Antitumor Activity of Asperphenin A, a Lipopeptidyl Benzophenone from Marine-Derived Aspergillus sp. Fungus, by Inhibiting Tubulin Polymerization in Colon Cancer Cells


Abstract
:1. Introduction
2. Results
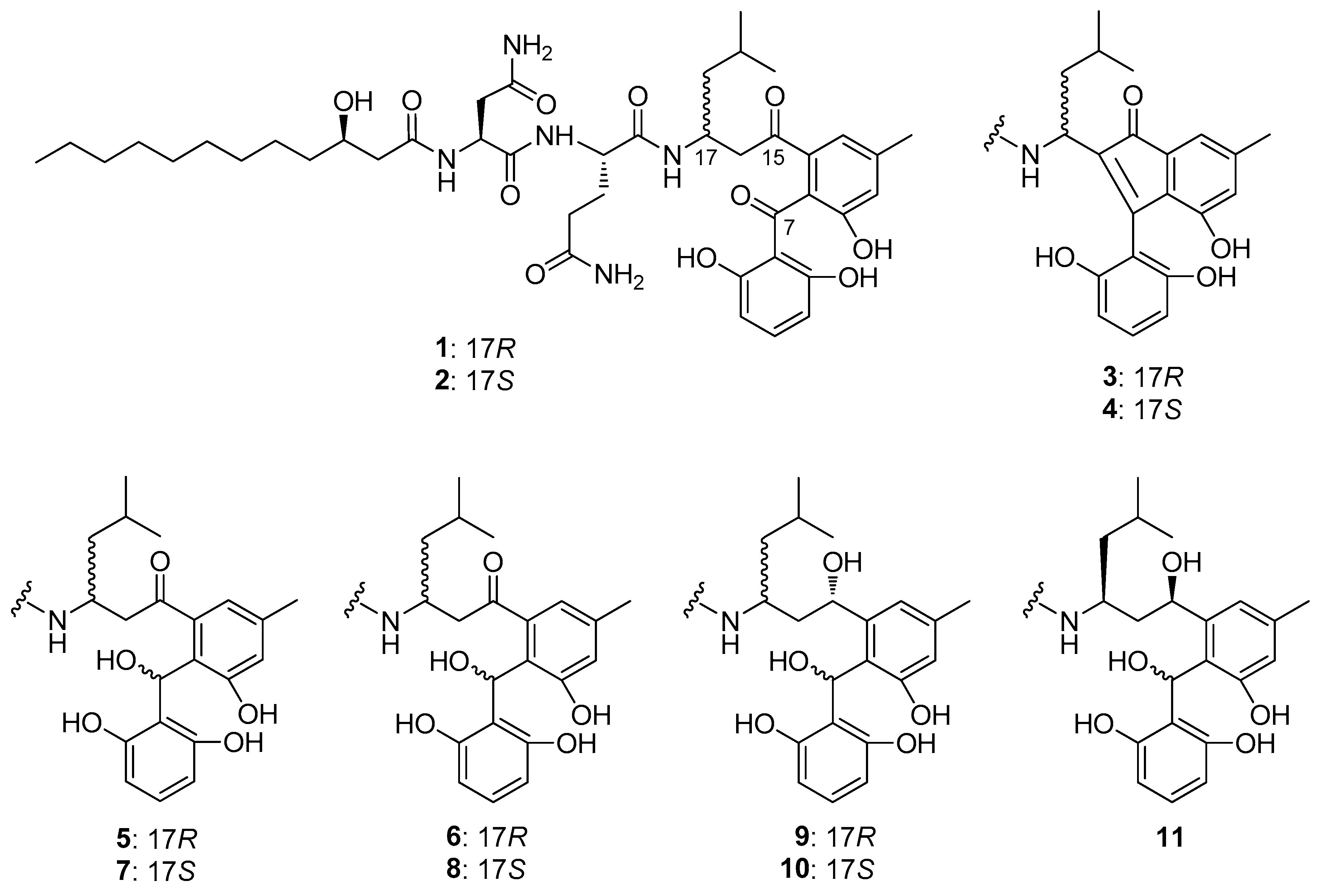
2.1. Aryl ketone at C-7 is Responsible for the Antiproliferative Effect of Asperphenins
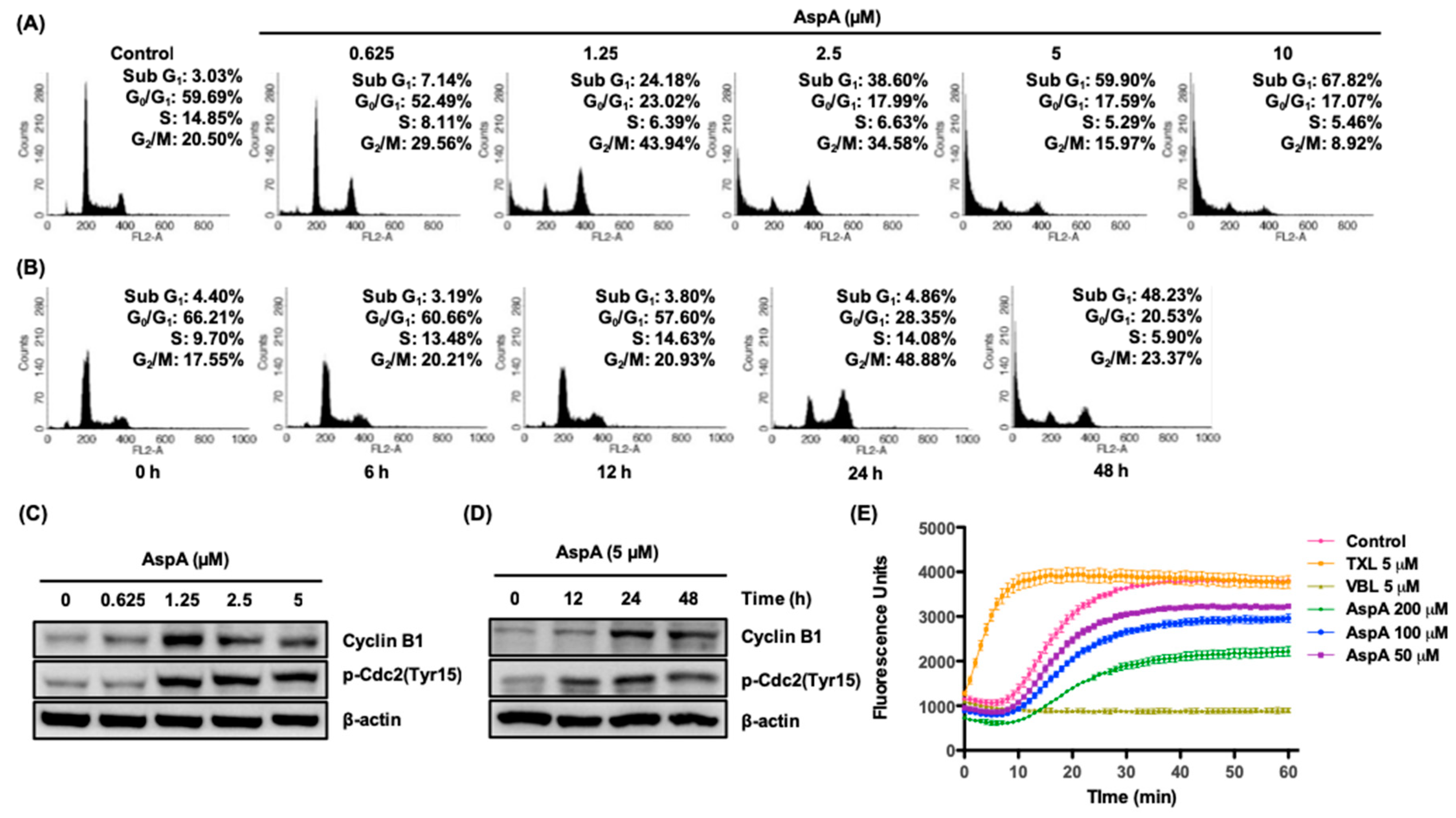
2.2. AspA Induces G2/M Phase Cell Cycle Arrest by Inhibiting Microtubule Assembly
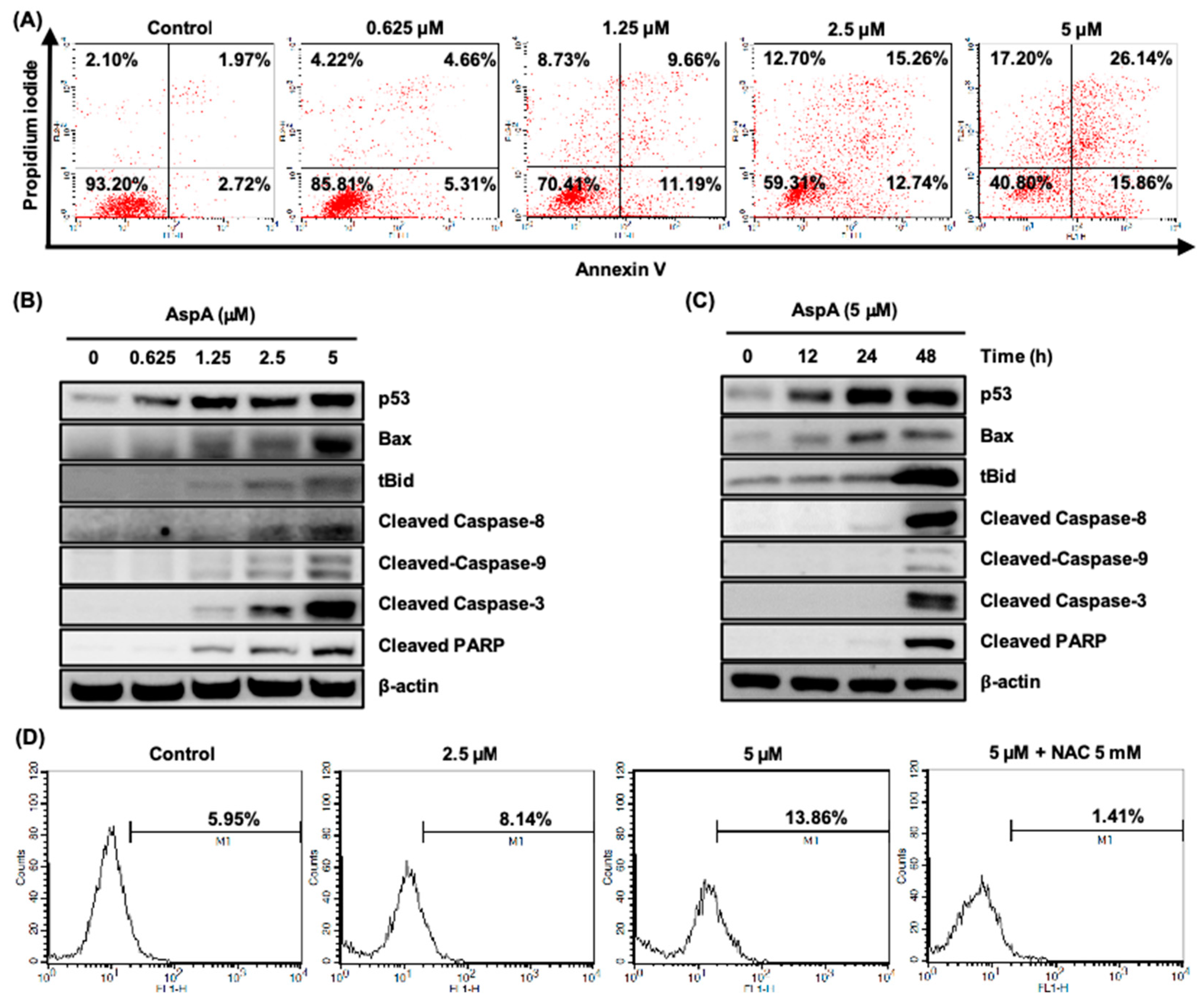
2.3. AspA Treated-Cells Undergo Apoptosis and Produce Intracellular Reactive Oxygen Species
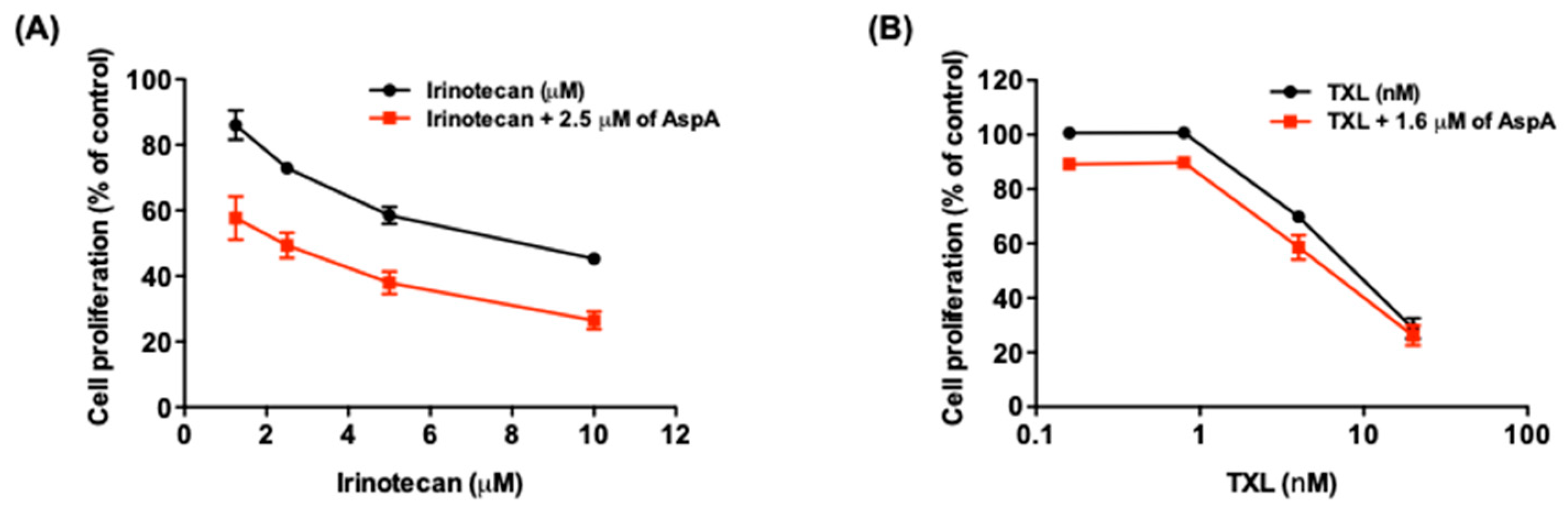
2.4. AspA Enhances the Effect of Irinotecan on Cell Growth Inhibition
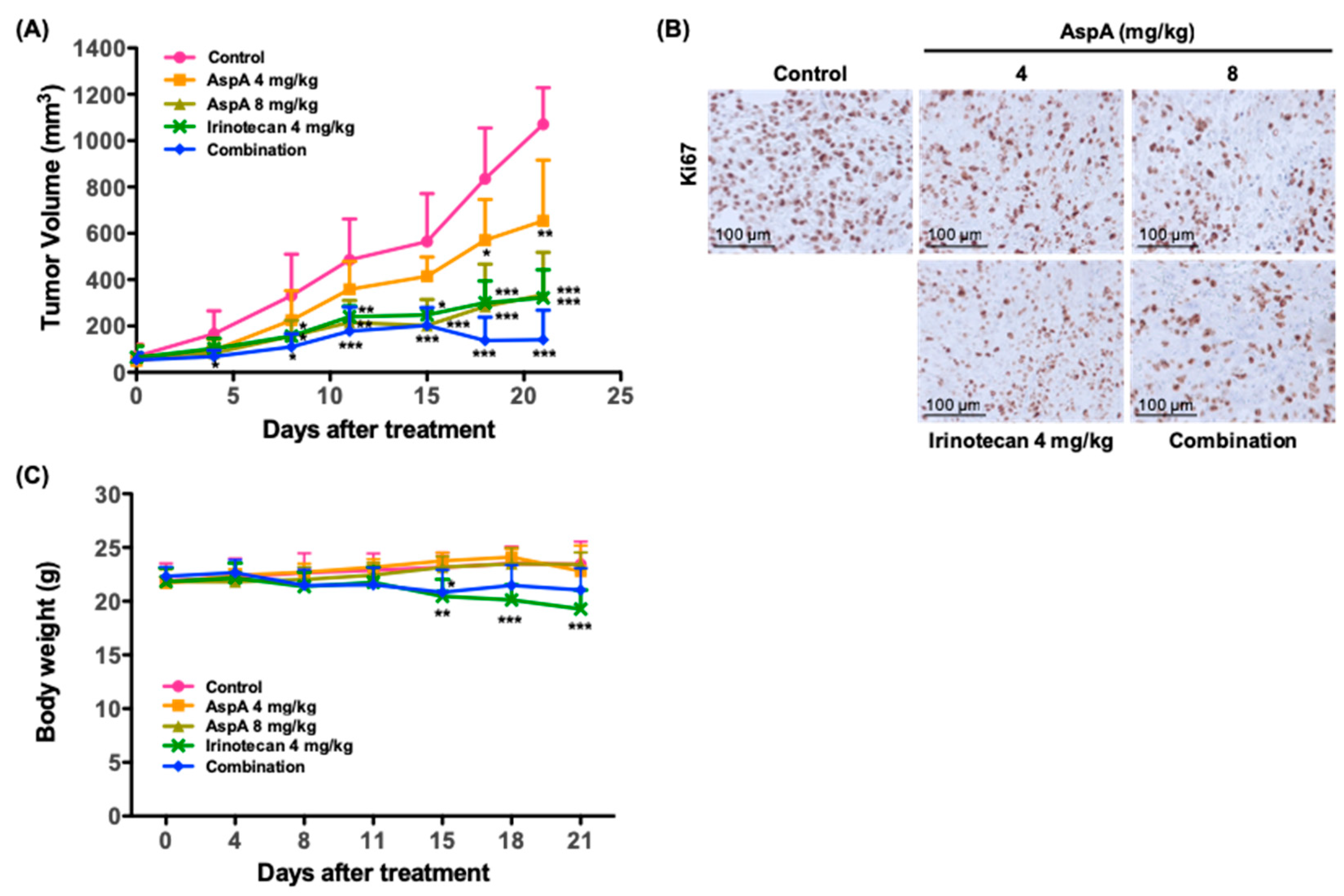
2.5. AspA Suppresses the Tumor Growth in RKO Cells-Implanted Nude Mouse Xenograft Models
3. Discussion
4. Materials and Methods
4.1. Preparation of Asperphenins and Synthetic Derivatives
4.2. Cell Proliferation Assay
4.3. Cell Cycle Analysis
4.4. Western Blot Analysis
4.5. Tubulin Polymerization Inhibition Assay
4.6. Annexin V/PI Staining
4.7. DCFH-DA Staining
4.8. In Vitro Drug Combination Analysis
4.9. In Vivo Tumor Xenograft Model
4.10. In Vivo Drug Combination Analysis
4.11. Immunohistochemistry
