Genome Sequencing and Analysis of Thraustochytriidae sp. SZU445 Provides Novel Insights into the Polyunsaturated Fatty Acid Biosynthesis Pathway



Abstract
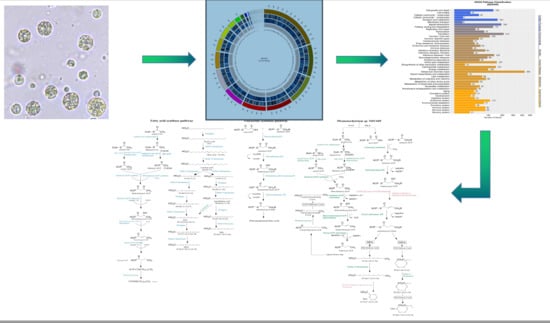
:
1. Introduction
2. Results
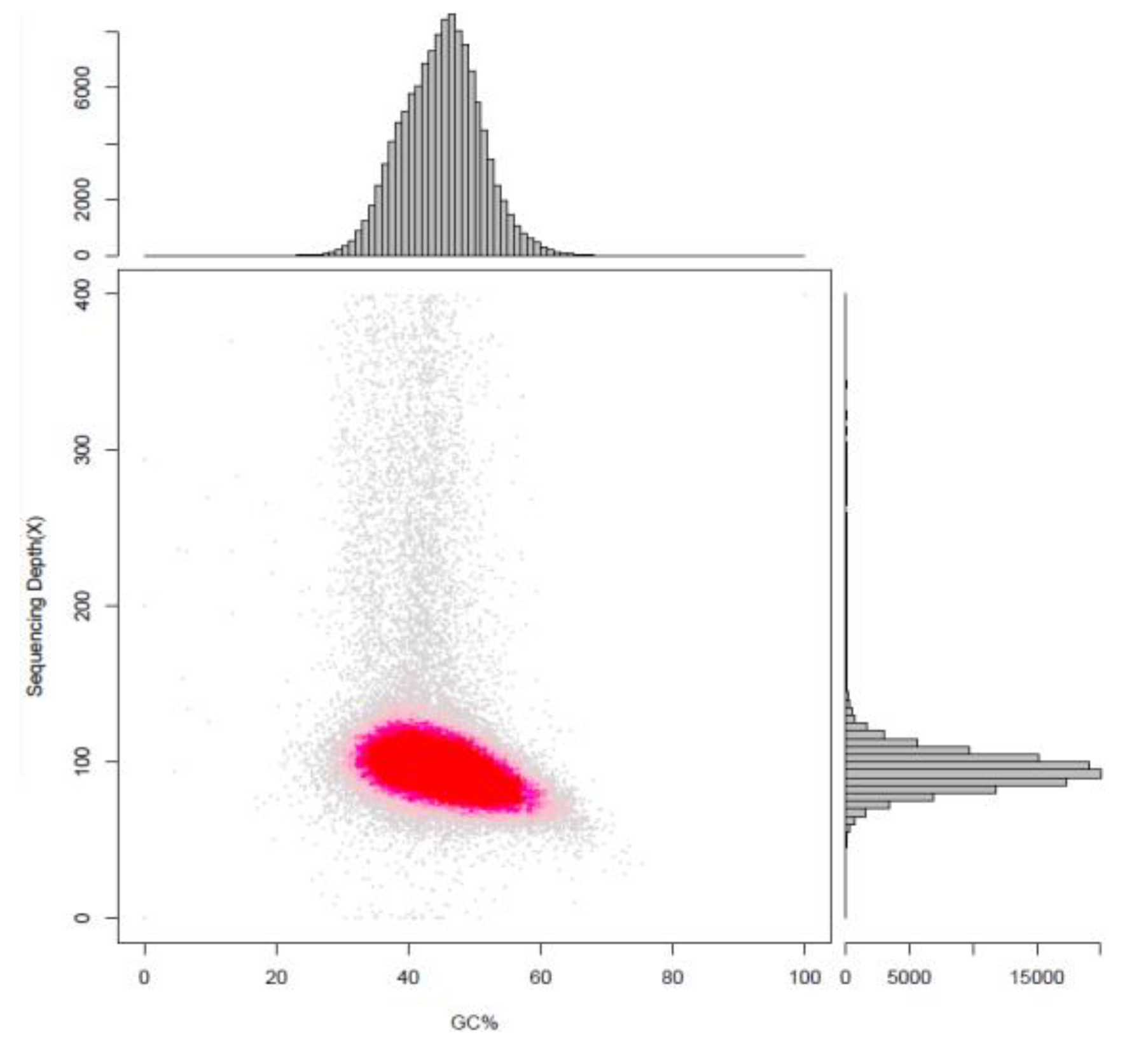
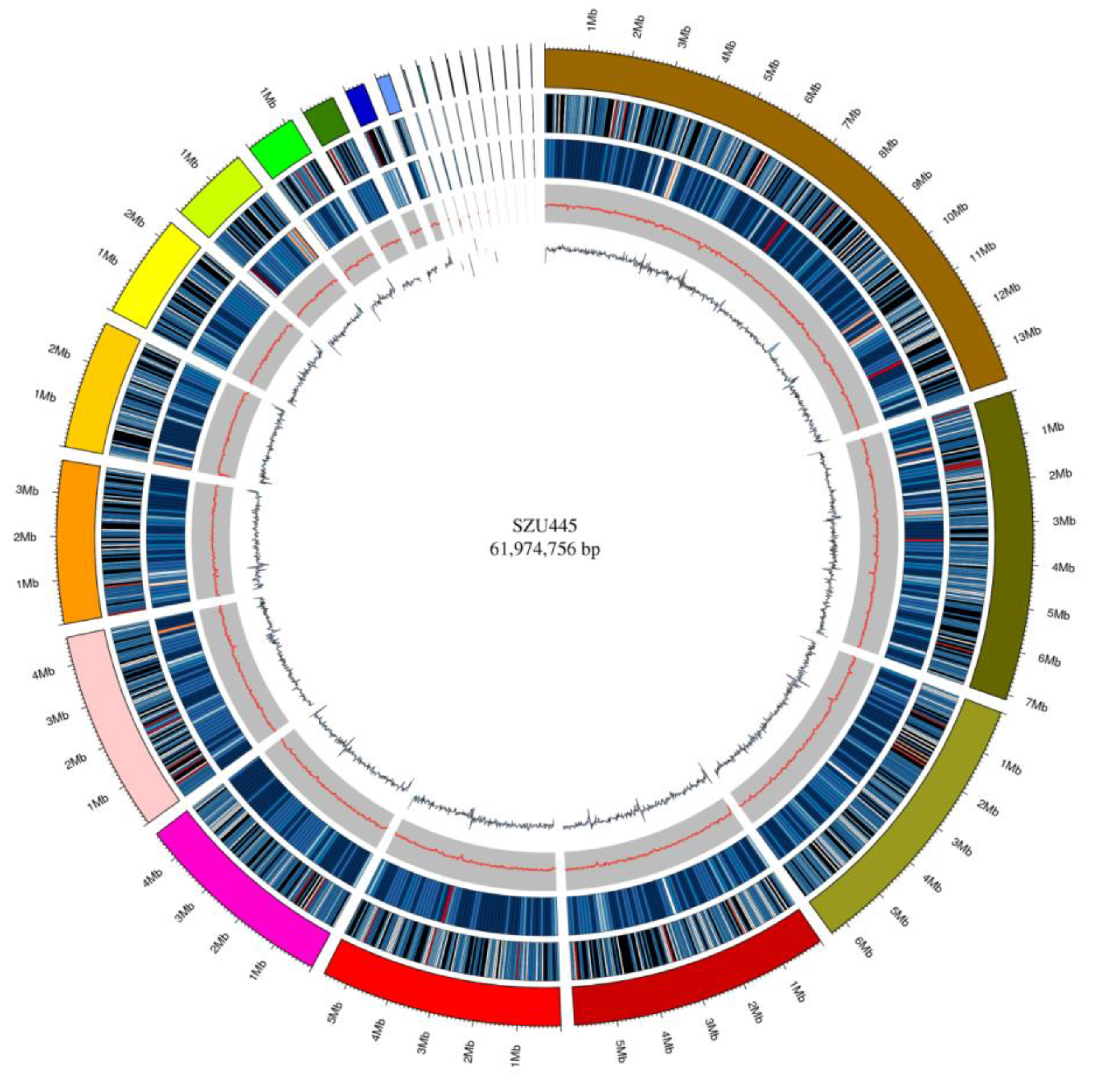
2.1. Genome Sequencing and De Novo Assembly
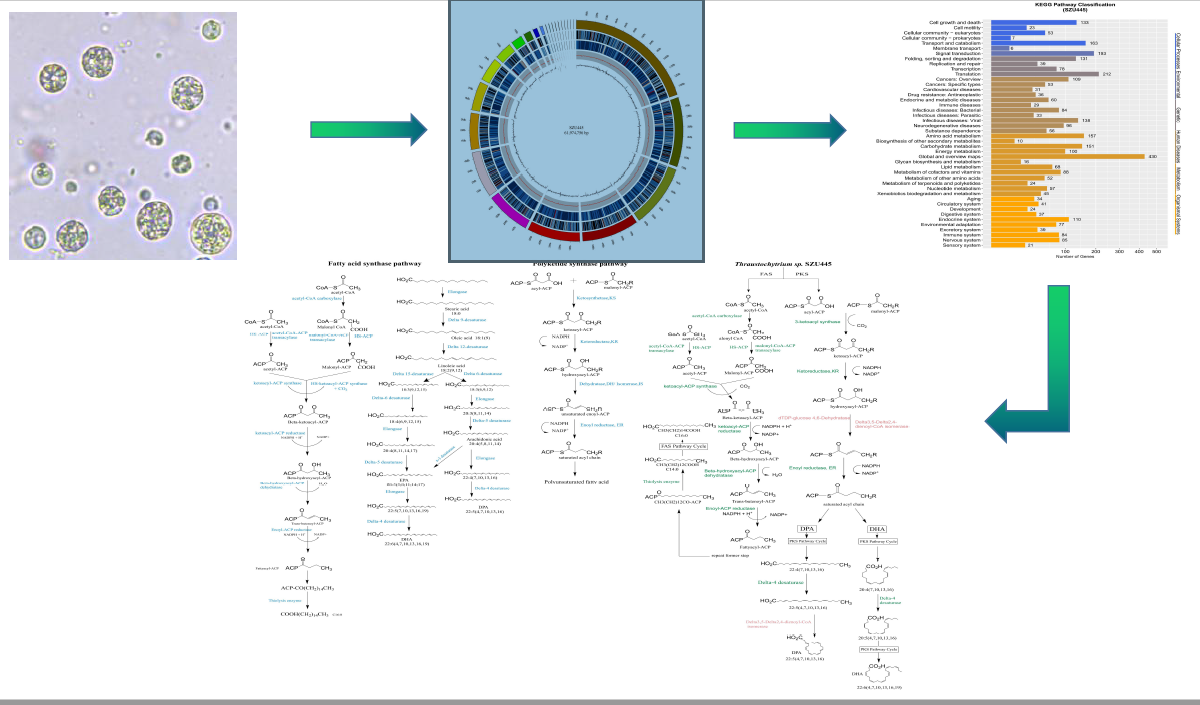
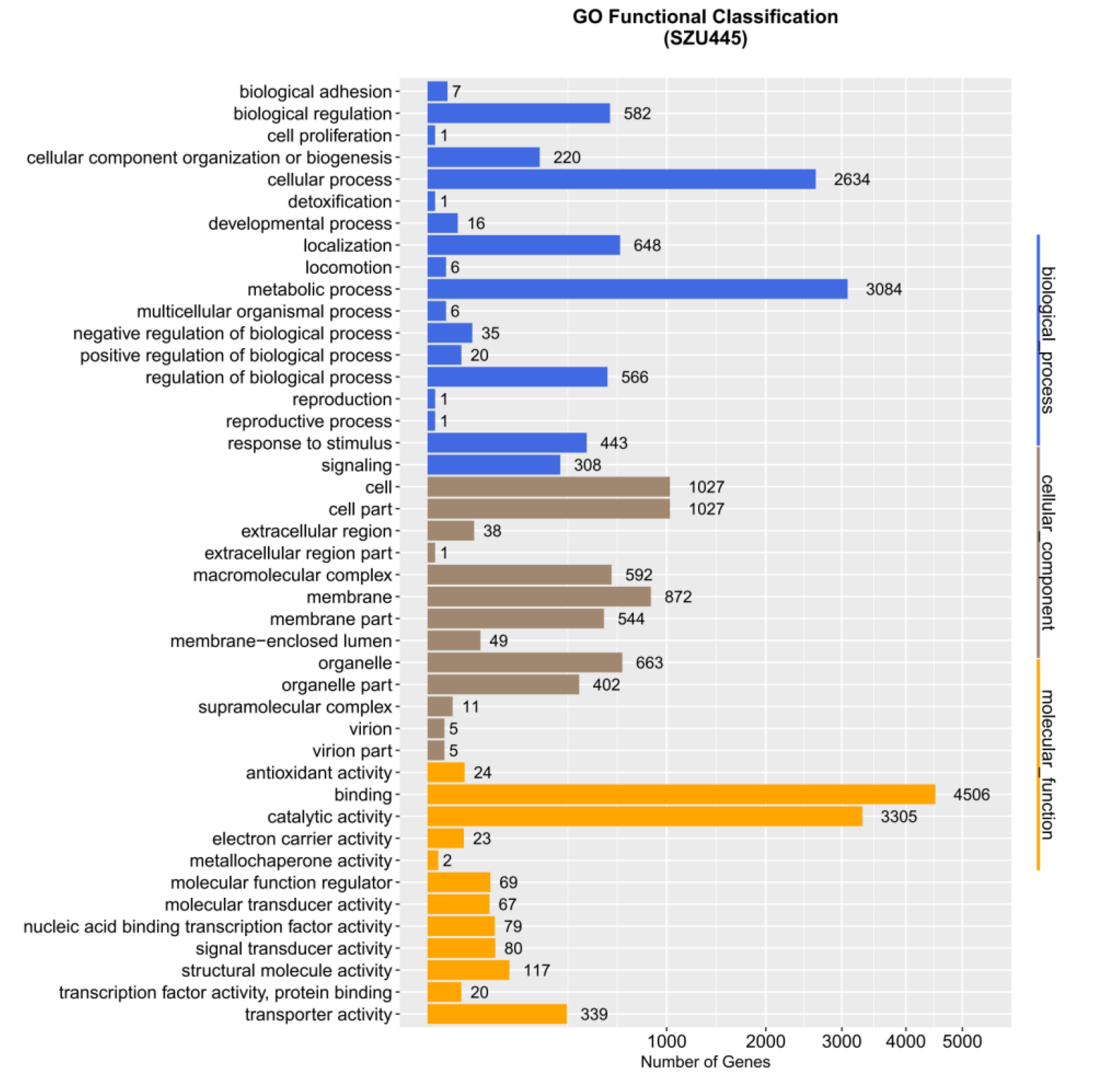
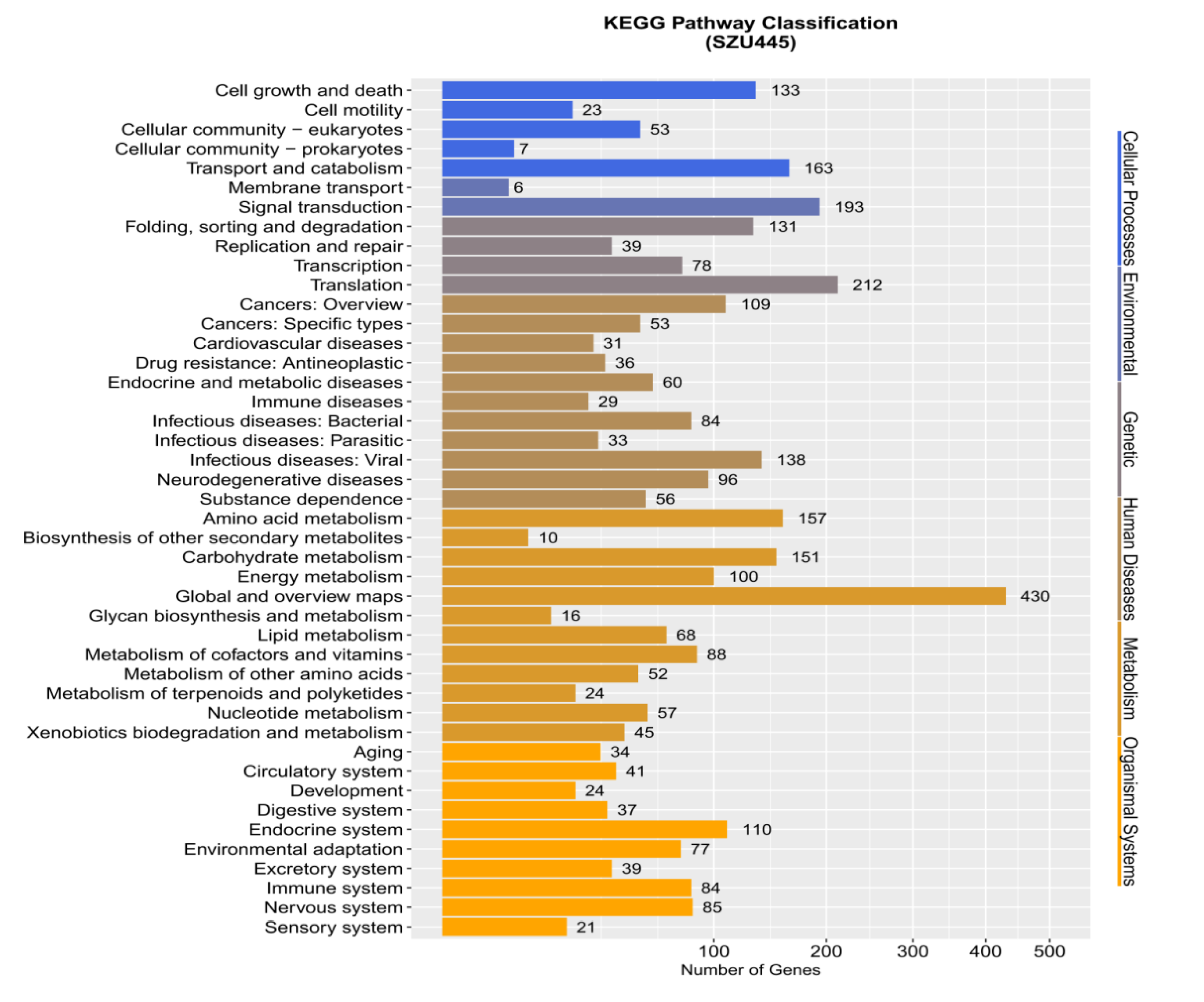
2.2. Genome Sequence Annotation
2.3. Phylogenetic Analysis of Thraustochytriidae sp. SZU445
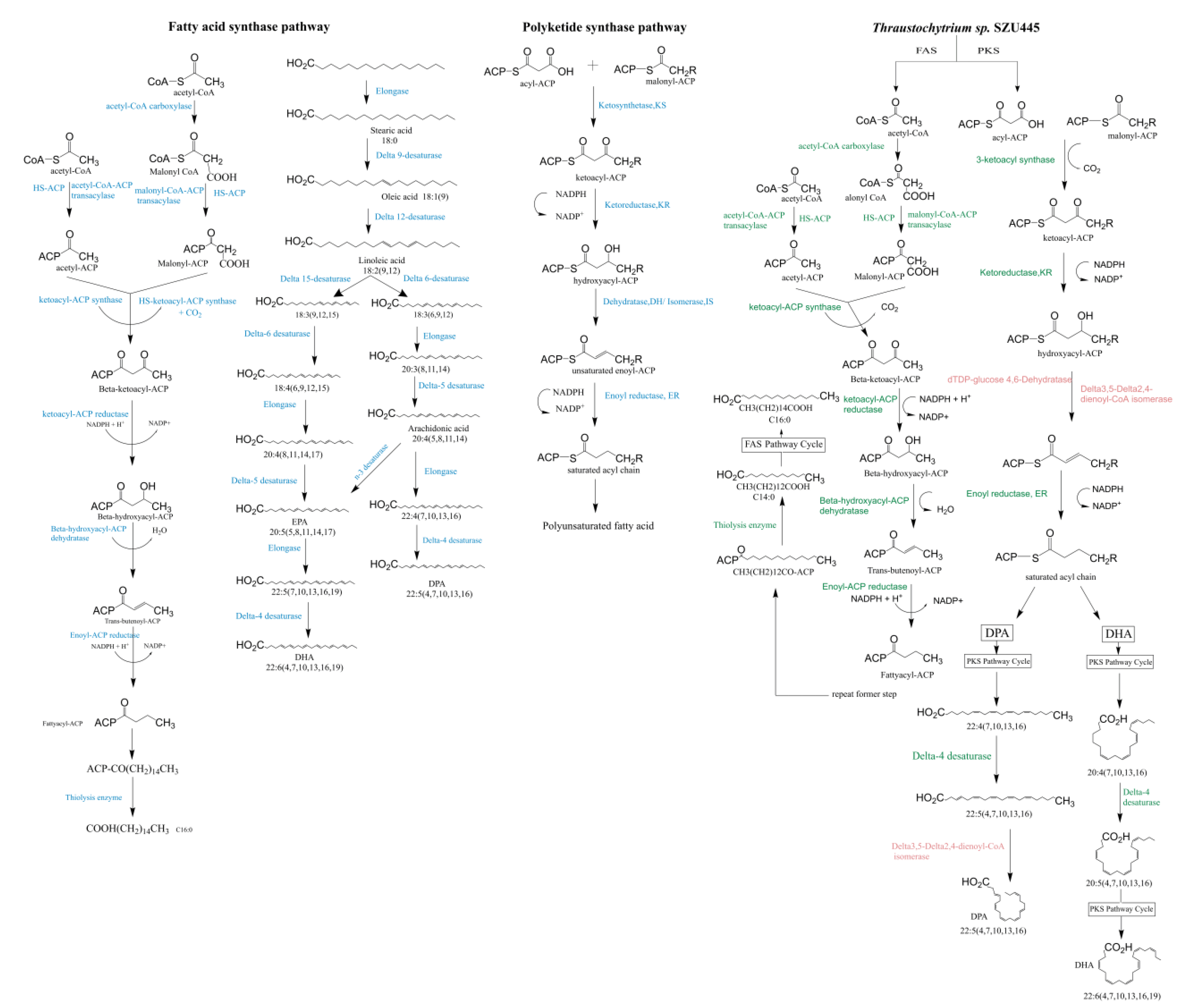
2.4. Analysis of Genes Involved in Long-Chain Fatty Acid (LCFA) Biosynthesis
3. Discussion
3.1. Fatty Acid Synthesis by the FAS Pathway
3.2. Fatty Acid Synthesis by the PKS Pathway
3.3. Putative Fatty Acid Synthesis Pathway in Thraustochytrium sp. SZU445
4. Materials and Methods
4.1. Microbes and Cultivation
4.2. DNA Preparation and Sequencing
4.3. Fragment Assembly and Gene Annotation
4.4. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Salem, N.; Litman, B.; Kim, H.-Y.; Gawrisch, K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids 2001, 36, 945–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, L. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]
- Von Schacky, C.; Harris, W.S. Cardiovascular benefits of omega-3 fatty acids. Cardiovasc. Res. 2007, 73, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Brown, R.E.; Zhang, P.C.; Zhao, Y.T.; Ju, X.H.; Song, C. DHA, EPA and their combination at various ratios differently modulated a beta(25-35)-induced neurotoxicity in sh-sy5y cells. Prostaglandins Leukot. Essent. Fatty Acids 2018, 136, 85–94. [Google Scholar] [CrossRef]
- Gillet, L.; Roger, S.; Bougnoux, P.; Le Guennec, J.-Y.; Besson, P. Beneficial effects of omega-3 long-chain fatty acids in breast cancer and cardiovascular diseases: Voltage-gated sodium channels as a common feature? Biochimie 2011, 93, 4–6. [Google Scholar] [CrossRef]
- Hashimoto, M.; Hossain, S.; Al Mamun, A.; Matsuzaki, K.; Arai, H. Docosahexaenoic acid: One molecule diverse functions. Crit. Rev. Biotechnol. 2017, 37, 579–597. [Google Scholar] [CrossRef]
