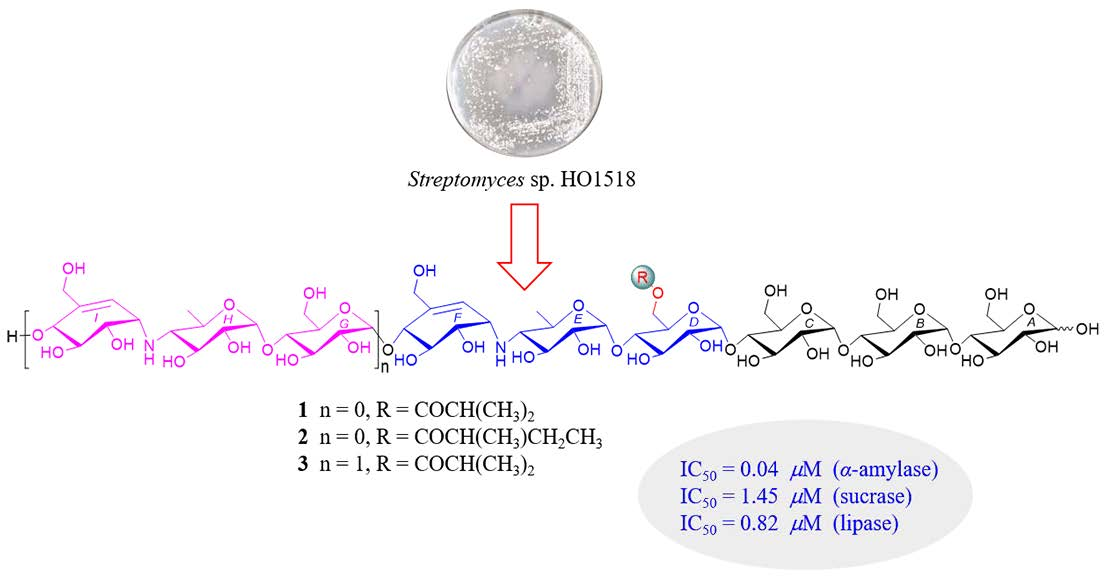
Acylated Aminooligosaccharides from the Yellow Sea Streptomyces sp. HO1518 as Both α-Glucosidase and Lipase Inhibitors

Abstract
:
1. Introduction
2. Results and Discussion
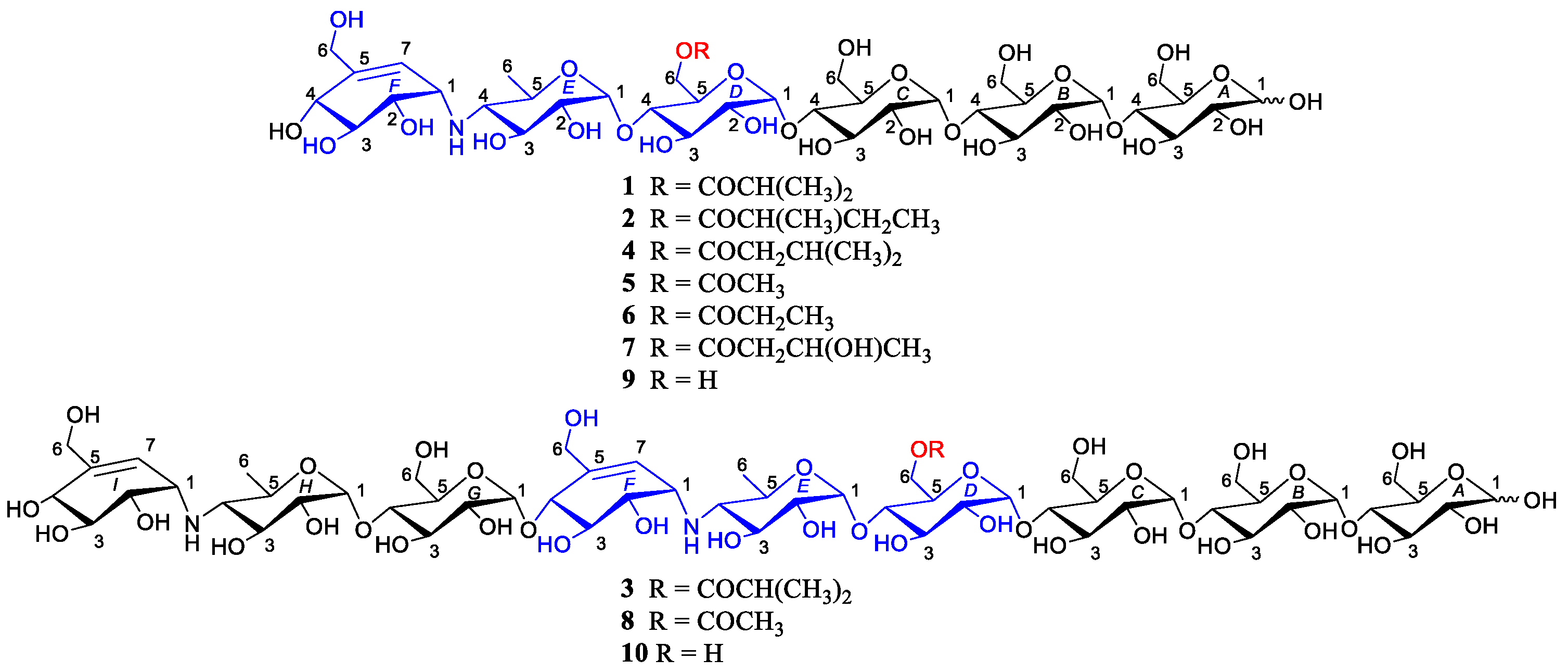
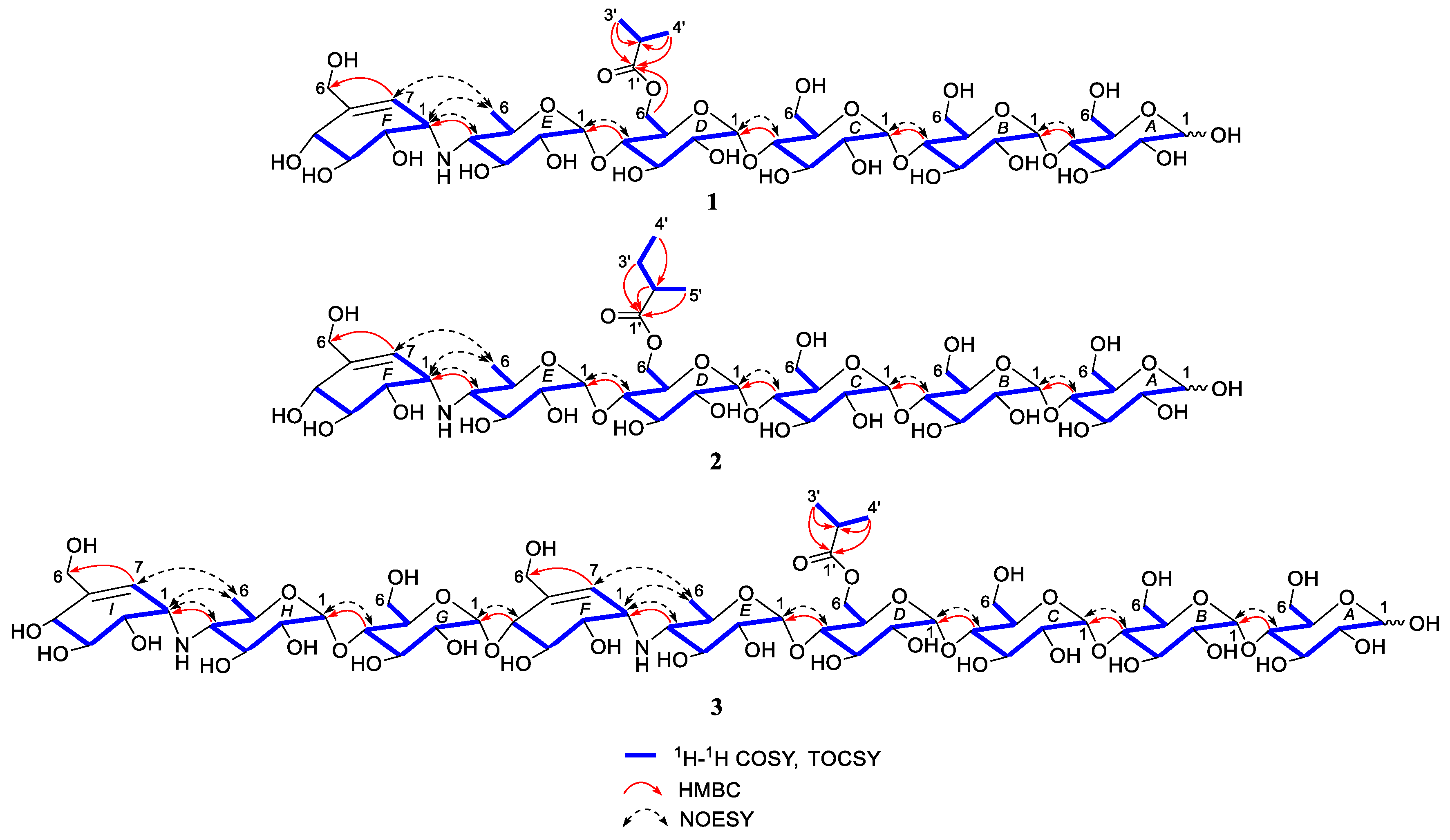
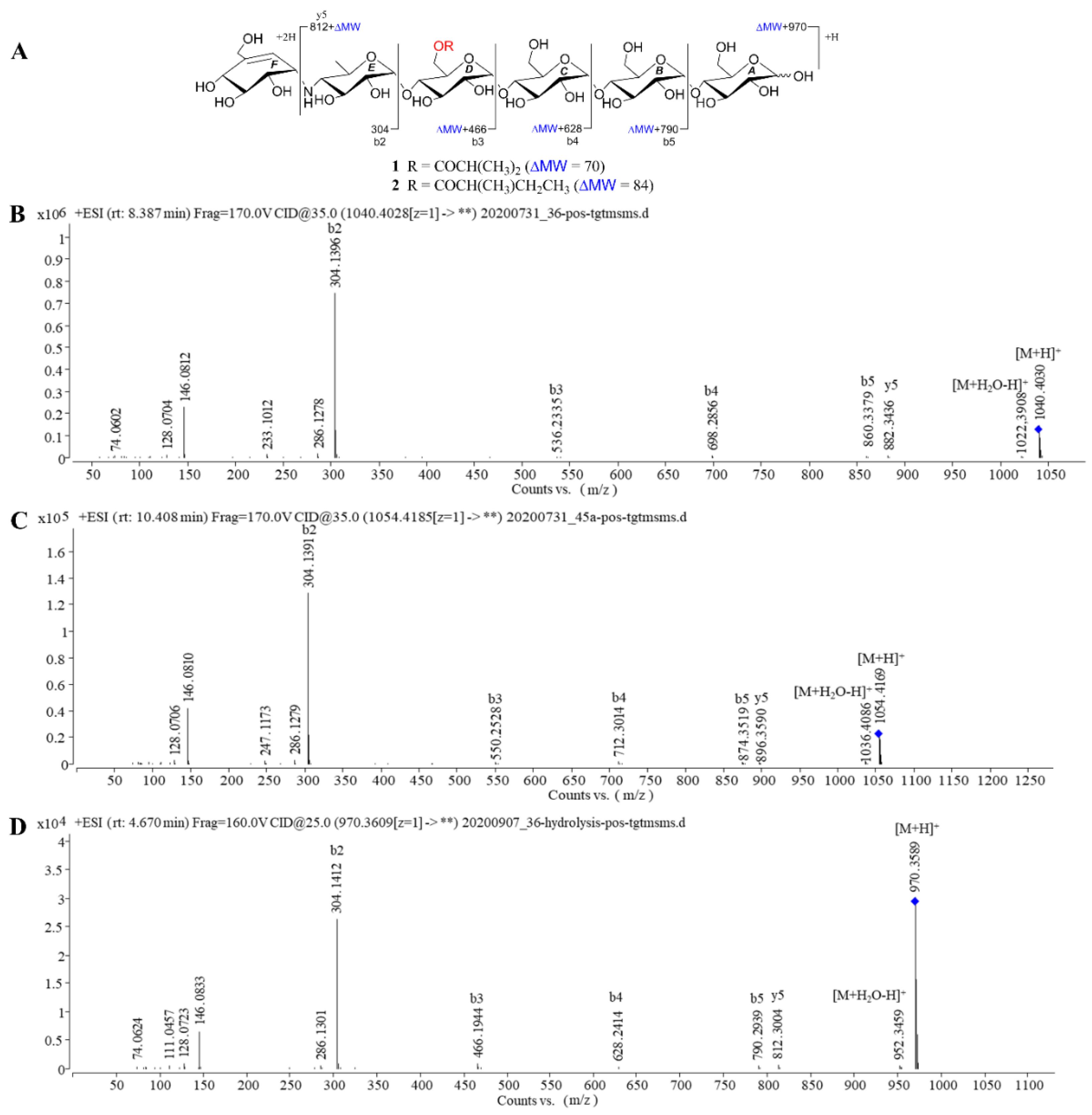
2.1. Structure Determination of New Compounds
2.2. Inhibitory Activities Against α-Glucosidase and Pancreatic Lipase
2.3. Structure-Activity Relationships (SAR) of Acylated Aminooligosaccharides
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Strain Isolation and Identification
3.3. Fermentation, Extraction and Isolation
3.4. Conversion of Compounds 1 and 2 to 9 and 3 to 10
3.5. α-Glucosidase Inhibition Assay
3.5.1. Porcine Pancreatic α-Amylase (PPA) Inhibition Assay
3.5.2. Sucrase Inhibition Assay
3.6. Pancreatic Lipase (PL) Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Bray, G.A.; Heisel, W.E.; Afshin, A.; Jensen, M.D.; Dietz, W.H.; Long, M.; Kushner, R.F.; Daniels, S.R.; Wadden, T.A.; Tsai, A.G.; et al. The science of obesity management: An endocrine society scientific statement. Endocr. Rev. 2018, 39, 79–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Li, C.J.; Chen, P.N.; Li, H.J.; Mahmud, T.; Wu, D.L.; Xu, J.; Lan, W.J. Potential antidiabetic fumiquinazoline alkaloids from the marine-derived fungus Scedosporium apiospermum F41-1. J. Nat. Prod. 2020, 83, 1082–1091. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Mellbye, F.B.; Jeppesen, P.B.; Shokouh, P.; Laustsen, C.; Hermansen, K.; Gregersen, S. Cafestol, a bioactive substance in coffee, has antidiabetic properties in KKAy mice. J. Nat. Prod. 2017, 80, 2353–2359. [Google Scholar] [CrossRef]