4.12. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council. Marine-Derived Pharmaceuticals and Related Bioactive Agents. In From Monsoons to Microbes: Understanding the Ocean’s Role in Human Health.; National Academies Press: Washington, DC, USA, 1999. [Google Scholar]
- Bhatnagar, I.; Kim, S.-K. Immense Essence of Excellence: Marine Microbial Bioactive Compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Barbash, O.; Diehl, J.A. 11—Regulation of the Cell Cycle. In The Molecular Basis of Cancer (Fourth Edition); Mendelsohn, J., Gray, J.W., Howley, P.M., Israel, M.A., Thompson, C.B., Eds.; Elsevier Inc.: Philadelphia, PA, USA, 2015; pp. 165–178.e2. ISBN 978-1-4557-4066-6. [Google Scholar]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [PubMed] [Green Version]
- Gookin, S.; Min, M.; Phadke, H.; Chung, M.; Moser, J.; Miller, I.; Carter, D.; Spencer, S.L. A map of protein dynamics during cell-cycle progression and cell-cycle exit. PLOS Biol. 2017, 15, e2003268. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, M.O.; Prota, A.E. Microtubule-Targeting Agents: Strategies to Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting Microtubules by Natural Agents for Cancer Therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Anderson, H.J.; Coleman, J.E.; Andersen, R.J.; Roberge, M. Cytotoxic peptides hemiasterlin, hemiasterlin A and hemiasterlin B induce mitotic arrest and abnormal spindle formation. Cancer Chemother. Pharmacol. 