A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemical Space of the SARS-CoV-2 Model
2.2. QSAR Classification Modeling
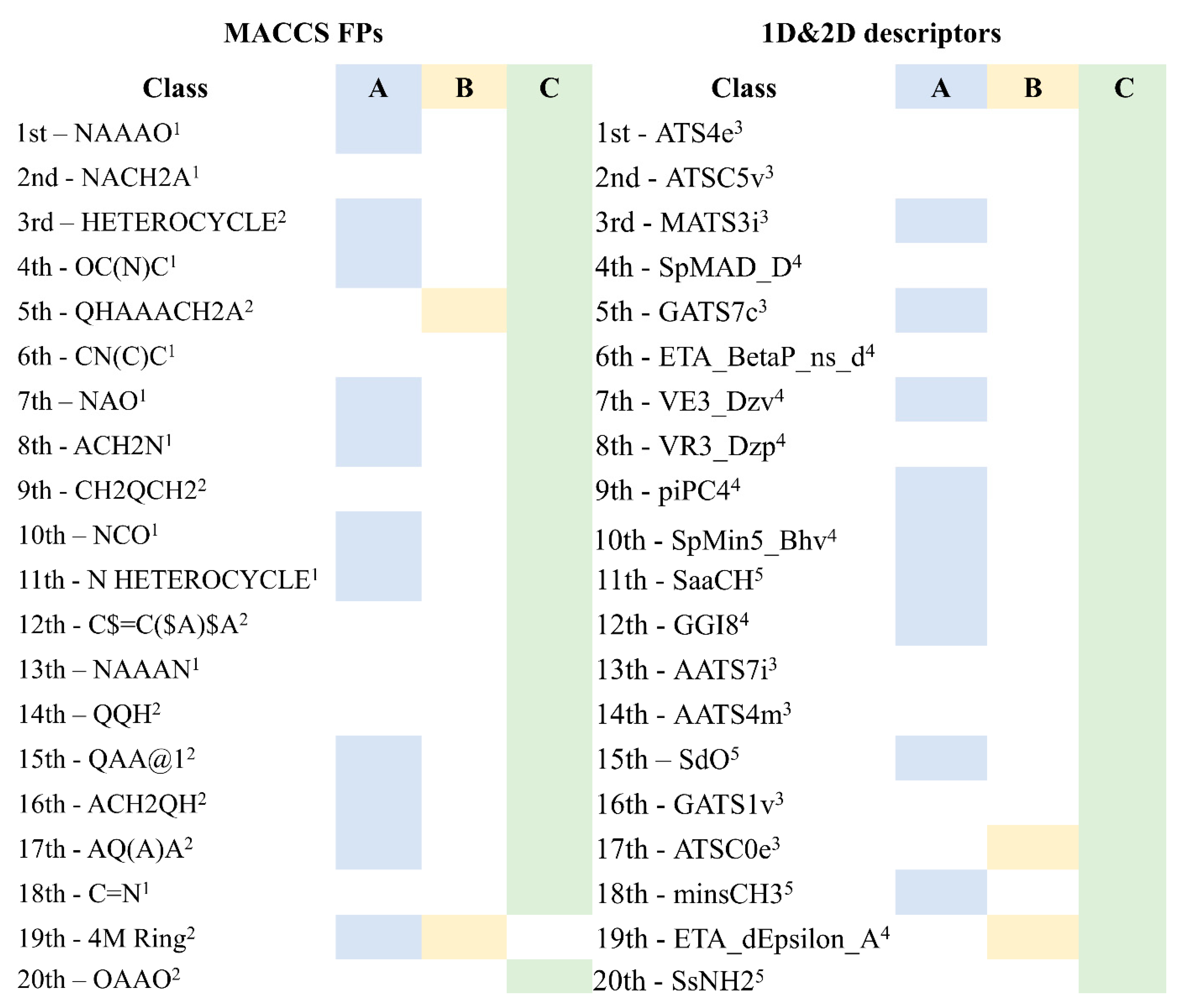
2.3. Analysis of MACCS FPs and 1D&2D Descriptors Identified as Relevant for Modeling the Antiviral Activity against SARS-CoV-2
2.4. Application of the In Silico Anti-Viral Model Against SARS-CoV-2 in Virtual Screening
2.5. Molecular Docking Against Mpro Enzyme
3. Materials and Methods
3.1. Data Sets and Selection of Training, Test, Test 2 Sets
3.2. Calculation of Molecular Descriptors and Fingerprints
3.3. Selection of Descriptors and Optimization of QSAR Models
3.4. Class Balancer
3.5. Machine Learning (ML) Method
Random Forest (RF)
3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coronavirus Resource Center. Global Tracking. Available online: https://coronavirus.jhu.edu/map.html (accessed on 9 December 2020).
