Antimalarial Peptide and Polyketide Natural Products from the Fijian Marine Cyanobacterium Moorea producens

 ,
,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
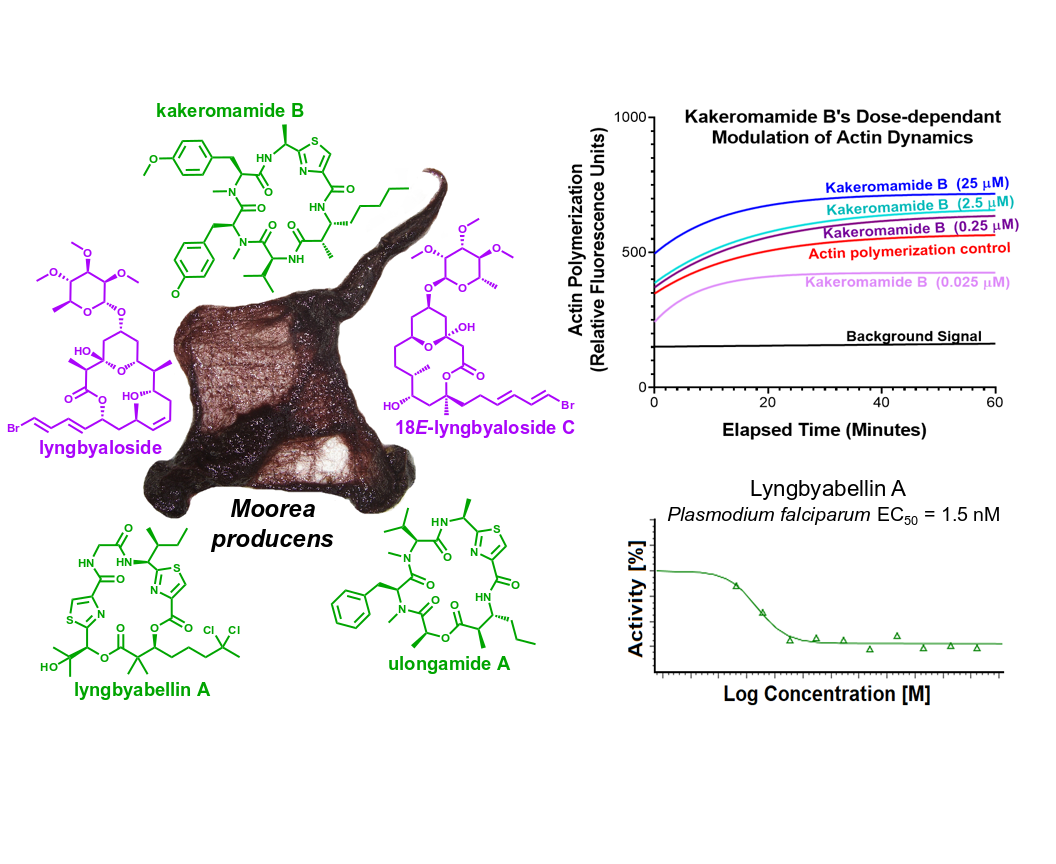
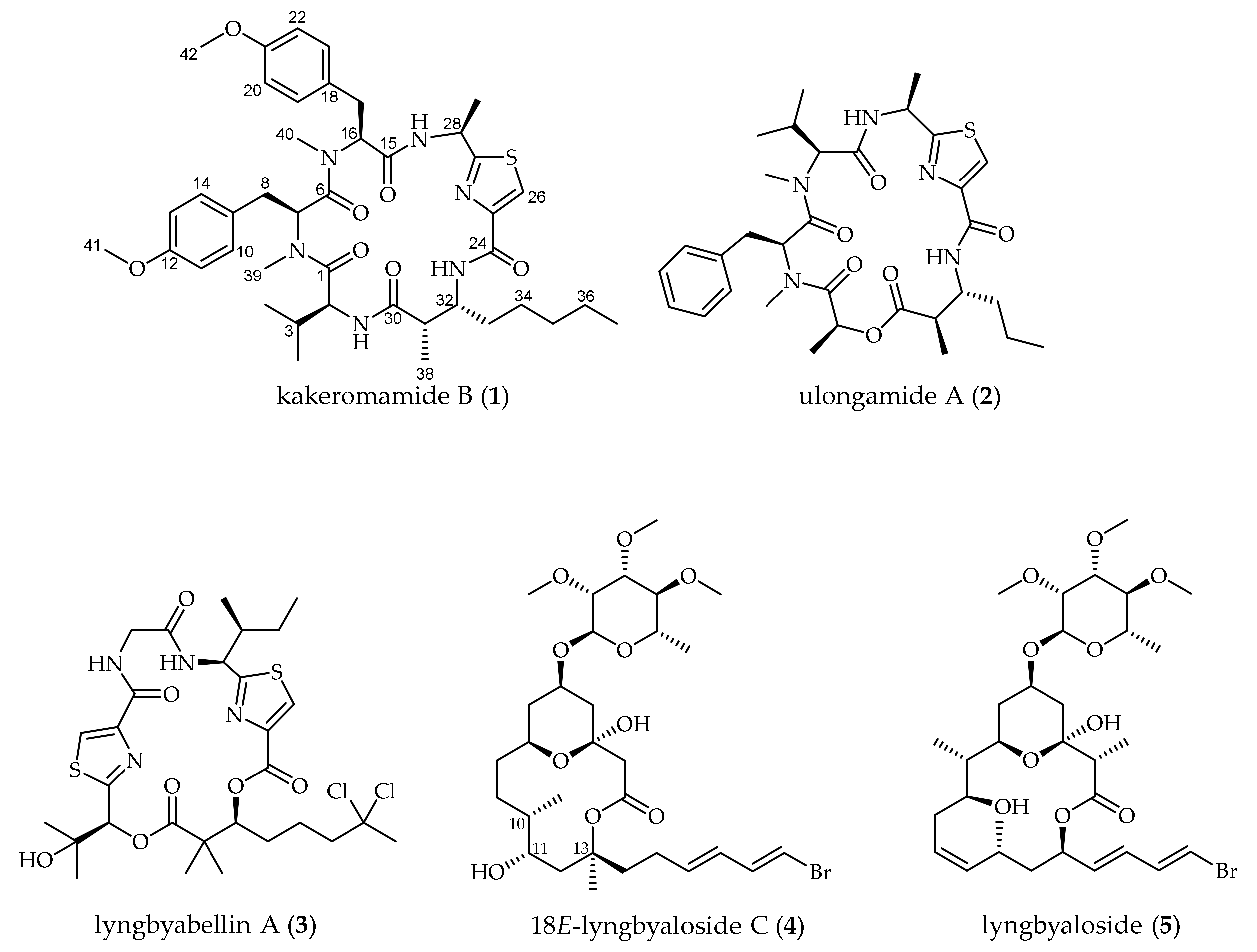
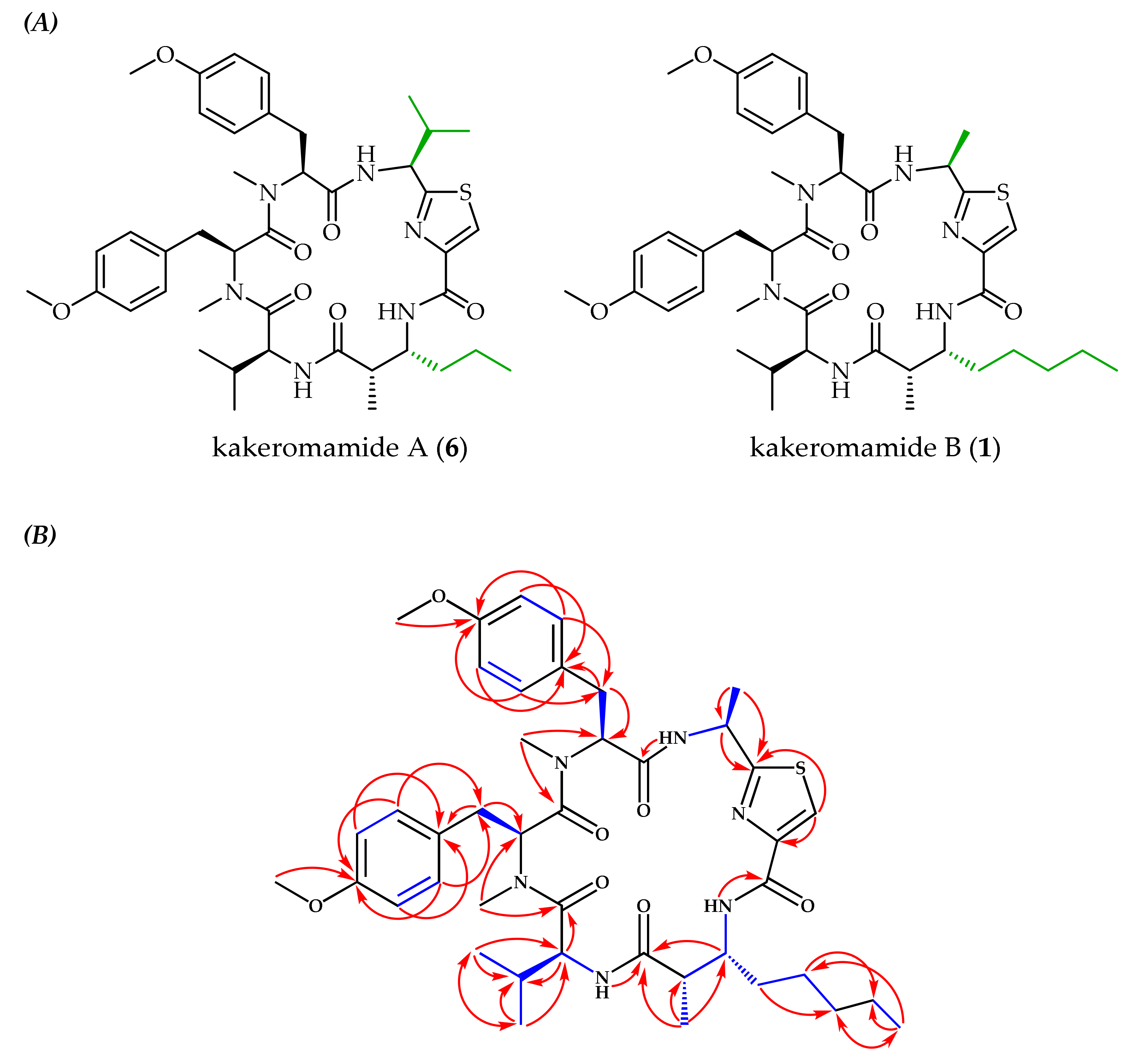
2.1. Molecular Structures of Natural Products from Moorea producens
2.2. Biological Activities of Cyanobacterial Natural Products
2.3. Putative Molecular Targets of Cyanobacterial Natural Products
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Material and Species Identification
3.3. Extraction and Isolation
3.4. Antimalarial and Cytotoxicity Assays
3.5. Computational Binding Predictions
3.6. Actin Polymerization Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Malaria Report 2019. World Health Organization. Available online: https://www.who.int/publications-detail/world-malaria-report-2019 (accessed on 18 October 2019).
