. J. Exp. Med. 1998, 188, 833–844. [Google Scholar] [CrossRef]
- Karur, V.G.; Lowell, C.A.; Besmer, P.; Aqosti, V.; Wojchowski, D.M. Lyn kinase promotes erythroblast expansion and late-stage development. Blood 2006, 108, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; Buitenhuis, M. The PI3K/PKB signaling module as key regulator of hematopoiesis: Implications for therapeutic strategies in leukemia. Blood 2012, 119, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, G.-B.; Kim, M.-J.; Vasileva, E.A.; Mishchenko, N.P.; Fedoreyev, S.A.; Stonik, V.A.; Han, J.; Lee, H.S.; Kim, D.; Jeong, J.-Y. Echinochrome A Promotes Ex Vivo Expansion of Peripheral Blood-Derived CD34+ Cells, Potentially through Downregulation of ROS Production and Activation of the Src-Lyn-p110δ Pathway. Mar. Drugs 2019, 17, 526. https://doi.org/10.3390/md17090526
Park G-B, Kim M-J, Vasileva EA, Mishchenko NP, Fedoreyev SA, Stonik VA, Han J, Lee HS, Kim D, Jeong J-Y. Echinochrome A Promotes Ex Vivo Expansion of Peripheral Blood-Derived CD34+ Cells, Potentially through Downregulation of ROS Production and Activation of the Src-Lyn-p110δ Pathway. Marine Drugs. 2019; 17(9):526. https://doi.org/10.3390/md17090526
Chicago/Turabian StylePark, Ga-Bin, Min-Jung Kim, Elena A. Vasileva, Natalia P. Mishchenko, Sergey A. Fedoreyev, Valentin A. Stonik, Jin Han, Ho Sup Lee, Daejin Kim, and Jee-Yeong Jeong. 2019. "Echinochrome A Promotes Ex Vivo Expansion of Peripheral Blood-Derived CD34+ Cells, Potentially through Downregulation of ROS Production and Activation of the Src-Lyn-p110δ Pathway" Marine Drugs 17, no. 9: 526. https://doi.org/10.3390/md17090526
APA StylePark, G. -B., Kim, M. -J., Vasileva, E. A., Mishchenko, N. P., Fedoreyev, S. A., Stonik, V. A., Han, J., Lee, H. S., Kim, D., & Jeong, J. -Y. (2019). Echinochrome A Promotes Ex Vivo Expansion of Peripheral Blood-Derived CD34+ Cells, Potentially through Downregulation of ROS Production and Activation of the Src-Lyn-p110δ Pathway. Marine Drugs, 17(9), 526. https://doi.org/10.3390/md17090526