1996, 39, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Monserrate, Z.; Vervoort, H.C.; Bai, R.; Newman, D.J.; Howell, S.B.; Los, G.; Mullaney, J.T.; Williams, M.D.; Pettit, G.R.; Fenical, W.; et al. Diazonamide A and a Synthetic Structural Analog: Disruptive Effects on Mitosis and Cellular Microtubules and Analysis of Their Interactions with Tubulin. Mol. Pharmacol. 2003, 63, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Edler, M.C.; Fernandez, A.M.; Lassota, P.; Ireland, C.M.; Barrows, L.R. Inhibition of tubulin polymerization by vitilevuamide, a bicyclic marine peptide, at a site distinct from colchicine, the vinca alkaloids, and dolastatin 10. Biochem. Pharmacol. 2002, 63, 707–715. [Google Scholar] [CrossRef]
- Schmidt, E.W.; Raventos-Suarez, C.; Bifano, M.; Menendez, A.T.; Fairchild, C.R.; Faulkner, D.J. Scleritodermin A, a Cytotoxic Cyclic Peptide from the Lithistid Sponge Scleritoderma nodosum. J. Nat. Prod. 2004, 67, 475–478. [Google Scholar] [CrossRef]
- Simmons, T.L.; Nogle, L.M.; Media, J.; Valeriote, F.A.; Mooberry, S.L.; Gerwick, W.H. Desmethoxymajusculamide C, a Cyanobacterial Depsipeptide with Potent Cytotoxicity in Both Cyclic and Ring-Opened Forms. J. Nat. Prod. 2009, 72, 1011–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, L.; Bae, S.Y.; Won, T.H.; You, M.; Kim, S.-H.; Oh, D.-C.; Lee, S.K.; Oh, K.-B.; Shin, J. Asperphenins A and B, Lipopeptidyl Benzophenones from a Marine-Derived Aspergillus sp. Fungus. Organic Lett. 2017, 19, 2066–2069. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Mitotic Arrest and Cell Fate: Why and How Mitotic Inhibition of Transcription Drives Mutually Exclusive Events. Cell Cycle 2007, 6, 70–74. [Google Scholar] [CrossRef]
- Schuler, M.; Bossy-Wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 Induces Apoptosis by Caspase Activation through Mitochondrial Cytochrome c Release. J. Biol. Chem. 2000, 275, 7337–7342. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, J.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel Action of Paclitaxel against Cancer Cells: Bystander Effect Mediated by Reactive Oxygen Species. Cancer Res. 2007, 67, 3512–3517. [Google Scholar] [CrossRef] [Green Version]
- Alexandre, J.; Batteux, F.; Nicco, C.; Chéreau, C.; Laurent, A.; Guillevin, L.; Weill, B.; Goldwasser, F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int. J. Cancer 2006, 119, 41–48. [Google Scholar] [CrossRef]