- Bailey, C.J.; Day, C. The future of new drugs for diabetes management. Diabetes Res. Clin. Pract. 2019, 155, 107785. [Google Scholar] [CrossRef]
- Liu, D.; Gao, H.; Tang, W.; Nie, S.P. Plant non-starch polysaccharides that inhibit key enzymes linked to type 2 diabetes mellitus. Ann. N. Y. Acad. Sci. 2017, 1401, 28–36. [Google Scholar] [CrossRef]
- Poongunran, J.; Perera, H.K.I.; Jayasinghe, L.; Fernando, I.T.; Sivakanesan, R.; Araya, H.; Fujimoto, Y. Bioassay-guided fractionation and identification of α-amylase inhibitors from Syzygium cumini leaves. Pharm. Biol. 2017, 55, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, D.D.; Frommer, W.; Junge, B.; Müller, L.; Wingender, W.; Truscheit, E.; Schäfer, D. α-Glucosidase inhibitors. New complex oligosaccharides of microbial origin. Naturwissenschaften 1977, 64, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Sun, T.; Zhong, Q.P.; Li, X.X.; Shi, L.Y.; Bai, F.; Bai, G. Two novel potent α-amylase inhibitors from the family of acarviostatins isolated from the culture of Streptomyces coelicoflavus ZG0656. Chem. Biodivers. 2013, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the β cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luyen, N.T.; Tram, L.H.; Hanh, T.T.H.; Binh, P.T.; Dang, N.H.; Minh, C.V.; Dat, N.T. Inhibitors of α-glucosidase, α-amylase and lipase from Chrysanthemum morifolium. Phytochem. Lett. 2013, 6, 322–325. [Google Scholar] [CrossRef]
- Saisho, Y.; Butler, A.E.; Manesso, E.; Elashoff, D.; Rizza, R.A.; Butler, P.C. β-Cell mass and turnover in humans. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Inthongkaew, P.; Chatsumpun, N.; Supasuteekul, C.; Kitisripanya, T.; Putalun, W.; Likhitwitayawuid, K.; Sritularak, B. α-Glucosidase and pancreatic lipase inhibitory activities and glucose uptake stimulatory effect of phenolic compounds from Dendrobium formosum. Rev. Bras. Farmacogn. 2017, 27, 480–487. [Google Scholar] [CrossRef]