- Rubio-Rodríguez, N.; Beltrán, S.; Jaime, I.; De Diego, S.M.; Sanz, M.T.; Carballido, J.R. Production of omega-3 polyunsaturated fatty acid concentrates: A review. Innov. Food Sci. Emerg. Technol. 2010, 11, 1–12. [Google Scholar] [CrossRef]
- Lewis, T.E.; Nichols, P.D.; McMeekin, T.A. The Biotechnological Potential of Thraustochytrids. Surg. Endosc. 1999, 1, 580–587. [Google Scholar] [CrossRef]
- She, Z.L.; Wu, L.; Wang, Q.; Gao, M.C.; Jin, C.J.; Zhao, Y.G.; Zhao, L.T.; Guo, L. Salinity effect on simultaneous nitrification and denitrification, microbial characteristics in a hybrid sequencing batch biofilm reactor. Bioproc. Biosyst. Eng. 2018, 41, 65–75. [Google Scholar] [CrossRef]
- Ma, Z.; Tan, Y.; Cui, G.; Feng, Y.; Cui, Q.; Song, X. Transcriptome and gene expression analysis of DHA producer Aurantiochytrium under low temperature conditions. Sci. Rep. 2015, 5, 14446. [Google Scholar] [CrossRef] [Green Version]
- Patil, K.P.; Gogate, P.R. Improved synthesis of docosahexaenoic acid (DHA) using Schizochytrium limacinum SR21 and sustainable media. Chem. Eng. J. 2015, 268, 187–196. [Google Scholar] [CrossRef]
- Metz, J.G.; Roessler, P.; Facciotti, D.; Levering, C.; Dittrich, F.; Lassner, M.; Valentine, R.; Lardizabal, K.; Domergue, F.; Yamada, A.; et al. Production of Polyunsaturated Fatty Acids by Polyketide Synthases in Both Prokaryotes and Eukaryotes. Science 2001, 293, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Anbu, P.; Kim, W.H.; Hur, B.K. Coexpression of elo-like enzyme and delta 5, delta 4-desaturases derived from thraustochytrium aureum atcc 34304 and the production of dha and dpa in pichia pastoris. Biotechnol. Bioproc. E 2008, 13, 483–490. [Google Scholar] [CrossRef]
- Matsuda, T. Analysis of delta 12-fatty acid desaturase function revealed that two distinct pathways are active for the synthesis of pufas in t Aureum atcc 34304. J. Lipid Res. 2012, 53, 1210–1222. [Google Scholar]