- Blumenthal, D.; Fowler, E.J.; Abrams, M.; Collins, S.R. Covid-19—Implications for the health care system. N. Engl. J. Med. 2020, 383, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative inhibitors of SARS-CoV-2 main protease from A Library of Marine Natural Products: A virtual screening and molecular modeling study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.T.; Ali, A.; Wang, Q.; Irfan, M.; Khan, A.; Zeb, M.T.; Zhang, Y.-J.; Chinnasamy, S.; Wei, D.-Q. Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2—a molecular dynamic study. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Khaerunnisa, S.; Kurniawan, H.; Awaluddin, R.; Suhartati, S.; Soetjipto, S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints 2020. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- El-Hoshoudy, A. Investigating the potential antiviral activity drugs against SARS-CoV-2 by molecular docking simulation. J. Mol. Liq. 2020, 318, 113968. [Google Scholar] [CrossRef]
- Pereira, F.; Aires-De-Sousa, J. Computational methodologies in the exploration of marine natural product leads. Mar. Drugs 2018, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F. Have marine natural product drug discovery efforts been productive and how can we improve their efficiency? Expert Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Boorla, V.S.; Maranas, C.D. Computational biophysical characterization of the SARS-CoV-2 spike protein binding with the ACE2 receptor and implications for infectivity. Comput. Struct. Biotechnol. J. 2020, 18, 2573–2582. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccio, G.; Ruocco, N.; Mutalipassi, M.; Costantini, M.; Zupo, V.; Coppola, D.; De Pascale, D.; Lauritano, C. Ten-Year Research Update Review: Antiviral Activities from Marine Organisms. Biomolecules 2020, 10, 1007. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Amin, S.A.; Gayen, S.; Jha, T. Chemical-informatics approach to COVID-19 drug discovery: Exploration of important fragments and data mining based prediction of some hits from natural origins as main protease (Mpro) inhibitors. J. Mol. Struct. 2020, 1224, 129026. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.M.; Bobrowski, T.; Melo-Filho, C.C.; Korn, D.; Auerbach, S.; Schmitt, C.; Muratov, E.N.; Tropsha, A. QSAR modeling of SARS-CoV Mpro inhibitors identifies Sufugolix, Cenicriviroc, Proglumetacin and other drugs as candidates for repurposing against SARS-CoV-2. Mol. Inform. 2020. [Google Scholar] [CrossRef]
- Kumar, V.; Roy, K. Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3C-like protease (3CLpro) enzyme inhibitors against SARS-CoV diseases. SAR QSAR Environ. Res. 2020, 31, 511–526. [Google Scholar] [CrossRef]
- Elzupir, A.O. Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J. Mol. Struct. 2020, 1222, 128878. [Google Scholar] [CrossRef]