- Yin, Z.H.; Zhang, W.; Feng, F.J.; Zhang, Y.; Kang, W.Y. α-Glucosidase inhibitors isolated from medicinal plants. Food. Sci. Hum. Wellness. 2014, 3, 136–174. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, D.; Zhu, J.; Liu, H.Y.; Liang, S.F.; Luo, Y.Z. Efficient multiplex genome editing in streptomyces via engineered CRISPR-Cas12a systems. Front. Bioeng. Biotechnol. 2020, 8, 726. [Google Scholar] [CrossRef]
- Robertsen, H.L.; Musiol-Kroll, E.M. Actinomycete-derived polyketides as a source of antibiotics and lead structures for the development of new antimicrobial drugs. Antibiotics 2019, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.Q.; Wang, J.F.; Hao, Y.Y.; Wang, Y. Recent advances in marine microbial natural products discovery and development. Mar. Drugs 2013, 11, 700–717. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.Q.; Liu, Q.X.; Pan, Z.L.; Zhao, N.; Feng, Z.X.; Wang, Y. Diversity and bioprospecting of culturable actinomycetes from marine sediment of the Yellow Sea, China. Arch. Microbiol. 2015, 197, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.Q.; Wang, Y. Draft genome sequence of the marine Streptomyces sp. strain AA1529, isolated from the Yellow Sea. J. Bacteriol. 2012, 194, 5474–5475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.Q.; Wang, Y. Draft genome sequence of marine-derived Streptomyces sp. strain AA0539, isolated from the Yellow Sea, China. J. Bacteriol. 2012, 194, 6622–6623. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.L.; Xie, D.A.; Cheng, W.B.; Tao, W.Q.; Wang, Y. Acylated aminooligosaccharides with inhibitory effects against α-amylase from Streptomyces sp. HO1518. Mar. Drugs 2018, 16, 403. [Google Scholar] [CrossRef] [Green Version]
