New Ophiobolins from the Deep-Sea Derived Fungus Aspergillus sp. WHU0154 and Their Anti-Inflammatory Effects


Abstract
:
1. Introduction
2. Results
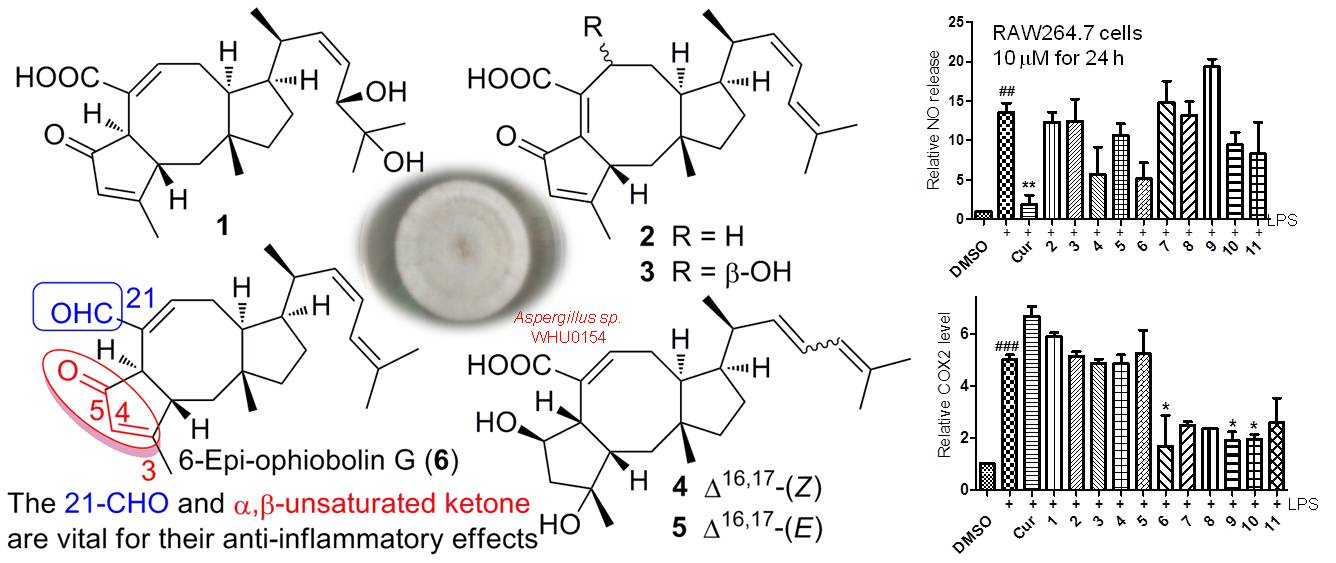
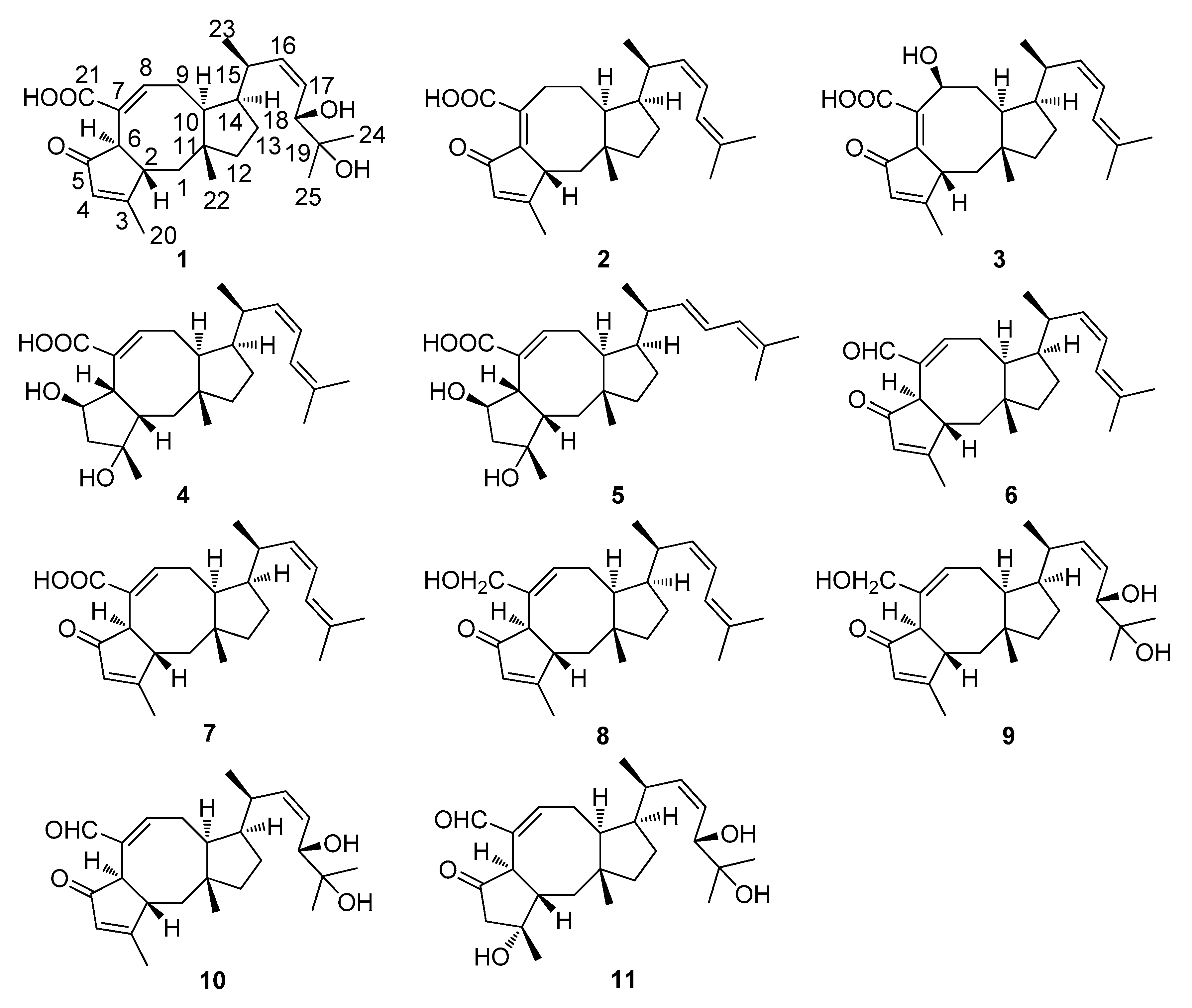
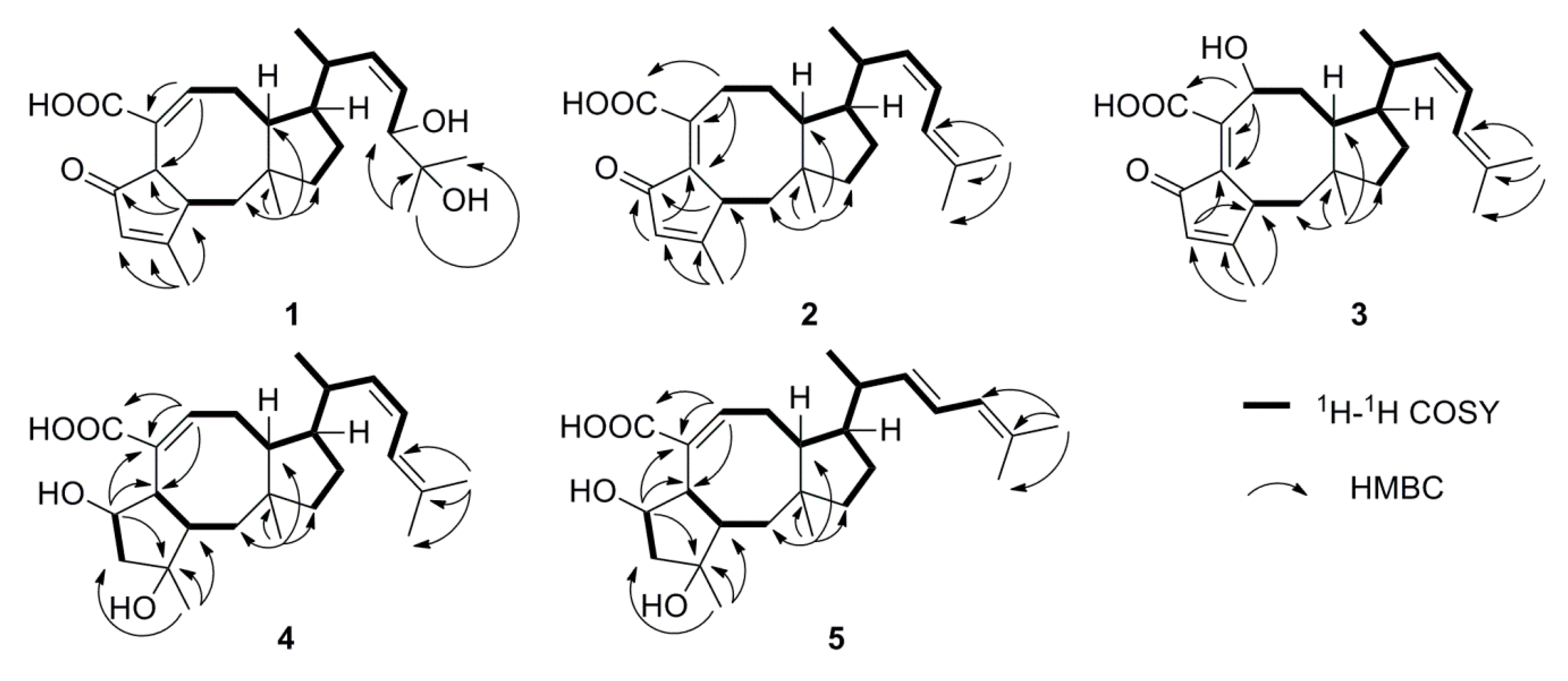
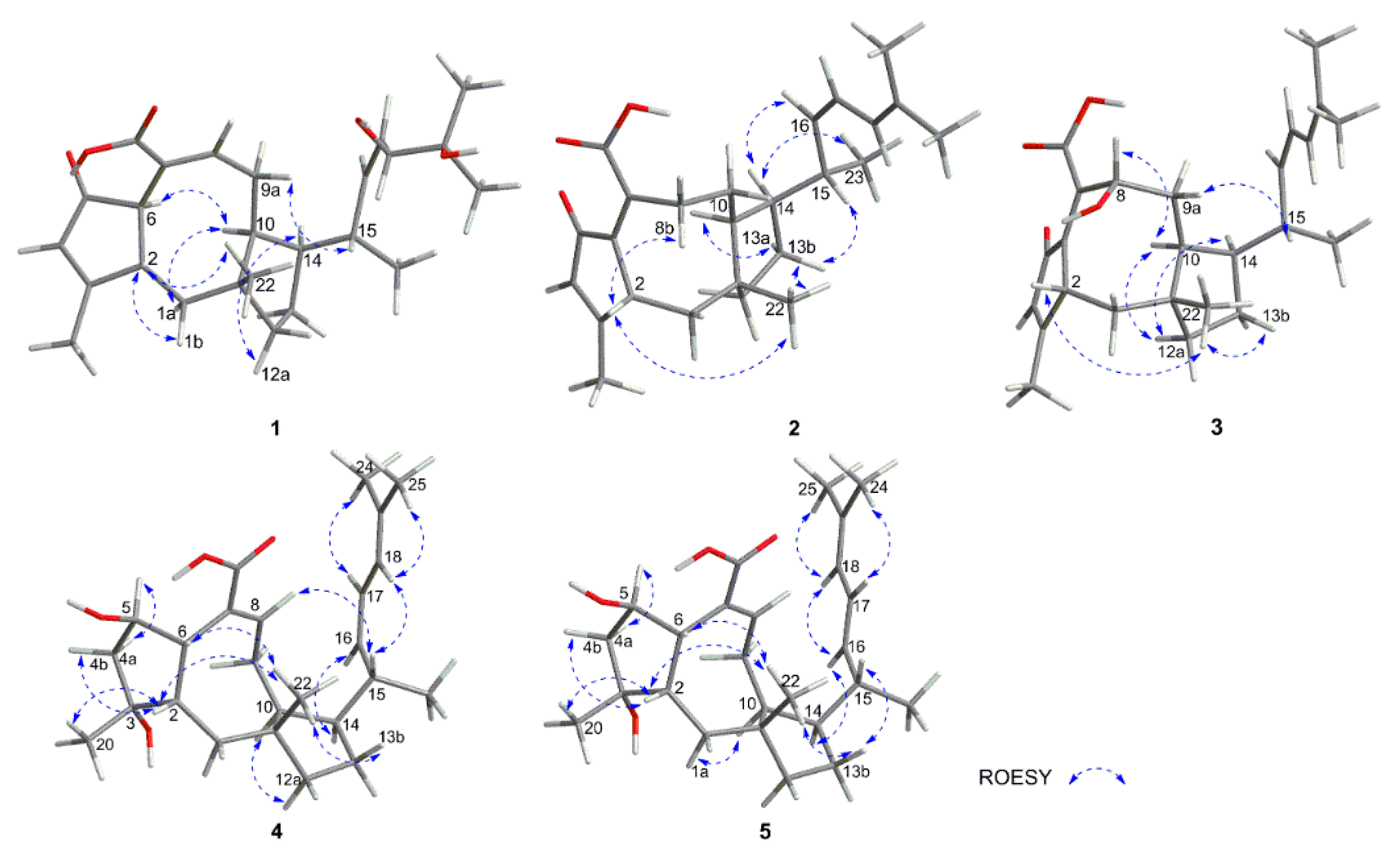
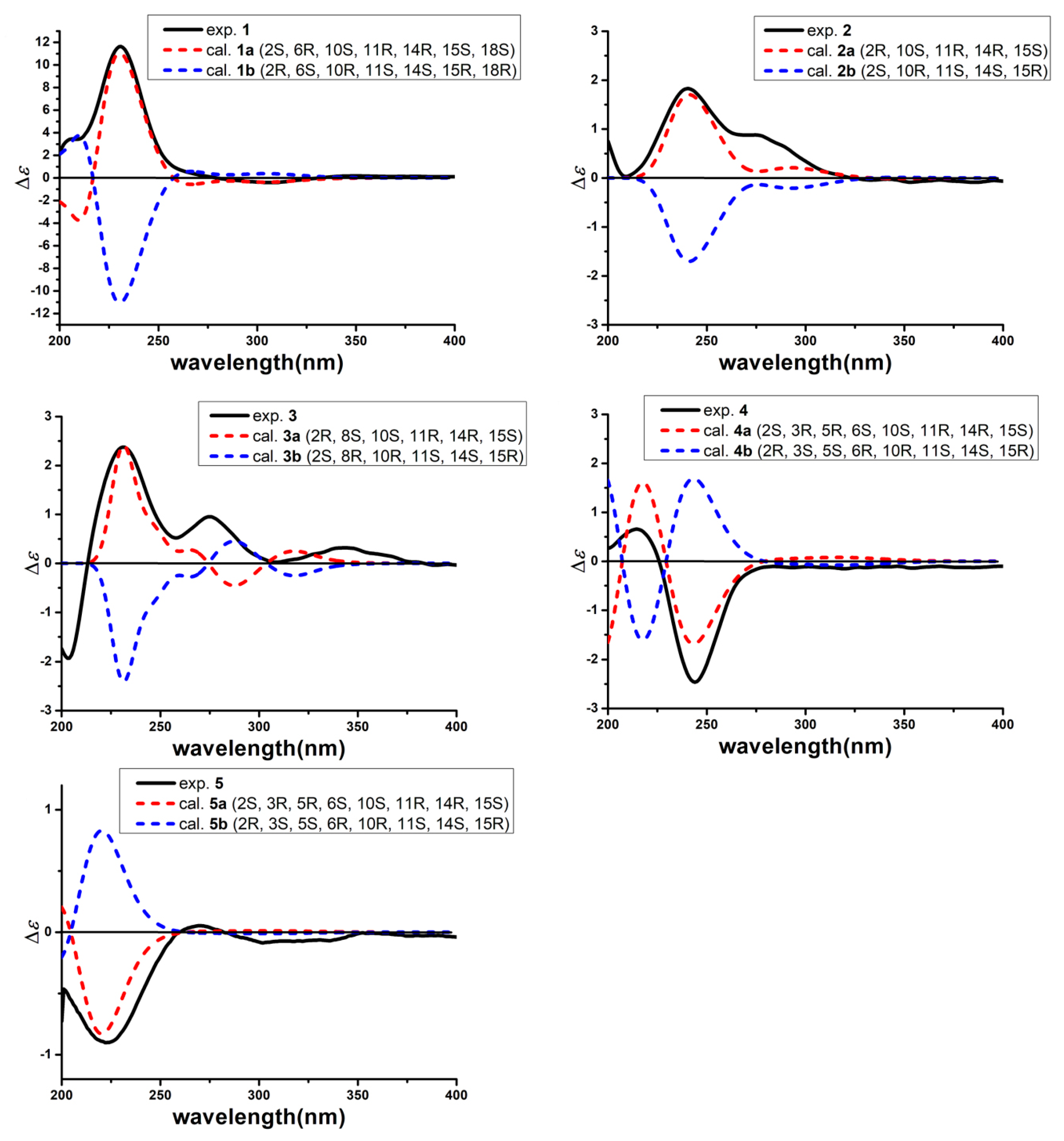
2.1. Structure Elucidation
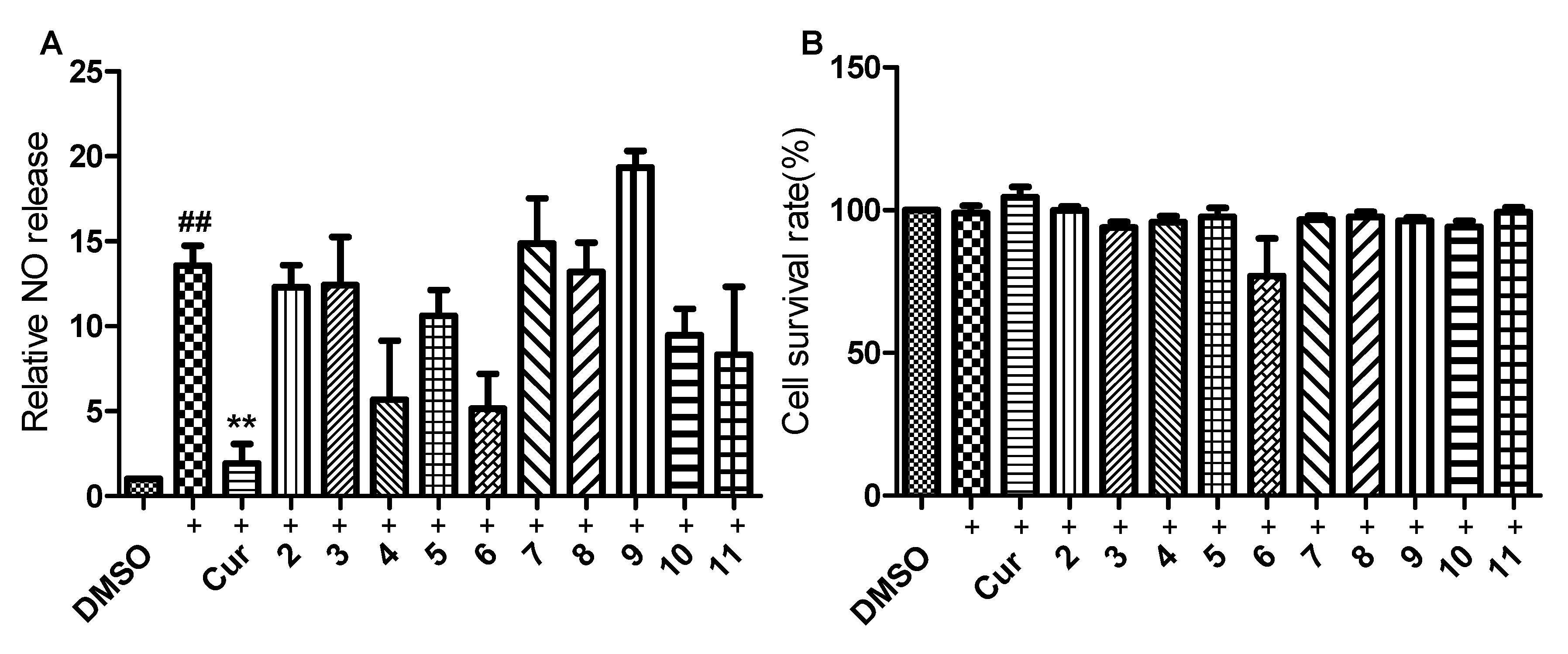
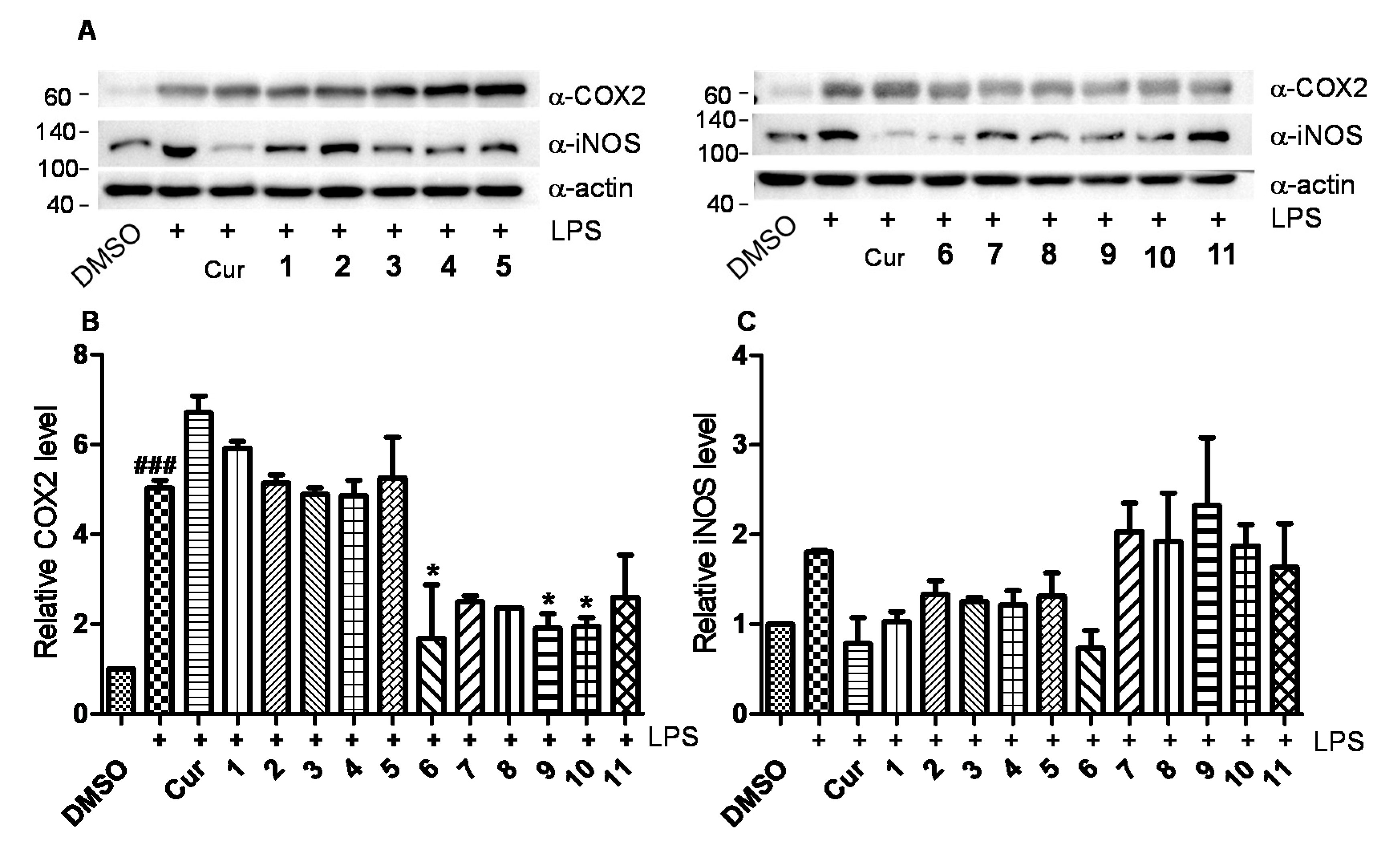
2.2. Anti-Inflammatory Bioactivities
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedure
4.2. Fungal Material and Identification
4.3. Cultivation, Extraction and Isolation
4.4. Extraction and Isolation
4.5. Quantum Chemical ECD Calculations of 1–5
4.6. Cell Culture
4.7. Measurement of Cell Viability
4.8. NO Assay
4.9. Western Blot
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arifeen, M.Z.; Ma, Y.N.; Xue, Y.R.; Liu, C.H. Deep-sea fungi could be the new arsenal for bioactive molecules. Mar. Drugs 2020, 18, 9. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.X.; Shah, M.; Zhang, Z.Z.; Feng, Y.Y.; Chang, Y.M.; Che, Q.; Gu, Q.Q.; Zhu, T.J.; Zhang, G.J.; Li, D.H. Secondary metabolites from deep-sea derived microorganisms. Curr. Med. Chem. 2019, 27, 6244–6273. [Google Scholar] [CrossRef]
- Skropeta, D.; Wei, L.Q. Recent advances in deep-sea natural products. Nat. Prod. Rep. 2014, 31, 999–1025. [Google Scholar] [CrossRef] [PubMed]