- Nagano, N.; Sakaguchi, K.; Taoka, Y.; Okita, Y.; Honda, D.; Ito, M.; Hayashi, M. Detection of genes involved in fatty acid elongation and desaturation in thraustochytrid marine eukaryotes. J. Oleo Sci. 2011, 60, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Yoshizawa, A.C.; Okuda, S.; Kuma, K.; Goto, S.; Kanehisa, M. The repertoire of desaturases and elongases reveals fatty acid variations in 56 eukaryotic genomes. J. Lipid Res. 2008, 49, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Ohara, J.; Sakaguchi, K.; Okita, Y.; Okino, N.; Ito, M. Two fatty acid elongases possessing c18-delta 6/c18-delta 9/c20-delta 5 or c16-delta 9 elongase activity in thraustochytrium sp. atcc 26185. Mar. Biotechnol. 2013, 15, 476–486. [Google Scholar] [CrossRef]
- Meesapyodsuk, D.; Qiu, X. Biosynthetic mechanism of very long chain polyunsaturated fatty acids in Thraustochytrium sp. 26185. J. Lipid Res. 2016, 57, 1854–1864. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Qiao, W.; Yu, X.-B.; Ji, X.-J.; Huang, H.; Collier, J.; Liu, L. Reconstruction and analysis of the genome-scale metabolic model of schizochytrium limacinum SR21 for docosahexaenoic acid production. BMC Genom. 2015, 16, 799. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Stajich, J.E.; Xie, Y.; Liu, X.; He, Y.; Chen, J.; Hicks, G.R.; Wang, G. Comparative analysis reveals unexpected genome features of newly isolated Thraustochytrids strains: On ecological function and PUFAs biosynthesis. BMC Genom. 2018, 19, 541. [Google Scholar] [CrossRef]
- Rismani-Yazdi, H.; Haznedaroglu, B.Z.; Bibby, K.; Peccia, J. Transcriptome sequencing and annotation of the microalgae Dunaliella tertiolecta: Pathway description and gene discovery for production of next-generation biofuels. BMC Genom. 2011, 12, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.M.; Dauenpen, M.; Qu, C.M.; Qiu, X. Genomic analysis of genes involved in the biosynthesis of very long chain polyunsaturated fatty acids in thraustochytrium sp. 26185. Lipids 2016, 51, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Kourmpetis, Y.A.; van der Burgt, A.; Bink, M.C.; ter Braak, C.J.; van Ham, R.C. The use of multiple hierarchically independent gene ontology terms in gene function prediction and genome annotation. Silico Boil. 2007, 7, 575–582. [Google Scholar]
- Marek, O.; Serge, E.; Antonio, D.S.; Joaquín, D. Evolutionary conservation and network structure characterize genes of phenotypic relevance for mitosis in human. Plos ONE 2012, 7, e36488. [Google Scholar]