- Tse, E.G.; Korsik, M.; Todd, M.H. The past, present and future of anti-malarial medicines. Malar. J. 2019, 18, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.Y.; Tan, R.X. Artemisinin, a miracle of traditional Chinese medicine. Nat. Prod. Rep. 2015, 32, 1617–1621. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maskey, R.P.; Helmke, E.; Kayser, O.; Fiebig, H.H.; Maier, A.; Busche, A.; Laatsch, H. Anti-cancer and antibacterial trioxacarcins with high anti-malaria activity from a marine Streptomycete and their absolute stereochemistry. J. Antibiot. 2004, 57, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, C.-L.; Linington, R.G.; Balunas, M.J.; Centeno, A.; Boudreau, P.; Zhang, C.; Engene, N.; Spadafora, C.; Mutka, T.S.; Kyle, D.E.; et al. Bastimolide A, a potent antimalarial polyhydroxy macrolide from the marine cyanobacterium Okeania hirsuta. J. Org. Chem. 2015, 80, 7849–7855. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Holmes, M.J.; Higa, T.; Hamann, M.T.; Kara, U.A. In vivo antimalarial activity of the beta-carboline alkaloid manzamine A. Antimicrob. Agents Chemother. 2000, 44, 1645–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.D.; Wang, H.; Gurrath, M.; König, G.M.; Kocak, G.; Neumann, G.; Loria, P.; Foley, M.; Tilley, L. Inhibition of heme detoxification processes underlies the antimalarial activity of terpene isonitrile compounds from marine sponges. J. Med. Chem. 2001, 44, 873–885. [Google Scholar] [CrossRef]
- Sanchez, C.P.; McLean, J.E.; Rohrbach, P.; Fidock, D.A.; Stein, W.D.; Lanzer, M. Evidence for a pfcrt-associated chloroquine efflux system in the human malarial parasite Plasmodium falciparum. Biochemistry 2005, 44, 9862–9870. [Google Scholar] [CrossRef]
- Shah, S.A.A.; Akhter, N.; Auckloo, B.N.; Khan, I.; Lu, Y.; Wang, K.; Wu, B.; Guo, Y.-W. Structural diversity, biological properties and applications of natural products from cyanobacteria. A review. Mar. Drugs. 2017, 15, 354. [Google Scholar] [CrossRef] [Green Version]
- Demay, J.; Bernard, C.; Reinhardt, A.; Marie, B. Natural products from cyanobacteria: Focus on beneficial activities. Mar. Drugs. 2019, 17, 320. [Google Scholar] [CrossRef] [Green Version]
- McPhail, K.L.; Correa, J.; Linington, R.G.; Gonzalez, J.; Ortega-Barria, E.; Capson, T.L.; Gerwick, W.H. Antimalarial linear lipopeptides from a Panamanian strain of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2007, 70, 984–988. [Google Scholar] [CrossRef] [Green Version]
- Linington, R.G.; Clark, B.R.; Trimble, E.E.; Almanza, A.; Urena, L.D.; Kyle, D.E.; Gerwick, W.H. Antimalarial peptides from marine cyanobacteria: Isolation and structural elucidation of gallinamide A. J. Nat. Prod. 2009, 72, 14–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linington, R.G.; Gonzalez, J.; Urena, L.D.; Romero, L.I.; Ortega-Barria, E.; Gerwick, W.H. Venturamides A and B: Antimalarial constituents of the Panamanian marine cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M.; Skolnick, J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc. Natl. Acad. Sci. USA 2008, 105, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Cao, H.; Skolnick, J. FINDSITEcomb2.0: A new approach for virtual ligand screening of proteins and virtual target screening of biomolecules. J. Chem. Inf. Model. 2018, 58, 2343–2354. [Google Scholar] [CrossRef]