- Świerczewska, A.; Buchholz, T.; Melzig, M.F.; Czerwińska, M.E. In vitro α-amylase and pancreatic lipase inhibitory activity of Cornus mas L. and Cornus alba L. fruit extracts. J. Food. Drug. Anal. 2019, 27, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Chang, S.K.C. Digestive enzyme inhibition activity of the phenolic substances in selected fruits, vegetables and tea as compared to black legumes. J. Funct. Foods 2017, 38, 644–655. [Google Scholar] [CrossRef]
- Meng, P.; Guo, Y.Q.; Zhang, Q.; Hou, J.; Bai, F.; Geng, P.; Bai, G. A novel amino-oligosaccharide isolated from the culture of Streptomyces strain PW638 is a potent inhibitor of α-amylase. Carbohydr. Res. 2011, 346, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Qiu, F.; Zhu, Y.Y.; Bai, G. Four acarviosin-containing oligosaccharides identified from Streptomyces coelicoflavus ZG0656 are potent inhibitors of α-amylase. Carbohydr. Res. 2008, 343, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Oda, S.; Yasuda, S.; Mineno, T.; Okawa, M.; Kinjo, J.; Miyashita, H.; Yoshimitsu, H.; Nohara, T.; Miyahara, K. Acylated glycosidic acid methyl esters generated from the convolvulin fraction of rhizoma jalapae braziliensis by treatment with indium(III) chloride in methanol. Chem. Pharm. Bull. 2017, 65, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Zhong, D.F.; Si, D.Y.; He, W.Y.; Zhao, L.M.; Xu, Q.M. Structural revision of isovalertatins M03, M13, and M23 isolated from the culture of Streptomyces luteogriseus. Carbohydr. Res. 2001, 331, 69–75. [Google Scholar] [CrossRef]
- Si, D.Y.; Zhong, D.F.; Chen, X.Y. Profiling of isovalertatin family aminooligosaccharides extracted from the culture of Streptomyces luteogriseus by using liquid chromatography/electrospray ionization mass spectrometry. Anal. Chem. 2001, 73, 3808–3815. [Google Scholar] [CrossRef]