- Middelboe, M.; Brussaard, C.P.D. Marine Viruses: Key Players in Marine Ecosystems. Viruses 2017, 9, 302. [Google Scholar] [CrossRef] [Green Version]
- Wigington, C.H.; Sonderegger, D.; Brussaard, C.P.D.; Buchan, A.; Finke, J.F.; Fuhrman, J.A.; Lennon, J.T.; Middelboe, M.; Suttle, C.A.; Stock, C.; et al. Re-examination of the relationship between marine virus and microbial cell abundances. Nat. Microbiol. 2016, 1, 15024. [Google Scholar] [CrossRef] [Green Version]
- Ordulj, M.; Krstulović, N.; Šantić, D.; Jozić, S.; Šolić, Μ. Distribution of marine viruses in the Central and South Adriatic Sea. Mediterr. Mar. Sci. 2015, 16, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Davó, A.; Dias, T.; Gomes, S.E.; Rodrigues, S.; Parera-Valadez, Y.; Borralho, P.M.; Pereira, F.; Rodrigues, C.M.P.; Santos-Sanches, I.; Gaudêncio, S.P. The Madeira Archipelago As a significant source of marine-derived Actinomycete diversity with Anticancer and Antimicrobial Potential. Front. Microbiol. 2016, 7, 1594. [Google Scholar] [CrossRef]
- Bauermeister, A.; Pereira, F.; Grilo, I.R.; Godinho, C.C.; Paulino, M.; Almeida, V.; Gobbo-Neto, L.; Prieto-Davó, A.; Sobral, R.; Lopes, N.P.; et al. Intra-clade metabolomic profiling of MAR4 Streptomyces from the Macaronesia Atlantic region reveals a source of anti-biofilm metabolites. Environ. Microbiol. 2019, 21, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Almeida, J.R.; Paulino, M.; Grilo, I.R.; Macedo, H.; Cunha, I.; Sobral, R.G.; Vasconcelos, V.; Gaudêncio, S.P. Antifouling Napyradiomycins from marine-derived Actinomycetes Streptomyces Aculeolatus. Mar. Drugs 2020, 18, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, F.; Latino, D.A.R.S.; Gaudêncio, S.P. A chemoinformatics approach to the discovery of lead-like molecules from marine and microbial sources en route to antitumor and antibiotic drugs. Mar. Drugs 2014, 12, 757–778. [Google Scholar] [CrossRef] [PubMed]
- Cruz, S.M.; Gaudêncio, S.P.; Pereira, F. A computational approach in the discovery of lead-like compounds for anticancer drugs. Front. Mar. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Dias, T.; Gaudêncio, S.P.; Pereira, F. A computer-driven approach to discover natural product leads for Methicillin-resistant Staphylococcus Aureus infection therapy. Mar. Drugs 2018, 17, 16. [Google Scholar] [CrossRef] [Green Version]
- Cruz, S.M.; Gomes, S.E.; Borralho, P.M.; Rodrigues, C.M.P.; Gaudêncio, S.P.; Pereira, F. In silico HCT116 human colon cancer cell-based models en route to the discovery of lead-like anticancer drugs. Biomolecules 2018, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Jain, S.; Kotsampasakou, E.; Ecker, G.F. Comparing the performance of meta-classifiers—a case study on selected imbalanced data sets relevant for prediction of liver toxicity. J. Comput. Aided Mol. Des. 2018, 32, 583–590. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; Available online: https://www.R-project.org (accessed on 24 October 2020).