- Luesch, H.; Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Ulongamides A−F, new β-amino acid-containing cyclodepsipeptides from Palauan collections of the marine cyanobacterium Lyngbya sp. J. Nat. Prod. 2002, 65, 996–1000. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615. [Google Scholar] [CrossRef]
- Matthew, S.; Salvador, L.A.; Schupp, P.J.; Paul, V.J.; Luesch, H. Cytotoxic halogenated macrolides and modified peptides from the apratoxin-producing marine cyanobacterium Lyngbya bouillonii from Guam. J. Nat. Prod. 2010, 73, 1544–1552. [Google Scholar] [CrossRef] [Green Version]
- Klein, D.; Braekman, J.C.; Daloze, D.; Hoffmann, L.; Demoulin, V. Lyngbyaloside, a novel 2,3,4-tri-O-methyl-6-deoxy-α-mannopyranoside macrolide from Lyngbya bouillonii (Cyanobacteria). J. Nat. Prod. 1997, 60, 1057–1059. [Google Scholar] [CrossRef]
- Nakamura, F.; Maejima, H.; Kawamura, M.; Arai, D.; Okino, T.; Zhao, M.; Ye, T.; Lee, J.; Chang, Y.-T.; Fusetani, N.; et al. Kakeromamide A, a new cyclic pentapeptide inducing astrocyte differentiation isolated from the marine cyanobacterium Moorea bouillonii. Bioorg. Med. Chem. Lett. 2018, 28, 2206–2209. [Google Scholar] [CrossRef]
- Alvarado, C.; Díaz, E.; Guzmán, Á. Total synthesis of ulongamide A, a cyclic depsipeptide isolated from marine cyanobacteria Lyngbya sp. Tetrahedron Lett. 2007, 48, 603–607. [Google Scholar] [CrossRef]
- Yokokawa, F.; Sameshima, H.; Shioiri, T. Total synthesis of lyngbyabellin A, a potent cytotoxic metabolite from the marine cyanobacterium Lyngbya majuscula. Tetrahedron Lett. 2001, 42, 4171–4174. [Google Scholar] [CrossRef]
- Chang, C.F.; Stefan, E.; Taylor, R.E. Total synthesis and structural reassignment of lyngbyaloside C highlighted by intermolecular ketene esterification. Chem. Eur. J. 2015, 21, 10681–10686. [Google Scholar] [CrossRef]
- Sone, H.; Kondo, T.; Kiryu, M.; Ishiwata, H.; Ojika, M.; Yamada, K. Dolabellin, a cytotoxic bisthiazole metabolite from the sea hare Dolabella auricularia: Structural determination and synthesis. J. Org. Chem. 1995, 60, 4774–4781. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Isolation and structure of the cytotoxin lyngbyabellin B and absolute configuration of lyngbyapeptin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 1437–1439. [Google Scholar] [CrossRef]
- Han, B.; McPhail, K.L.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Isolation and structure of five lyngbyabellin derivatives from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. Tetrahedron 2005, 61, 11723–11729. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Structurally diverse new alkaloids from Palauan collections of the apratoxin-producing marine cyanobacterium Lyngbya sp. Tetrahedron. 2002, 58, 7959–7966. [Google Scholar] [CrossRef]