- Daletos, G.; Ebrahim, W.; Ancheeva, E.; El-Neketi, M.; Song, W.G.; Lin, W.H.; Proksch, P. Natural products from deep-sea-derived fungi-a new source of novel bioactive compounds? Curr. Med. Chem. 2018, 25, 186–207. [Google Scholar] [CrossRef] [PubMed]
- Ogaki, M.B.; Coelho, L.C.; Vieira, R.; Neto, A.A.; Zani, C.L.; Alves, T.M.A.; Junior, P.A.S.; Murta, S.M.F.; Barbosa, E.C.; Oliveira, J.G.; et al. Cultivable fungi present in deep-sea sediments of Antarctica: Taxonomy, diversity, and bioprospecting of bioactive compounds. Extremophiles 2020, 24, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Au, T.K.; Chick, W.S.H.; Leung, P.C. The biology of ophiobolins. Life Sci. 2000, 67, 733–742. [Google Scholar] [CrossRef]
- Hong, W.; Takuya, I.; Masahiro, K.; Yasuhide, N.; Mineko, K.; Motomasa, K. Cytotoxic sesterterpenes, 6-epi-ophiobolin G and 6-epi-ophiobolin N, from marine derived fungus Emercella variecolor GF10. Tetrahedron 2004, 60, 6015–6019. [Google Scholar]
- Liu, H.B.; Edrada-Ebel, R.; Ebel, R.; Wang, Y.; Schulz, B.; Draeger, S.; Muller, W.E.G.; Wray, V.; Lin, W.H.; Proksch, P. Ophiobolin Sesterterpenoids and Pyrrolidine Alkaloids from the Sponge-Derived Fungus Aspergillus ustus. Helv. Chim. Acta 2011, 94, 623–631. [Google Scholar] [CrossRef]