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random Forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and regression by randomforest. R News 2002, 2, 18–22. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clusters 1 | # 2 (A class) 3 | Average MW 4 | Average ALogP 5 | |||
|---|---|---|---|---|---|---|
| Tr Set | Te Set | Tr Set | Te Set | Tr Set | Te Set | |
I—Indole derivative | 410 (41) | 186 (17) | 377.71 | 389.00 | 2.79 | 3.10 |
II—Benzoate derivative | 333 (35) | 149 (14) | 404.26 | 412.74 | 3.59 | 3.24 |
III—γ-Lactone derivative | 294 (10) | 112 (7) | 296.10 | 302.50 | 3.05 | 3.14 |
IV—Benzimidazole derivative | 516 (57) | 224 (32) | 404.77 | 408.17 | 2.86 | 3.10 |
V—α-Amino acid ester derivative | 270 (23) | 110 (8) | 464.60 | 445.25 | 2.83 | 2.76 |
VI—Quinoline derivative | 258 (28) | 132 (9) | 400.27 | 412.95 | 2.92 | 2.63 |
VII—Piperidine derivative | 279 (13) | 131 (13) | 383.15 | 355.98 | 3.07 | 2.81 |
VIII—Acyclic derivative | 588 (31) | 254 (20) | 329.16 | 336.52 | 1.89 | 1.63 |
IX—Oxazole derivative | 285 (29) | 136 (13) | 422.11 | 397.43 | 3.62 | 3.75 |
X—Piperidine derivative | 266 (35) | 98 (12) | 395.85 | 389.66 | 3.68 | 3.61 |
| Model | MACCS | Sub | SubC | PubChem | CDK | CDKExt | 1D&2D |
|---|---|---|---|---|---|---|---|
| TA 1 | 156 | 185 | 193 | 164 | 132 | 131 | 164 |
| TB 2 | 43 | 64 | 49 | 37 | 33 | 37 | 24 |
| TC 3 | 2017 | 1183 | 1574 | 1960 | 2170 | 2208 | 2100 |
| FA_B 4 | 57 | 94 | 76 | 58 | 53 | 51 | 55 |
| FA_C 5 | 567 | 1029 | 827 | 568 | 508 | 494 | 646 |
| FB_A 6 | 29 | 38 | 37 | 34 | 17 | 19 | 14 |
| FB_C 7 | 351 | 723 | 534 | 407 | 257 | 233 | 186 |
| FC_A 8 | 117 | 79 | 72 | 104 | 153 | 152 | 124 |
| FC_B 9 | 165 | 107 | 140 | 170 | 179 | 177 | 186 |
| SE 10 | 0.52 | 0.61 | 0.64 | 0.54 | 0.44 | 0.43 | 0.54 |
| SP_B 11 | 0.16 | 0.24 | 0.18 | 0.14 | 0.12 | 0.14 | 0.09 |
| SP_C 12 | 0.69 | 0.40 | 0.54 | 0.67 | 0.74 | 0.75 | 0.72 |
| Q 13 | 0.63 | 0.41 | 0.52 | 0.62 | 0.67 | 0.68 | 0.65 |