- Wang, L.Q.; Cui, Q.X.; Hou, Y.Y.; Bai, F.; Sun, J.X.; Cao, X.F.; Liu, P.; Jiang, M.; Bai, G. An integrated strategy of ultra-high-performance liquid chromatography/quadrupole-time-of-flight mass spectrometry and virtual screening for the identification of α-glucosidase inhibitors in acarviostatin-containing complex. J. Chromatogr. A 2013, 1319, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Li, W.T.; Chuang, Y.H.; Hsieh, J.F. Characterization of maltase and sucrase inhibitory constituents from Rhodiola crenulata. Foods 2019, 8, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDougall, G.J.; Kulkarni, N.N.; Stewart, D. Berry polyphenols inhibit pancreatic lipase activity in vitro. Food. Chem. 2009, 115, 193–199. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| A1α | 91.9, CH | 5.24, d (3.5) | 91.9, CH | 5.21, d (3.5) |
| A2α | 71.3, CH | 3.58, m | 71.3, CH | 3.58, m |
| A3α | 73.1, CH | 3.98, m | 73.1, CH | 3.96, m |
| A4α | 76.2, CH | 3.66, m | 76.2, CH | 3.69, m |
| A5α | 69.9, CH | 3.98, m | 69.9, CH | 3.96, m |
| A6α | 60.4, CH2 | 3.84, m | 60.4, CH2 | 3.88, m |
| A1β | 95.8, CH | 4.66, d (8.0) | 95.8, CH | 4.63, d (8.0) |
| A2β | 74.0, CH | 3.28, m | 74.0, CH | 3.25, m |
| A3β | 77.0, CH | 3.78, t (9.0) | 76.9, CH | 3.75, m |
| A4β | 76.8, CH | 3.66, m | 76.8, CH | 3.63, m |
| A5β | 74.5, CH | 3.61, m | 74.5, CH | 3.61, m |
| A6β | 60.4, CH2 | 3.91, m | 60.4, CH2 | 3.89, m |
| B1 | 99.4, CH | 5.42, overlapped, (3.5) | 99.4, CH | 5.38, overlapped, (3.5) |
| B2 | 71.5, CH | 3.64, m | 71.6, CH | 3.60, m |
| B3 | 73.2, CH | 3.96, m | 73.2, CH | 3.94, m |
| B4 | 76.8, CH | 3.67, m | 76.8, CH | 3.64, m |
| B5 | 71.1, CH | 3.84, m | 71.1, CH | 3.82, m |
| B6 | 60.5, CH2 | 3.84, m | 60.5, CH2 | 3.82, m |