- Bladt, T.J.; Durr, C.; Knudsen, P.B.; Kildgaard, S.; Frisvad, J.C.; Gotfredsen, C.H.; Seiffert, M.; Larsen, T.O. Bio-activity and dereplication-based discovery of ophiobolins and other fungal secondary metabolites targeting leukemia cells. Molecules 2013, 18, 14629–14650. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.H.; Lu, Z.Y.; Fan, J.; Wang, L.P.; Zhu, G.L.; Wang, Y.; Li, X.; Hong, K.; Piyachaturawat, P.; Chairoungdua, A.; et al. Ophiobolins from the mangrove fungus Aspergillus ustus. J. Nat. Prod. 2018, 81, 2–9. [Google Scholar] [CrossRef]
- Choi, B.K.; Trinh, P.T.H.; Lee, H.S.; Choi, B.W.; Kang, J.S.; Ngoc, N.T.D.; Van, T.T.T.; Shin, H.J. New ophiobolin derivatives from the marine fungus Aspergillus flocculosus and their cytotoxicities against cancer cells. Mar. Drugs 2019, 17, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, R.L.; Jiang, H.M.; Mo, Y.L.; Guo, H.X.; Li, C.Y.; Long, Y.H.; Zang, Z.M.; She, Z.G. Ophiobolin-type sesterterpenoids from the mangrove endophytic fungus Aspergillus sp. ZJ-68. J. Nat. Prod. 2019, 82, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, C.X.; Lu, J.; Wu, J.; Li, C.H.; Hu, Z.Y.; Tian, W.; Yang, L.; Xiang, J.; Zhou, H.B.; et al. Sesterterpene MHO7 suppresses breast cancer cells as a novel estrogen receptor degrader. Pharmacol. Res. 2019, 146, 104294. [Google Scholar] [CrossRef] [PubMed]
- Li, E.G.; Clark, A.M.; Rotella, D.P.; Hufford, C.D. Microbial metabolites of ophiobolin A and antimicrobial evaluation of ophiobolins. J. Nat. Prod. 1995, 58, 74–81. [Google Scholar] [CrossRef]