- Masi, G.; Allegrini, G.; Cupini, S.; Marcucci, L.; Cerri, E.; Brunetti, I.; Fontana, E.; Ricci, S.; Andreuccetti, M.; Falcone, A. First-line treatment of metastatic colorectal cancer with irinotecan, oxaliplatin and 5-fluorouracil/leucovorin (FOLFOXIRI): Results of a phase II study with a simplified biweekly schedule. Ann. Oncol. 2004, 15, 1766–1772. [Google Scholar] [CrossRef] [PubMed]
- Negi, B.; Kumar, D.; Rawat, D.S. Marine Peptides as Anticancer Agents: A Remedy to Mankind by Nature. Curr. Protein Pept. Sci. 2017, 18, 885–904. [Google Scholar] [PubMed]
- Checchi, P.M.; Nettles, J.H.; Zhou, J.; Snyder, J.P.; Joshi, H.C. Microtubule-interacting drugs for cancer treatment. Trends Pharmacol. Sci. 2003, 24, 361–365. [Google Scholar] [CrossRef]
- Mitchison, T.J. Microtubule Dynamics and Kinetochore Function in Mitosis. Ann. Rev. Cell Biol. 1988, 4, 527–545. [Google Scholar]
- Conklin, K.A. Chemotherapy-Associated Oxidative Stress: Impact on Chemotherapeutic Effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar]
- Goodman, M.; Bostick, R.M.; Kucuk, O.; Jones, D.P. Clinical trials of antioxidants as cancer prevention agents: Past, present, and future. Free Rad. Biol. Med. 2011, 51, 1068–1084. [Google Scholar]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334ra51. [Google Scholar] [CrossRef] [Green Version]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar]