- Speer, N.; Spieth, C.; Zell, A. Spectral Clustering Gene Ontology Terms to Group Genes by Function. Comput. Vision 2005, 3692, 1–12. [Google Scholar]
- Yao, D.; Zhang, C.; Zhao, X.; Liu, C.; Wang, C.; Zhang, Z.; Zhang, C.; Wei, Q.; Wang, Q.; Yan, H.; et al. Transcriptome analysis reveals salt-stress-regulated biological processes and key pathways in roots of cotton (Gossypium hirsutum L.). Genomics 2011, 98, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Zuo, G.; Chen, Z.; Jiang, Y.; Zhu, Z.; Ding, C.; Zhang, Z.; Chen, Y.; Zhou, C.; Li, Q. Structural insights into repression of the Pneumococcal fatty acid synthesis pathway by repressor FabT and co-repressor acyl-ACP. FEBS Lett. 2019, 593, 2730–2741. [Google Scholar] [CrossRef]
- Lee, J.Y.; Ha, J.; Yi, J.; Jang, J.; Lee, W.; Lee, Y.; Oh, D.-Y.; Han, K. Superior single nucleotide polymorphisms that contribute to two main routes of the fatty acid synthesis pathway in Korean cattle. Genes Genom. 2018, 40, 945–954. [Google Scholar] [CrossRef]
- Arthur, C.; Williams, C.; Pottage, K.; Płoskoń, E.; PFindlow, S.C.; Burston, S.G.; Simpson, T.J.; Crump, M.P.; Crosby, J. Structure and Malonyl CoA-ACP Transacylase Binding of Streptomyces coelicolor Fatty Acid Synthase Acyl Carrier Protein.E. J. ACS Chem. Boil. 2009, 4, 625–636. [Google Scholar]
- Seddiki, K.; Godart, F.O.; Aiese Cigliano, R.; Sanseverino, W.; Barakat, M.; Ortet, P.; Rébeillé, F.; Maréchal, E.; Cagnac, O.; Amato, A. Sequencing, de novo assembly, and annotation of the complete genome of a new thraustochytrid species, strain ccap_4062/3. Genome Announc. 2018, 6, e01335-17. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Hong, H.; MacKenzie, S.L. Identification of a delta 4 fatty acid desaturase from thraustochytrium sp. Involved in the biosynthesis of docosahexanoic acid by heterologous expression in Saccharomyces cerevisiae and Brassica juncea. J. Biol. Chem. 2001, 276, 31561–31566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-Y.; Ohashi, T.; Wu, C.