- Leung, P.C.; Taylor, W.A.; Wang, J.H.; Tipton, C.L. Role of calmodulin inhibition in the mode of action of ophiobolin A. Plant Physiol. 1985, 77, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Brill, Z.G.; Grover, H.K.; Maimone, T.J. Enantioselective synthesis of an ophiobolin sesterterpene via a programmed radical cascade. Science 2016, 352, 1078–1082. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.P.; Liu, Q.; Gao, H.; Lin, H.P.; Tian, H.Y.; Hong, K.; Li, J.; Jiang, R.W.; Yao, X.S.; Tang, J.S. Streptospirodienoic acids A and B, 6,6-spiroketal polyketides from Streptomyces sp. RSC Adv. 2014, 4, 63324–63327. [Google Scholar] [CrossRef]
- Gou, X.S.; Jia, J.; Xue, Y.X.; Ding, W.J.; Dong, Z.T.; Tian, D.M.; Chen, M.; Bi, H.K.; Hong, K.; Tang, J.S. New pyrones and their analogs from the marine mangrove-derived Aspergillus sp. DM94 with antibacterial activity against Helicobacter pylori. Appl. Microbiol. Biotechnol. 2020, 104, 7971–7978. [Google Scholar] [CrossRef]
- Bari, L.D.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1. Snatzke’s method revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef]
- Liu, X.H.; Miao, F.P.; Qiao, M.F.; Cichewicz, R.H.; Ji, N.Y. Terretonin, ophiobolin, and drimane terpenes with absolute configurations from an algiconus Aspergillus ustus. RSC Adv. 2013, 3, 588–595. [Google Scholar] [CrossRef]