| C1 | 99.5, CH | 5.42, overlapped, (3.5) | 99.5, CH | 5.38, overlapped, (3.5) |
| C2 | 71.6, CH | 3.64, m | 71.5, CH | 3.60, m |
| C3 | 73.3, CH | 3.96, m | 73.3, CH | 3.94, m |
| C4 | 77.0, CH | 3.67, m | 77.0, CH | 3.64, m |
| C5 | 71.2, CH | 3.84, m | 71.2, CH | 3.82, m |
| C6 | 60.7, CH2 | 3.84, m | 60.7, CH2 | 3.82, m |
| D1 | 99.6, CH | 5.41, overlapped, (3.5) | 99.6, CH | 5.38, overlapped, (3.5) |
| D2 | 72.2, CH | 3.65, m | 72.2, CH | 3.61, m |
| D3 | 73.3, CH | 3.95, m | 73.3, CH | 3.93, m |
| D4 | 78.2, CH | 3.65, m | 78.4, CH | 3.63, m |
| D5 | 69.0, CH | 4.06, d (11.6) | 69.0, CH | 4.01, m |
| D6a | 63.5, CH2 | 4.44, d (11.6) | 63.4, CH2 | 4.44, d (10.0) |
| D6b | 4.23, dd (11.6, 5.0) | 4.20, d (10.0) | ||
| E1 | 100.7, CH | 5.27, d (3.4) | 100.7, CH | 5.23, d (3.4) |
| E2 | 71.3, CH | 3.59, m | 71.3, CH | 3.56, m |
| E3 | 72.7, CH | 3.62, m | 72.8, CH | 3.60, m |
| E4 | 65.0, CH | 2.46, m | 65.0, CH | 2.43, m |
| E5 | 69.8, CH | 3.73, m | 69.8, CH | 3.72, m |
| E6 | 17.3, CH3 | 1.31, d (6.0) | 17.3, CH3 | 1.29, d (6.6) |
| F1 | 56.0, CH | 3.53, m | 56.0, CH | 3.52, m |
| F2 | 72.9, CH | 3.65, m | 73.0, CH | 3.62, m |
| F3 | 73.0, CH | 3.75, m | 73.0, CH | 3.73, m |
| F4 | 70.8, CH | 4.06, d (4.6) | 70.9, CH | 4.01, d (4.8) |
| F5 | 139.0, C | 139.0, C | ||
| F6a | 61.6, CH2 | 4.23, brd (14.1) | 61.6, CH2 | 4.20, brd (14.2) |
| F6b | 4.12, brd (14.1) | 4.09, brd (14.2) | ||
| F7 | 123.7, CH | 5.90, s | 123.7, CH | 5.87, s |
| 1′ | 180.1, C=O | 179.8, C=O | ||
| 2′ | 33.8, CH | 2.71, m | 40.9, CH | 2.52, m |
| 3′ | 18.1, CH3 | 1.19, d (3.8) | 26.4, CH2 | 1.51, m |
| 1.64, m | ||||
| 4′ | 18.2, CH3 | 1.19, d (3.8) | 10.8, CH3 | 0.88, t (7.0) |
| 5′ | 15.7, CH3 | 1.14, d (7.0) | ||
| No. | 3 | No. | 3 | ||
|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | ||