- Williams, P.G.; Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Continuing studies on the cyanobacterium Lyngbya sp.: Isolation and structure determination of 15-norlyngbyapeptin A and lyngbyabellin D. J. Nat. Prod. 2003, 66, 595–598. [Google Scholar] [CrossRef]
- Choi, H.; Mevers, E.; Byrum, T.; Valeriote, F.A.; Gerwick, W.H. Lyngbyabellins K–N from two Palmyra Atoll collections of the marine cyanobacterium Moorea bouillonii. Eur. J. Org. Chem. 2012, 2012, 5141–5150. [Google Scholar] [CrossRef]
- Petitbois, J.G.; Casalme, L.O.; Lopez, J.A.V.; Alarif, W.M.; Abdel-Lateff, A.; Al-Lihaibi, S.S.; Yoshimura, E.; Nogata, Y.; Umezawa, T.; Matsuda, F.; et al. Serinolamides and lyngbyabellins from an Okeania sp. cyanobacterium collected from the Red Sea. J. Nat. Prod. 2017, 80, 2708–2715. [Google Scholar] [CrossRef]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; Hooft van Huijsduijnen, R.; Kaszubska, W.; Macintyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 26. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Lemgruber, L.; Tay, C.L.; Baum, J.; Meissner, M. Multiple essential functions of Plasmodium falciparum actin-1 during malaria blood-stage development. BMC Biol. 2017, 15, 70. [Google Scholar] [CrossRef] [Green Version]
- Hallee, S.; Counihan, N.A.; Matthews, K.; de Koning-Ward, T.F.; Richard, D. The malaria parasite Plasmodium falciparum Sortilin is essential for merozoite formation and apical complex biogenesis. Cell. Microbiol. 2018, 20, e12844. [Google Scholar] [CrossRef]
- Mailu, B.M.; Li, L.; Arthur, J.; Nelson, T.M.; Ramasamy, G.; Fritz-Wolf, K.; Becker, K.; Gardner, M.J. Plasmodium apicoplast Gln-tRNAGln biosynthesis utilizes a unique GatAB amidotransferase essential for erythrocytic stage parasites. J. Biol. Chem. 2015, 290, 29629–29641. [Google Scholar] [CrossRef] [Green Version]
- Plaimas, K.; Wang, Y.; Rotimi, S.O.; Olasehinde, G.; Fatumo, S.; Lanzer, M.; Adebiyi, E.; König, R. Computational and experimental analysis identified 6-diazo-5-oxonorleucine as a potential agent for treating infection by Plasmodium falciparum. Infect. Genet. Evol. 2013, 20, 389–395. [Google Scholar] [CrossRef]
- Wieland, T. Modification of actins by phallotoxins. Naturwissenschaften 1977, 64, 303–309. [Google Scholar] [CrossRef]
- Morton, W.M.; Ayscough, K.R.; McLaughlin, P.J. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2000, 2, 376–378. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.A. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 1987, 105, 1473–1478. [Google Scholar] [CrossRef] [Green Version]
- Bubb, M.R.; Senderowicz, A.M.; Sausville, E.A.; Duncan, K.L.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871. [Google Scholar]
- Douglas, R.G.; Nandekar, P.; Aktories, J.-E.; Kumar, H.; Weber, R.; Sattler, J.M.; Singer, M.; Lepper, S.; Sadiq, S.K.; Wade, R.C.; et al. Inter-subunit interactions drive divergent dynamics in mammalian and Plasmodium actin filaments. PLoS Biol. 2018, 16, e2005345. [Google Scholar] [CrossRef] [Green Version]