- Masayoshi, A.; Hiroki, N.; Motomasa, K. Marine-derived fungal sesterpenes, ophiobolins, inhibit biofilm formation of Mycobacterium species. J. Nat. Med. 2013, 67, 271–275. [Google Scholar]
- Xiao, J.Z.; Tsuda, M.; Doke, N.; Nishimura, S. Phytotoxins produced by germinating spores of Bipolaris oryzae. Phytopathology 1991, 81, 58–64. [Google Scholar] [CrossRef]
- Antonio, E.; Anna, A.; Alessio, C.; Maurizio, V.; Mariano, F.; Raghavan, C.; Andrea, M. Ophiobolin E and 8-epi-ophiobolin J produced by Drechslera gigantean, a potential mycoherbicide of weedy grasses. Phytochemistry 2006, 67, 2281–2287. [Google Scholar]
- Zhang, J.; Zhao, S.S.; Xie, J.; Yang, J.; Chen, G.D.; Hu, D.; Zhang, W.G.; Wang, C.X.; Yao, X.S.; Gao, H. N-methoxy-β-carboline alkaloids with inhibitory activities against Aβ42 aggregation and acetylcholinesterase from the stems of Picrasma quassioides. Bioorg. Chem. 2020, 101, 104043. [Google Scholar] [CrossRef] [PubMed]
- Torsten, B.; Anu, S.; Yasmin, H.; Gerhard, B. SpecDics: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar]
- Pósa, A.; Szabó, R.; Szalai, Z.; Kupai, K.; Deim, Z.; Murlasits, Z.; Bencsik, O.; Szekeres, A.; Vágvölgyi, C.; Laszlo, B.; et al. The effect of acute ophiobolin A treatment on HO-mediated inflammatory processes. Hum. Exp. Toxicol. 2016, 36, 594–602. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 47.1 | 2.16(1H,m) 1.20(1H,m) | 50.7 | 2.12(1H,m); 0.99(1H,dd, 15.4, 10.5) | 49.8 | 2.06(1H,d, 12.3); 0.94(1H) | 36.7 | 1.54(1H,dd, 12.1, 8.7); 1.11(1H,t, 13.5) | 37 | 1.58(1H,m) 1.13; (1H,ddd, 16.0, 13.6, 9.0) |
| 2 | 51.2 | 2.88(1H,d,12.9) | 47.2 | 3.37(1H,d,12.3) | 45.1 | 3.95(1H,d, 11.1) | 51.8 | 2.31(1H,m) | 51.9 | 2.35(1H,m) |
| 3 | 181.7 | 180.9 | 181.5 | 82.9 | 82.9 | |||||
| 4 | 130.3 | 5.93(1H,s) | 131.5 | 6.03(1H,m) | 130.5 | 6.06(1H,s) | 53.1 | 2.44(1H,dd,14.9, 7.8); 1.79(1H,m) | 53.2 | 2.46(1H,dd,14.9, 7.8); 1.81(1H,m) |
| 5 | 211.6 | 196.7 | 198 | 75.1 | 4.52(1H,m) | 75.2 | 4.55(1H,m) | |||
| 6 | 52.9 | 3.44(1H,d, 3.4) | 140.8 | 138.7 | 52.3 | 3.07(1H,d, 10.1) | 52.3 | 3.11(1H,d, 10.1) | ||
| 7 | 130.6 | 139.5 | 139.7 | 134 | 134.4 | |||||
| 8 | 146.5 | 7.00(1H,s) | 32.3 | 2.52(1H,td,13.2, 6.1); 2.40 (1H,dd,12.1, 5.1) | 73.4 | 4.39(1H,t,6.5) | 147.7 | 6.88(1H,t,8.8) | 147.1 | 6.89(1H,t,8.7) |
| 9 | 30.9 | 3.00(1H,d,19.8) 2.20(1H,m) | 26.9 | 1.58(1H,m) 2.01(1H,m) | 32.3 | 2.24(1H,ddd,15.0, 5.9,3.7); 1.85 (1H,dd,14.9, 7.4) | 25.7 | 2.68(1H,ddd,15.1, 13.5, 5.8) 2.07(1H,m) | 26.9 | 2.53 (1H,dd,12.9, 8.4); 2.09 (1H,dd,11.7,10.3) |
| 10 | 45.2 | 2.65(1H,m) | 45.2 | 2.07(1H,m) | 45.4 | 1.91(1H,m) | 55.5 | 1.54(1H,dd,12.1, 8.7) | 55.7 | 1.58(1H,m) |
| 11 | 46.4 | 44.9 | 44.7 | 44.9 | 44.8 | |||||