- Cui, Q.; Wang, J.-Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Modern Natural Products Drug Discovery and its Relevance to Biodiversity Conservation. J. Nat. Prod. 2011, 74, 496–511. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.; Hong, J.-Y.; Bae, S.Y.; Kang, S.S.; Park, H.J.; Lee, S.K. Antitumor Activity of Americanin A Isolated from the Seeds of Phytolacca americana by Regulating the ATM/ATR Signaling Pathway and the Skp2–p27 Axis in Human Colon Cancer Cells. J. Nat. Prod. 2015, 78, 2983–2993. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Dings, R.P.M.; Yokoyama, Y.; Ramakrishnan, S.; Griffioen, A.W.; Mayo, K.H. The Designed Angiostatic Peptide Anginex Synergistically Improves Chemotherapy and Antiangiogenesis Therapy with Angiostatin. Cancer Res. 2003, 63, 382–385. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µM), 72 h | |||
|---|---|---|---|---|
| RKO | SNU638 | SK-HEP-1 | MDA-MB-231 | |
| Asperphenin A (1) | 0.84 ± 0.26 | 4.31 ± 0.83 | 2.89 ± 0.03 | 6.48 ± 0.74 |
| Asperphenin B (2) | 1.26 ± 0.43 | 7.59 ± 1.77 | 3.08 ± 0.18 | 9.43 ± 0.39 |
| Cycloasperphenin A (3) | >50 | >50 | >50 | >50 |
| Cycloasperphenin B (4) | >50 | >50 | >50 | >50 |
| 7-Hydroxyasperphenin A (5) | 24.23 ± 1.05 | 42.55 ± 4.59 | 37.58 ± 0.96 | >50 |