| A1α | 94.8, CH | 5.26, d (3.3) | E5 | 72.6, CH | 3.73, m |
| A2α | 74.2, CH | 3.56, m | E6 | 20.2, CH3 | 1.33, d (6.0) |
| A3α | 76.1, CH | 4.18, m | F1 | 57.9, CH | 3.56, m |
| A4α | 79.6, CH | 3.72, m | F2 | 73.5, CH | 3.82, m |
| A5α | 72.8, CH | 3.93, m | F3 | 73.6, CH | 4.15, m |
| A6α | 63.3, CH2 | 3.76, m | F4 | 79.0, CH | 4.07, m |
| A1β | 98.6, CH | 4.68, d (7.8) | F5 | 139.3, C | |
| A2β | 76.8, CH | 3.30, dd (9.0, 7.8) | F6a | 64.8, CH2 | 4.25, m |
| A3β | 79.1, CH | 3.79, m | F6b | 4.15, m | |
| A4β | 79.7, CH | 3.66, m | F7 | 129.2, CH | 6.01, d (4.5) |
| A5β | 77.4, CH | 3.60, m | G1 | 100.4, CH | 5.40, d (3.5) |
| A6β | 63.4, CH2 | 3.92, m | G2 | 76.0, CH | 3.65, m |
| B1 | 102.4, CH | 5.43, d (3.3) | G3 | 76.3, CH | 3.92, m |
| B2 | 74.4, CH | 3.64, m | G4 | 73.7, CH | 3.63, m |
| B3 | 76.2, CH | 3.97, m | G5 | 74.0, CH | 3.94, m |
| B4 | 79.9, CH | 3.69, m | G6 | 63.3, CH2 | 3.82, m |
| B5 | 74.0, CH | 3.86, m | H1 | 102.7, CH | 5.34, d (3.3) |
| B6 | 63.5, CH2 | 3.86, m | H2 | 74.2, CH | 3.65, m |
| C1 | 102.4, CH | 5.43, d (3.3) | H3 | 75.4, CH | 3.65, m |
| C2 | 74.3, CH | 3.64, m | H4 | 67.8, CH | 2.49, m |
| C3 | 76.2, CH | 3.97, m | H5 | 72.4, CH | 3.81, m |
| C4 | 79.7, CH | 3.69, m | H6 | 20.2, CH3 | 1.37, d (6.0) |
| C5 | 74.1, CH | 3.86, m | I1 | 58.8, CH | 3.56, t (5.0) |
| C6 | 65.3, CH2 | 3.86, m | I2 | 75.6, CH | 3.69, m |
| D1 | 102.2, CH | 5.43, d (3.3) | I3 | 75.8, CH | 3.79, m |
| D2 | 73.9, CH | 3.68, m | I4 | 73.8, CH | 4.07, m |
| D3 | 76.0, CH | 3.97, m | I5 | 141.8, C | |
| D4 | 81.0, CH | 3.68, m | I6a | 64.4, CH2 | 4.25, m |
| D5 | 71.8, CH | 4.07, m | I6b | 4.15, m | |
| D6a | 66.3, CH2 | 4.46, m | I7 | 126.6, CH | 5.93, d (2.5) |
| D6b | 4.25, q (5.4) | 1′ | 182.9, C=O | ||
| E1 | 103.6, CH | 5.29, d (3.3) | 2′ | 36.7, CH | 2.73, m |