| MCC 14 | 0.27 | 0.20 | 0.25 | 0.25 | 0.26 | 0.27 | 0.26 |
| Model | SE 1 | SP_B 2 | SP_C 3 | Q 4 | MCC 5 |
|---|---|---|---|---|---|
| ExtCDK FP | |||||
| 50 6 | 0.41 | 0.19 | 0.65 | 0.59 | 0.21 |
| 100 6 | 0.44 | 0.14 | 0.70 | 0.64 | 0.23 |
| 150 6 | 0.46 | 0.17 | 0.71 | 0.64 | 0.27 |
| 200 6 | 0.44 | 0.14 | 0.71 | 0.65 | 0.23 |
| 1D&2D descriptors | |||||
| 50 6 | 0.54 | 0.11 | 0.69 | 0.63 | 0.25 |
| 100 6 | 0.59 | 0.09 | 0.70 | 0.64 | 0.25 |
| 150 6 | 0.55 | 0.12 | 0.69 | 0.63 | 0.26 |
| 200 6 | 0.55 | 0.08 | 0.71 | 0.65 | 0.22 |
| CM | SE 1 | SP_B 2 | SP_C 3 | Q 4 | MCC 5 |
|---|---|---|---|---|---|
| Training set 6 | 0.51 | 0.14 | 0.74 | 0.68 | 0.31 |
| Test Set | 0.48 | 0.08 | 0.74 | 0.67 | 0.19 |
| Clusters | # | SE 1 | SP_B 2 | SP_C 3 | Q 4 | MCC 5 |
|---|---|---|---|---|---|---|
| I | 186 | 0.71 | --- | 0.73 | 0.68 | 0.32 |
| II | 149 | 0.36 | 0.13 | 0.75 | 0.68 | 0.29 |
| III | 112 | --- | --- | 0.81 | 0.71 | 0.37 |
| IV | 224 | 0.69 | --- | 0.58 | 0.55 | 0.21 |
| V | 110 | 0.25 | 0.20 | 0.74 | 0.68 | 0.05 |
| VI | 132 | 0.67 | 0.29 | 0.70 | 0.67 | 0.38 |
| VII | 131 | 0.38 | --- | 0.82 | 0.74 | 0.25 |
| VIII | 254 | 0.30 | 0.11 | 0.81 | 0.72 | 0.25 |
| IX | 136 | 0.38 | --- | 0.79 | 0.66 | 0.13 |
| X | 98 | 0.50 | 0.40 | 0.64 | 0.61 | 0.21 |
| All | 1533 | 0.48 | 0.08 | 0.74 | 0.67 | 0.19 |
| Code | Chemical Structure | Structural Category | Natural Source | Prob_A | ∆GB (kcal/mol) |
|---|---|---|---|---|---|
| 22947654 1 |  | carbazole | marine derived bacteria | 0.42 | −9.9 6/−7.6 7 |
| 22947655 1 |  | carbazole | marine derived bacteria | 0.42 | −9.9 6/−7.6 7 |
| 22435742 1 |  | anthraquinone | marine derived bacteria | 0.42 | −9.4 6/−7.8 7 |
| 22435744 1 |  | anthraquinone | marine derived bacteria | 0.41 | −9.4 6/−7.8 7 |
| 30380251 1 |  | phenoxazinone | marine derived bacteria | 0.68 | −9.1 6/−6.9 7 |
| 19600610 1 |  | quinoxaline | marine derived bacteria | 0.62 | −8.9 6/−8.9 7 |