| 7-epi-Hydroxyasperphenin A (6) | 28.12 ± 4.36 | 49.26 ± 0.93 | 40.93 ± 0.66 | >50 |
| 7-Hydroxyasperphenin B (7) | 36.32 ± 1.45 | 42.40 ± 1.27 | 48.09 ± 0.01 | >50 |
| 7-epi-Hydroxyasperphenin B (8) | 27.75 ± 2.98 | >50 | >50 | >50 |
| 7,15(S)-Dihydroxyasperphenin A (9) | >50 | >50 | >50 | >50 |
| 7,15(S)-Dihydroxyasperphenin B (10) | >50 | >50 | >50 | >50 |
| 7,15(R)-Dihydroxyasperphenin B (11) | >50 | >50 | >50 | >50 |
| Etoposide 1 | 3.82 ± 0.74 | 0.30 ± 0.05 | 0.49 ± 0.12 | 10.72 ± 0.88 |
| Irinotecan (μM) | CI Value | Description |
|---|---|---|
| 1.25 | 0.811 ± 0.204 | Moderate synergism |
| 2.5 | 0.756 ± 0.100 | Moderate synergism |
| 5 | 0.694 ± 0.096 | Synergism |
| 10 | 0.652 ± 0.087 | Synergism |
| TXL (nM) | CI Value | Description |
|---|---|---|
| 0.16 | 1.673 ± 0.106 | Antagonism |
| 0.8 | 1.925 ± 0.305 | Antagonism |
| 4 | 0.742 ± 0.086 | Moderate synergism |
| 20 | 1.185 ± 0.102 | Slight antagonism |
| Group | AspA (mg/kg) | 4 mg/kg of Irinotecan | Combination | |
|---|---|---|---|---|
| 4 | 8 | |||
| Inhibition rate (%) | 38.9 ± 24.5 | 68.7 ± 17.1 | 70.0 ± 11.3 | 86.9 ± 11.9 |
| FTV 1 | Expected FTV | Observed FTV | Combination Ratio | |
|---|---|---|---|---|
| 4 mg/kg of AspA | 4 mg/kg of Irinotecan | |||