- Boddey, J.A.; Hodder, A.N.; Gunther, S.; Gilson, P.R.; Patsiouras, H.; Kapp, E.A.; Pearce, J.A.; de Koning-Ward, T.F.; Simpson, R.J.; Crabb, B.S.; et al. An aspartyl protease directs malaria effector proteins to the host cell. Nature 2010, 463, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.T.; Fairlie, D.P.; Madala, P.K.; Ray, J.; Wyatt, D.M.; Hilton, P.M.; Melville, L.A.; Beattie, L.; Gardiner, D.L.; Reid, R.C.; et al. Potencies of human immunodeficiency virus protease inhibitors in vitro against Plasmodium falciparum and in vivo against murine malaria. Antimicrob. Agents Chemother. 2006, 50, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR spectroscopy. Trends Analyt. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Frank, J.A.; Reich, C.I.; Sharma, S.; Weisbaum, J.S.; Wilson, B.A.; Olsen, G.J. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 2008, 74, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Plouffe, D.; Brinker, A.; McNamara, C.; Henson, K.; Kato, N.; Kuhen, K.; Nagle, A.; Adrian, F.; Matzen, J.T.; Anderson, P.; et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. USA 2008, 105, 9059–9064. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.; Corey, V.; Scherer, C.A.; Kato, N.; Comer, E.; Maetani, M.; Antonova-Koch, Y.; Reimer, C.; Gagaring, K.; Ibanez, M.; et al. High-throughput luciferase-based assay for the discovery of therapeutics that prevent malaria. ACS Infect. Dis. 2016, 2, 281–293. [Google Scholar] [CrossRef]
- Love, M.S.; Beasley, F.C.; Jumani, R.S.; Wright, T.M.; Chatterjee, A.K.; Huston, C.D.; Schultz, P.G.; McNamara, C.W. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl. Trop. Dis. 2017, 11, e0005373. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc. Natl. Acad. Sci. USA 2004, 101, 7594–7599. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer Jr, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank. Europ. J. Biochem. 1977, 80, 319–324. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| no. | δC (mult., J in Hz) | δH (mult., J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 176.3 | |||
| 2 | 56.9 | 4.30 t (7.1) | H-3,2-NH | C-1 w,C-3,C-4 w |
| 2-NH | 6.32 d (6.6) | H-2 | C-30 w | |
| 3 | 30.8 | 1.80 m | H-2,H-4,H-5 | |
| 4 | 18.6 | 0.78 d (6.9) | H-3 | C-2,C-3,C-5 |
| 5 | 19.0 | 0.80 d (6.8) | H-3 | C-2,C-3,C-4 |
| 6 | 172.1 | |||
| 7 | 50.3 | 5.63 dd (4.9, 10.9) | H-8a,H-8b | |
| 8a | 33.2 | 1.37 m | H-7,H-8b | C-9 w |
| 8b | 2.74 dd (11.0, 16.1) | H-7,H-8a | C-7,C-9 | |
| 9 | 129.8 | |||
| 10/14 | 130.0 | 6.89 d (8.5) | H-11/13 | C-8 w,C-12 |
| 11/13 | 114.7 | 6.78 d (8.7) | H-10/14 | C-9 |
| 12 | 159.1 | |||
| 15 | 168.9 | |||
| 16 | 62.9 | 5.23 dd (5.0, 10.0) | H-17a,H-17b | |
| 17a | 34.8 | 2.67 dd (10.1, 14.4) | H-16,H-17b | C-16 |
| 17b | 2.92 dd (4.9, 14.4) | H-16,H-17a | C-18 | |
| 18 | 130.7 | |||
| 19/23 | 131.4 | 7.00 d (8.6) | H-20/22 | C-17,C-21 |
| 20/22 | 114.9 | 6.60 d (8.7) | H-19/23 | C-18 |
| 21 | 159.4 | |||
| 24 | 169.5 | |||
| 25 | 150.2 | |||
| 26 | 123.2 | 8.02 s | C-25 w,C-27 | |
| 27 | 171.6 | |||
| 28 | 48.1 | 5.45 p (7.4) | 28-NH,H-29 | C-27 w |
| 28-NH | 8.61 d (7.8) | H-28 | C-15 w | |
| 29 | 24.0 | 1.40 d (6.9) | H-28 | C-27,C-28 |
| 30 | 173.6 | |||
| 31 | 44.5 | 2.63 qd (3.7, 6.8) | H-32,H-38 | |
| 32 | 53.1 | 4.02 m | H-31,32-NH,H-33a,b | C-30 w |