-C.; Bataa, D.; Misaki, R.; Limtong, S.; Fujiyama, K. Delta-9 fatty acid desaturase overexpression enhanced lipid production and oleic acid content in Rhodosporidium toruloides for preferable yeast lipid production. J. Biosci. Bioeng. 2019, 127, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Chen, G.; Chen, J.; Zheng, M. Identification of a Long-Chain Fatty Acid Elongase from Nannochloropsis sp. Involved in the Biosynthesis of Fatty Acids by Heterologous Expression in Saccharomyces cerevisiae. J. Ocean Univ. China 2019, 18, 1199–1206. [Google Scholar] [CrossRef]
- Kaulmann, U.; Hertweck, C.J.A.C.I.E. Cheminform abstract: Biosynthesis of polyunsaturated fatty acids by polyketide synthases. Angew. Chem. Int. Ed. 2002, 41, 1866–1869. [Google Scholar] [CrossRef]
- Podell, S.; Blanton, J.M.; Neu, A.; Agarwal, V.; Biggs, J.S.; Moore, B.S.; Allen, E.E. Pangenomic comparison of globally distributed poribacteria associated with sponge hosts and marine particles. ISME J. 2019, 13, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Maeda, Y.; Veluchamy, A.; Tanaka, M.; Abida, H.; Maréchal, E.; Bowler, C.; Muto, M.; Sunaga, Y.; Tanaka, M.; et al. Oil Accumulation by the Oleaginous Diatom Fistulifera solaris as Revealed by the Genome and Transcriptome. Plant Cell 2015, 27, 162–176. [Google Scholar] [CrossRef] [Green Version]
- Schonauer, M.S.; Kastaniotis, A.J.; Hiltunen, J.K.; Dieckmann, C.L. Intersection of RNA Processing and the Type II Fatty Acid Synthesis Pathway in Yeast Mitochondria. Mol. Cell. Boil. 2008, 28, 6646–6657. [Google Scholar] [CrossRef] [Green Version]
- Horrobin, D. Nutritional and medical importance of gamma-linolenic acid. Prog. Lipid Res. 1992, 31, 163–194. [Google Scholar] [CrossRef]
- Prescott, D.J.; Vagelos, P.R. Acyl carrier protein. Adv. Enzymol. Relat. Areas Mol. Biol. 1972, 36, 269–311. [Google Scholar]
- Semenkovich, C.F. Regulation of fatty acid synthase (fas). Prog. Lipid Res. 1997, 36, 43–53. [Google Scholar] [CrossRef]
- Kumaresan, V.; Sannasimuthu, A.; Arasu, M.V.; Al-Dhabi, N.A.; Arockiaraj, J. Molecular insight into the metabolic activities of a protein-rich micro alga, Arthrospira platensis by de novo transcriptome analysis. Mol. Boil. Rep. 2018, 45, 829–838. [Google Scholar] [CrossRef]