| 0.61 | 0.30 | 0.18 | 0.13 | 1.39 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, S.Y.; Liao, L.; Park, S.H.; Kim, W.K.; Shin, J.; Lee, S.K. Antitumor Activity of Asperphenin A, a Lipopeptidyl Benzophenone from Marine-Derived Aspergillus sp. Fungus, by Inhibiting Tubulin Polymerization in Colon Cancer Cells. Mar. Drugs 2020, 18, 110. https://doi.org/10.3390/md18020110
Bae SY, Liao L, Park SH, Kim WK, Shin J, Lee SK. Antitumor Activity of Asperphenin A, a Lipopeptidyl Benzophenone from Marine-Derived Aspergillus sp. Fungus, by Inhibiting Tubulin Polymerization in Colon Cancer Cells. Marine Drugs. 2020; 18(2):110. https://doi.org/10.3390/md18020110
Chicago/Turabian StyleBae, Song Yi, Lijuan Liao, So Hyun Park, Won Kyung Kim, Jongheon Shin, and Sang Kook Lee. 2020. "Antitumor Activity of Asperphenin A, a Lipopeptidyl Benzophenone from Marine-Derived Aspergillus sp. Fungus, by Inhibiting Tubulin Polymerization in Colon Cancer Cells" Marine Drugs 18, no. 2: 110. https://doi.org/10.3390/md18020110
APA StyleBae, S. Y., Liao, L., Park, S. H., Kim, W. K., Shin, J., & Lee, S. K. (2020). Antitumor Activity of Asperphenin A, a Lipopeptidyl Benzophenone from Marine-Derived Aspergillus sp. Fungus, by Inhibiting Tubulin Polymerization in Colon Cancer Cells. Marine Drugs, 18(2), 110. https://doi.org/10.3390/md18020110