| 32-NH | 8.71 d (10.2) | H-32 | C-24 w | |
| 33a | 41.8 | 1.13 m | H-32,H-33b,H-34a,b | C-35 w |
| 33b | 1.72 m | H-32,H-33a,H-34a,b | ||
| 34a | 30.4 | 1.29 m | H-33a,b | C-36 w |
| 34b | 1.45 m | H-33a,b | ||
| 35 | 33.7 | 1.35 m | C-37 w | |
| 36 | 24.5 | 1.35 m | H-37 | |
| 37 | 14.9 | 0.88 t (6.6) | H-36 | C-34,C-35,C-36 |
| 38 | 19.6 | 1.09 d (6.9) | H-31 | C-30,C-31,C-32 |
| 39 | 31.4 | 3.02 s | C-1,C-7 | |
| 40 | 28.7 | 2.87 s | C-6,C-16 | |
| 41 | 56.0 | 3.74 s | C-12 | |
| 42 | 55.7 | 3.49 s | C-21 |
| Blood-Stage P. falciparum a | Liver-Stage P. berghei a | HEK293T Cytotoxicity b | HepG2 Cytotoxicity b | |
|---|---|---|---|---|
| 1 | 0.89 | 1.1 d, >1.2 e | >2.3 | >2.3 |
| 2 | 0.99 | >4.0 c | >4.8 | not tested |
| 3 | 0.00015 | >1.0 d,e | 1.9 | 0.33 |
| 4 | >1.9 | 0.71 d, >1.6 e | >3.1 | 1.7 |
| 5 | >0.79 | 0.45 d, >0.63 e | >1.3 | >1.3 |
| lyngbyabellin-like 1 (LYN1) | 0.0073 | >0.32 e,d | >0.65 | 0.45 |
| lyngbyabellin-like 2 (LYN2) | 0.11 | >0.26 e,d | >0.52 | >0.52 |
| Atovaquone (+ control) | 0.0061 | <0.00028 c, 0.0017 d, 0.0037 e | >2.0 | not tested |
| Accession Number | Protein Description | mTC | Precision |
|---|---|---|---|
| Q8ILW9 | Actin-2 | 0.50 | 0.77 |
| Q8ILM5 | Actin-related protein homolog, arp4 | 0.50 | 0.77 |
| Q8IIW6 | Actin-like protein, putative | 0.50 | 0.77 |
| Q8IIQ4 | Actin-like protein homolog, ALP1 | 0.50 | 0.77 |
| Q8IBH5 | Actin-like protein, putative | 0.50 | 0.77 |
| Q8I4X0 | Actin-1 | 0.50 | 0.77 |
| Q8I450 | Actin-like protein, putative | 0.50 | 0.77 |
| Q8I345 | Actin-like protein, putative | 0.50 | 0.77 |
| Q8I2A2 | Actin-related protein, ARP1 | 0.50 | 0.77 |
| Q8I1V9 | Actin-like protein, putative | 0.50 | 0.77 |
| Q8IKV8 | Sortilin, putative | 0.48 | 0.45 |
| Q8I1S6 | Glutamyl-tRNA(Gln) amidotransferase subunit A, putative | 0.46 | 0.26 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sweeney-Jones, A.M.; Gagaring, K.; Antonova-Koch, J.; Zhou, H.; Mojib, N.; Soapi, K.; Skolnick, J.; McNamara, C.W.; Kubanek, J. Antimalarial Peptide and Polyketide Natural Products from the Fijian Marine Cyanobacterium Moorea producens. Mar. Drugs 2020, 18, 167. https://doi.org/10.3390/md18030167
Sweeney-Jones AM, Gagaring K, Antonova-Koch J, Zhou H, Mojib N, Soapi K, Skolnick J, McNamara CW, Kubanek J. Antimalarial Peptide and Polyketide Natural Products from the Fijian Marine Cyanobacterium Moorea producens. Marine Drugs. 2020; 18(3):167. https://doi.org/10.3390/md18030167
Chicago/Turabian StyleSweeney-Jones, Anne Marie, Kerstin Gagaring, Jenya Antonova-Koch, Hongyi Zhou, Nazia Mojib, Katy Soapi, Jeffrey Skolnick, Case W. McNamara, and Julia Kubanek. 2020. "Antimalarial Peptide and Polyketide Natural Products from the Fijian Marine Cyanobacterium Moorea producens" Marine Drugs 18, no. 3: 167. https://doi.org/10.3390/md18030167
APA StyleSweeney-Jones, A. M., Gagaring, K., Antonova-Koch, J., Zhou, H., Mojib, N., Soapi, K., Skolnick, J., McNamara, C. W., & Kubanek, J. (2020). Antimalarial Peptide and Polyketide Natural Products from the Fijian Marine Cyanobacterium Moorea producens. Marine Drugs, 18(3), 167. https://doi.org/10.3390/md18030167