- Hoang, M.H.; Nguyen, C.; Pham, H.Q.; Duc, L.H.; Van Son, L.; Hai, T.N.; Ha, C.H.; Nhan, L.D.; Anh, H.T.L.; Thom, L.T.; et al. Transcriptome sequencing and comparative analysis of Schizochytrium mangrovei PQ6 at different cultivation times. Biotechnol. Lett. 2016, 38, 1781–1789. [Google Scholar] [CrossRef]
- Shah, S.; Sahoo, D.; Shukla, R.N.; Mishra, G. De novo transcriptome sequencing of monodopsis subterranea ccala 830 and identification of genes involved in the biosynthesis of eicosapentanoic acid and triacylglycerol. Vegetos 2019, 32, 600–608. [Google Scholar]
- Wei, Z.; Liu, X.; Zhou, Z.; Xu, J. De novo transcriptomic analysis of gonad of Strongylocentrotus nudus and gene discovery for biosynthesis of polyunsaturated fatty acids. Genes Genom. 2019, 41, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R.; Bennett, J.W. Constructing Polyketides: From Collie to Combinatorial Biosynthesis. Annu. Rev. Microbiol. 1999, 53, 411–446. [Google Scholar] [CrossRef] [PubMed]
- Von Wettstein-Knowles, P.; Olsen, J.; McGuire, K.A.; Henriksen, A. Fatty acid synthesis. Role of active site histidines and lysine in Cys-His-His-type beta-ketoacyl-acyl carrier protein synthases. FEBS J. 2006, 273, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X. Biosynthesis of docosahexaenoic acid (DHA, 22:6-4, 7,10,13,16,19): Two distinct pathways. Prostaglandins Leukot. Essent. Fat. Acids 2003, 68, 181–186. [Google Scholar] [CrossRef]
- Gross, J.W.; Hegeman, A.D.; Vestling, M.M.; Frey, P.A. Characterization of Enzymatic Processes by Rapid Mix−Quench Mass Spectrometry: The Case of dTDP-glucose 4,6-Dehydratase. Biochemistry 2000, 39, 13633–13640. [Google Scholar] [CrossRef]
- Geisbrecht, B.V.; Zhu, D.; Schulz, K.; Nau, K.; Morrell, J.C.; Geraghty, M.; Schulz, H.; Erdmann, R.; Gould, S.J. Molecular characterization of Saccharomyces cerevisiae delta3, delta2-enoyl-coa isomerase. J. Biol. Chem. 1998, 273, 33184–33191. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Seq Type (#) | Total Number (#) | Total Length (Mb) | N50 Length (Mb) | N90 Length (Mb) | Max Length (Mb) | Min Length (Mb) | Gap Number (Mb) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|---|
| SZU445 | Scaffold | 25 | 61.97 | 5.98 | 2.41 | 13.75 | 0.0054 | 0.091 | 45.04 |