| 22435741 1 |  | anthraquinone | marine derived bacteria | 0.40 | −8.8 6/−7.8 7 |
| 7450892 1 |  | benzo[f]pyrano[4,3-b]chromene | marine derived fungus | 0.41 | −8.4 6/−6.9 7 |
| 19384758 1 |  | prenylated indole alkaloids | marine derived fungus | 0.40 | −8.4 6/−7.4 7 |
| 26845562 1 |  | indoloditerpenes | marine derived fungus | 0.41 | −8.2 6/−6.9 7 |
| 19384759 1 |  | prenylated indole alkaloids | marine derived fungus | 0.39 | −8.1 6/−7.3 7 |
| 22435737 1 |  | anthraquinone | marine derived bacteria | 0.41 | −8.0 6/−7.0 7 |
| 30380253 1 |  | phenoxazinone | marine derived bacteria | 0.59 | −8.0 6/−8.5 7 |
| 10714788 1 |  | bromo deoxytopsentin | sponge | 0.38 | −7.6 6/−8.3 7 |
| 10720065 1 |  | dibromodeoxytopsentin | sponge | 0.38 | −7.6 6/−8.5 7 |
| PTM346F6F45 2 |  | marinone | marine derived bacteria | 0.30 | −7.0 6/−5.5 7 |
| nelarabine (Arranon®) 3 |  | purine | sponge | 0.31 | −5.4 6/−5.5 7 |
| fludarabine phosphate (Fludara®) 3 |  | purine | sponge | 0.31 | −5.8 6/−6.5 7 |
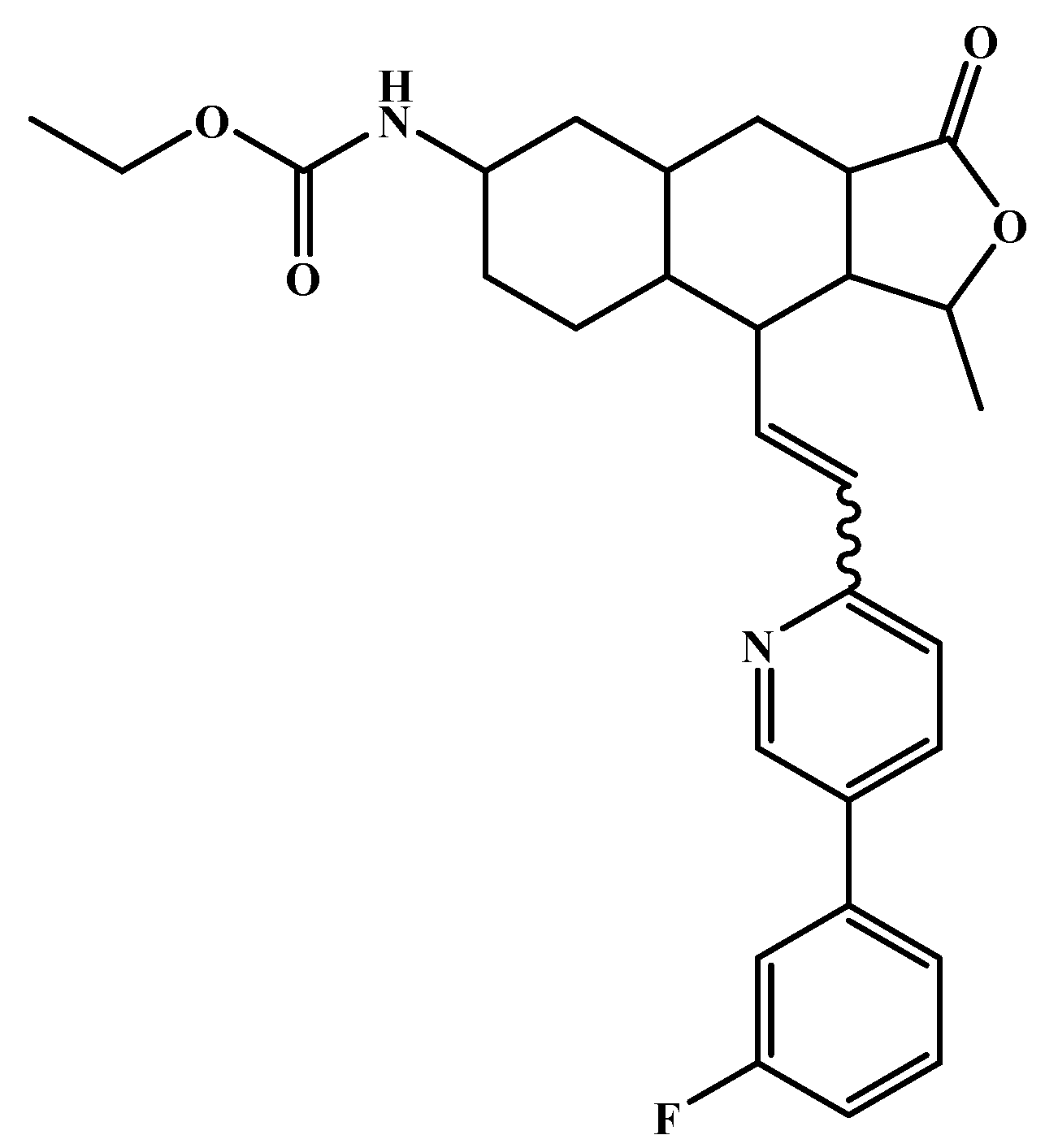
| nelfinavir 4 |  | octahydro 1H-isoquinoline | --- | --- | −7.4 6/−6.7 7 |
| lopinavir 4 |  | 2-oxotetrahydro pyrimidine | --- | --- | −6.5 6/−6.0 7 |
| allicin 5 |  | diallyl thiosulfinate | --- | --- | −3.3 6/−2.9 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaudêncio, S.P.; Pereira, F. A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition. Mar. Drugs 2020, 18, 633. https://doi.org/10.3390/md18120633
Gaudêncio SP, Pereira F. A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition. Marine Drugs. 2020; 18(12):633. https://doi.org/10.3390/md18120633
Chicago/Turabian StyleGaudêncio, Susana P., and Florbela Pereira. 2020. "A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition" Marine Drugs 18, no. 12: 633. https://doi.org/10.3390/md18120633
APA StyleGaudêncio, S. P., & Pereira, F. (2020). A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition. Marine Drugs, 18(12), 633. https://doi.org/10.3390/md18120633