| E2 | 74.1, CH | 3.55, m | 3′ | 21.0, CH | 1.20, d (2.7) |
| E3 | 75.8, CH | 3.57, m | 4′ | 21.1, CH | 1.22, d (2.7) |
| E4 | 67.1, CH | 2.49, m | |||
| Compounds | IC50 Values (μM) a | ||
|---|---|---|---|
| Against PPA | Against Sucrase | Against PL | |
| 1 | 0.22 ± 0.04 | 5.50 ± 0.09 | 2.16 ± 0.60 |
| 2 | 0.15 ± 0.01 | 3.12 ± 0.10 | 4.92 ± 0.20 |
| 3 | 0.04 ± 0.01 | 1.45 ± 0.07 | 0.82 ± 0.08 |
| 4 | 0.34 ± 0.07 | 9.34 ± 0.12 | 3.20 ± 0.10 |
| 5 | n.t. b | 2.98 ± 0.10 | 19.70 ± 1.00 |
| 6 | n.t. b | 2.36 ± 0.12 | 15.90 ± 0.60 |
| 7 | n.t. b | 3.68 ± 0.13 | 8.10 ± 0.60 |
| 8 | n.t. b | 0.41 ± 0.05 | 2.00 ± 0.18 |
| acarbose | 3.80 ± 0.15 | 11.27 ± 0.20 | 207.57 ± 9.77 |
| orlistat | n.t. | n.t. | 0.58 ± 0.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.-L.; Liu, H.-L.; Liu, Z.-F.; Ren, Y.-H.; Wang, Y. Acylated Aminooligosaccharides from the Yellow Sea Streptomyces sp. HO1518 as Both α-Glucosidase and Lipase Inhibitors. Mar. Drugs 2020, 18, 576. https://doi.org/10.3390/md18110576
Xu J-L, Liu H-L, Liu Z-F, Ren Y-H, Wang Y. Acylated Aminooligosaccharides from the Yellow Sea Streptomyces sp. HO1518 as Both α-Glucosidase and Lipase Inhibitors. Marine Drugs. 2020; 18(11):576. https://doi.org/10.3390/md18110576
Chicago/Turabian StyleXu, Jian-Lin, Hai-Li Liu, Zhi-Feng Liu, Yu-Hong Ren, and Yong Wang. 2020. "Acylated Aminooligosaccharides from the Yellow Sea Streptomyces sp. HO1518 as Both α-Glucosidase and Lipase Inhibitors" Marine Drugs 18, no. 11: 576. https://doi.org/10.3390/md18110576
APA StyleXu, J. -L., Liu, H. -L., Liu, Z. -F., Ren, Y. -H., & Wang, Y. (2020). Acylated Aminooligosaccharides from the Yellow Sea Streptomyces sp. HO1518 as Both α-Glucosidase and Lipase Inhibitors. Marine Drugs, 18(11), 576. https://doi.org/10.3390/md18110576