| SZU445 | Contig | 54 | 61.88 | 2.55 | 1.39 | 4.00 | 0.0054 | - | 45.04 |
| Sample Name (#) | Type (#) | Total Number (#) | Total Length (bp) | Average Length (bp) | Length/Genome Length (%) |
|---|---|---|---|---|---|
| SZU445 | Gene Stat | 14,145 | 26,947,341 | 1905.08 | 43.48 |
| Exons Stat | 18,768 | 25,518,500 | 1359.68 | 41.18 | |
| CDS Stat | 14,145 | 25,518,500 | 1804.07 | 41.18 | |
| Intron Stat | 4623 | 1,428,841 | 309.07 | 2.31 |
| Sample Name (#) | Type | Copy# | Avg_Len | Total_Len | % in Genome |
|---|---|---|---|---|---|
| tRNA | 493 | 77.81 | 38,362 | 0.0619 | |
| SZU445 | rRNA | 235 | 1683.92 | 395,723 | 0.6385 |
| snRNA | 77 | 70.15 | 5402 | 0.0087 |
| Total | CAZY | TCDB | IPR | SWISS-PROT | GO | KEGG | KOG | COG | P450 | TF | EKPD | NOG | CARD | CWDE | NR | DBCAN | PHI | PHOSPHATASE | Overall |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14,145 | 34 (0.24%) | 232 (1.64%) | 9707 (68.62%) | 2122 (15%) | 7255 (51.29%) | 1625 (11.48%) | 1866 (13.19%) | 1324 (9.36%) | 740 (5.23%) | 364 (2.57%) | 369 (2.60%) | 3550 (25.09%) | 7 (0.04%) | 1 (0.7%) | 2629 (18.58%) | 207 (1.46%) | 412 (2.91%) | 85 (0.60%) | 9852 (69.65%) |
| Enzyme | EC Number | Number of Transcripts |
|---|---|---|
| Fatty Acid Desaturation and Elongation | ||
| delta7-sterol 5-desaturase | 1.14.19.20 | 1 |
| sphingolipid 8-(E)-desaturase | 1.14.19.18 | 1 |
| sphingolipid 4-desaturase | 1.14.19.17 1.14.18.5 | 1 |
| aldehyde dehydrogenase (NAD+) | 1.2.1.3 | 4 |
| 17beta-estradiol 17-dehydrogenase | 1.1.1.62 1.1.1.330 | 1 |
| acyl-CoA dehydrogenase | 1.3.8.7 | 145 |
| glycerol-3-phosphate dehydrogenase | 1.1.5.3 | 26 |
| S-(hydroxymethyl)glutathione dehydrogenase/alcohol dehydrogenase | 1.1.1.284 1.1.1.1 | 2 |
| glycerol-3-phosphate dehydrogenase | 1.1.5.3 | 26 |
| glutaryl-CoA dehydrogenase | 1.3.8.6 | 5 |
| glycerol-3-phosphate dehydrogenase (NAD+) | 1.1.1.8 | 1 |
| alcohol dehydrogenase (NADP+) | 1.1.1.2 | 9 |
| aldehyde dehydrogenase family 7 member A1 | 1.2.1.31 1.2.1.8 1.2.1.3 | 2 |
| 3-hydroxyacyl-CoA dehydrogenase | 1.1.1.35 | 29 |
| glycerol 2-dehydrogenase (NADP+) | 1.1.1.156 | 1 |