| 12 | 45.3 | 1.54(1H,m) 1.48(1H,m) | 46.1 | 1.52(1H,m) 1.38(1H,m) | 44.3 | 1.55(1H,m) 1.29(1H,m) | 43.9 | 1.40(2H,m) | 44.1 | 1.43(2H,dd,9.5, 4.4) |
| 13 | 28.6 | 1.68(1H,m) 1.36(1H,m) | 29.3 | 1.59(1H,m) 1.47(1H,m) | 29.3 | 1.68(1H,m) 1.55(1H,m) | 27.7 | 1.60(1H,m) 1.40(1H,m) | 27.5 | 1.58(2H,m) |
| 14 | 53.2 | 1.95(1H,m) | 52.3 | 1.94(1H,dd,17.3,8.2) | 50.5 | 2.06(1H,d, 12.3) | 48.6 | 2.07(1H,m) | 48.6 | 2.18(1H,ddd,25.8, 12.2, 6.9) |
| 15 | 33.8 | 2.63(1H,m) | 34.1 | 2.64(1H,dd,16.0, 9.2) | 35.4 | 2.64(1H,m) | 37.2 | 2.74(1H,ddd,15.1, 13.5, 5.8) | 42.9 | 2.36(1H,m) |
| 16 | 141.9 | 5.48(1H,m) | 138.9 | 5.20(1H,t,9.8) | 138.0 | 5.21(1H,m) | 138.7 | 5.22(1H,m) | 140.3 | 5.47(1H,dd, 15.1, 8.6) |
| 17 | 127.7 | 5.41(1H,m) | 123.4 | 6.03(1H,m) | 123.8 | 6.09(1H,m) | 123.2 | 6.03(1H,m) | 125.9 | 6.16(1H,dd, 15.1, 10.8) |
| 18 | 75.3 | 4.17(1H,d,9.6) | 121.7 | 6.03(1H,m) | 121.6 | 6.09(1H,m) | 121.5 | 6.03(1H,m) | 126.5 | 5.77(1H,d, 10.7) |
| 19 | 73.6 | 135.7 | 136.2 | 136 | 133.3 | |||||
| 20 | 17.3 | 2.11(3H,s) | 17.6 | 2.17(3H,s) | 17.5 | 2.17(3H,s) | 26.5 | 1.22(3H,s) | 26.5 | 1.24(3H,s) |
| 21 | 172 | 174.7 | 174.5 | 174.7 | 175.2 | |||||
| 22 | 23.1 | 0.99(3H,s) | 21.9 | 1.20(3H,s) | 22.4 | 1.19(3H,s) | 19.1 | 0.99(3H,s) | 19.3 | 0.99(3H,s) |
| 23 | 21.5 | 0.99(3H,d,6.0) | 21.2 | 0.91(3H,d,6.5) | 20.7 | 0.95(3H,d,6.5) | 20.8 | 0.89(3H,d,6.6) | 21 | 0.96(3H,d,6.6) |