| S-(hydroxymethyl)glutathione dehydrogenase/alcohol dehydrogenase | 1.1.1.284 1.1.1.1 | 2 |
| 17beta-estradiol 17-dehydrogenase/very-long-chain 3-oxoacyl-CoA reductase | 1.1.1.62 1.1.1.330 | 1 |
| delta14-sterol reductase | 1.3.1.70 | 1 |
| Fatty Acid Biosynthesis | ||
| acetyl-CoA acyltransferase 2 | 2.3.1.16 | 2 |
| acetyl-CoA acyltransferase 1 | 2.3.1.16 | 2 |
| hydroxymethylglutaryl-CoA synthase | 2.3.3.10 | 6 |
| fatty acid synthase subunit alpha | 2.3.1.86 | 2 |
| 3-oxoacyl-[acyl-carrier-protein] synthase II | 2.3.1.179 | 1 |
| acetyl-CoA carboxylase/biotin carboxylase 1 | 6.4.1.2 6.3.4.14 2.1.3.15 | 1 |
| Reference Strains | NCBI Accession |
|---|---|
| Aurantiochytrium sp. HS399 | MH319338.1 |
| Aurantiochytrium sp. ST-2012 | JQ982490.1 |
| Aurantiochytrium sp. LY-2012 | JX847377.1 |
| Schizochytrium sp. SH104 | KX379459.1 |
| Thraustochytriidae sp. NIOS-1 | AY705769.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.; Li, S.; Liu, L.; Li, S.; Luo, Y.; Lv, C.; Wang, B.; Cheng, C.H.K.; Chen, H.; Yang, X. Genome Sequencing and Analysis of Thraustochytriidae sp. SZU445 Provides Novel Insights into the Polyunsaturated Fatty Acid Biosynthesis Pathway. Mar. Drugs 2020, 18, 118. https://doi.org/10.3390/md18020118
Zhu X, Li S, Liu L, Li S, Luo Y, Lv C, Wang B, Cheng CHK, Chen H, Yang X. Genome Sequencing and Analysis of Thraustochytriidae sp. SZU445 Provides Novel Insights into the Polyunsaturated Fatty Acid Biosynthesis Pathway. Marine Drugs. 2020; 18(2):118. https://doi.org/10.3390/md18020118
Chicago/Turabian StyleZhu, Xingyu, Shuangfei Li, Liangxu Liu, Siting Li, Yanqing Luo, Chuhan Lv, Boyu Wang, Christopher H. K. Cheng, Huapu Chen, and Xuewei Yang. 2020. "Genome Sequencing and Analysis of Thraustochytriidae sp. SZU445 Provides Novel Insights into the Polyunsaturated Fatty Acid Biosynthesis Pathway" Marine Drugs 18, no. 2: 118. https://doi.org/10.3390/md18020118
APA StyleZhu, X., Li, S., Liu, L., Li, S., Luo, Y., Lv, C., Wang, B., Cheng, C. H. K., Chen, H., & Yang, X. (2020). Genome Sequencing and Analysis of Thraustochytriidae sp. SZU445 Provides Novel Insights into the Polyunsaturated Fatty Acid Biosynthesis Pathway. Marine Drugs, 18(2), 118. https://doi.org/10.3390/md18020118