| 24 | 24.9 | 1.17(3H,s) | 18.1 | 1.75(3H,s) | 18.1 | 1.75(3H,s) | 18.1 | 1.72(3H,s) | 18.2 | 1.74(3H,s) |
| 25 | 26.2 | 1.19(3H,s) | 26.5 | 1.82(3H,s) | 26.5 | 1.82(3H,s) | 26.5 | 1.79(3H,s) | 26 | 1.76(3H,s) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, W.; Uvarani, C.; Wang, F.; Xue, Y.; Wu, N.; He, L.; Tian, D.; Chen, M.; Zhang, Y.; Hong, K.; et al. New Ophiobolins from the Deep-Sea Derived Fungus Aspergillus sp. WHU0154 and Their Anti-Inflammatory Effects. Mar. Drugs 2020, 18, 575. https://doi.org/10.3390/md18110575
Ding W, Uvarani C, Wang F, Xue Y, Wu N, He L, Tian D, Chen M, Zhang Y, Hong K, et al. New Ophiobolins from the Deep-Sea Derived Fungus Aspergillus sp. WHU0154 and Their Anti-Inflammatory Effects. Marine Drugs. 2020; 18(11):575. https://doi.org/10.3390/md18110575
Chicago/Turabian StyleDing, Wenjuan, Chokkalingam Uvarani, Fangfang Wang, Yaxin Xue, Ning Wu, Liming He, Danmei Tian, Mei Chen, Youwei Zhang, Kui Hong, and et al. 2020. "New Ophiobolins from the Deep-Sea Derived Fungus Aspergillus sp. WHU0154 and Their Anti-Inflammatory Effects" Marine Drugs 18, no. 11: 575. https://doi.org/10.3390/md18110575
APA StyleDing, W., Uvarani, C., Wang, F., Xue, Y., Wu, N., He, L., Tian, D., Chen, M., Zhang, Y., Hong, K., & Tang, J. (2020). New Ophiobolins from the Deep-Sea Derived Fungus Aspergillus sp. WHU0154 and Their Anti-Inflammatory Effects. Marine Drugs, 18(11), 575. https://doi.org/10.3390/md18110575