Design, Synthesis and Biological Evaluation of Jahanyne Analogs as Cell Cycle Arrest Inducers

Abstract
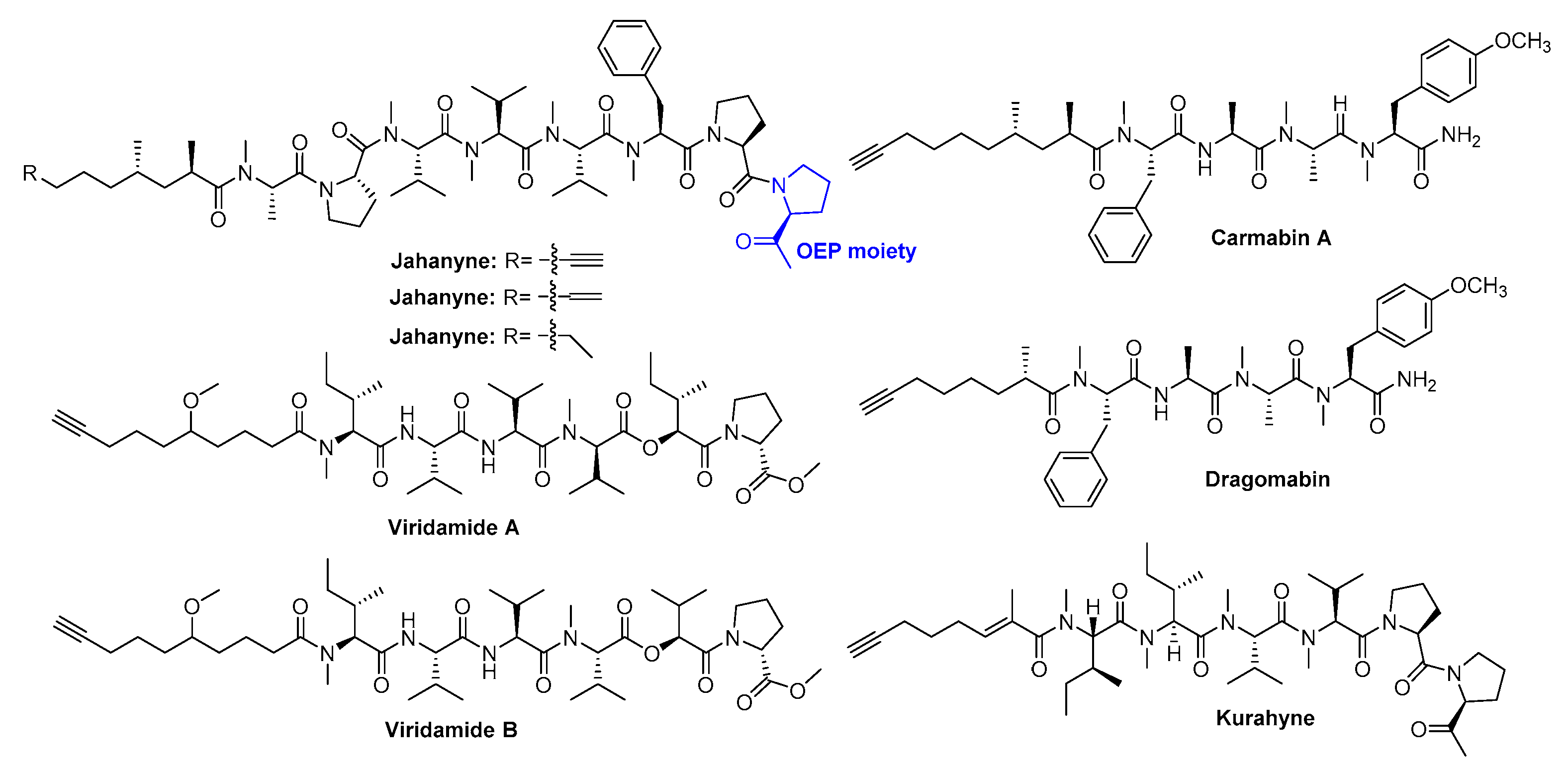
:1. Introduction
2. Results
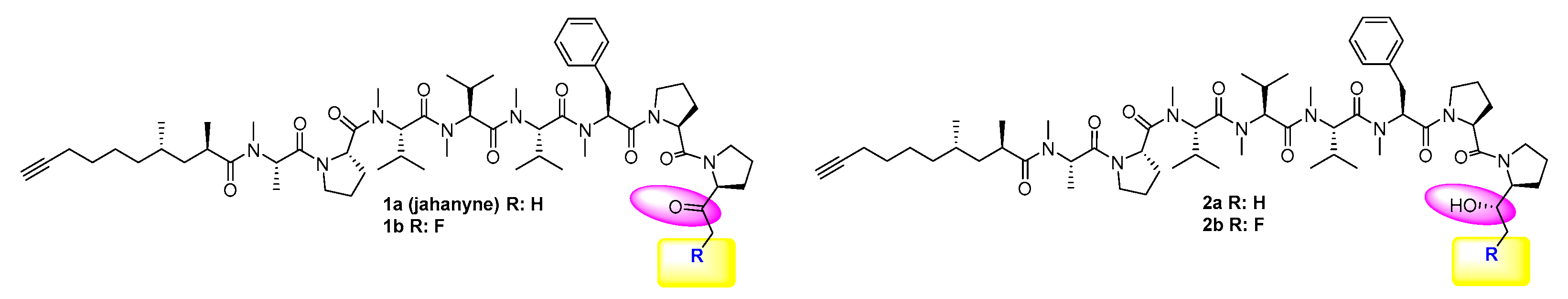
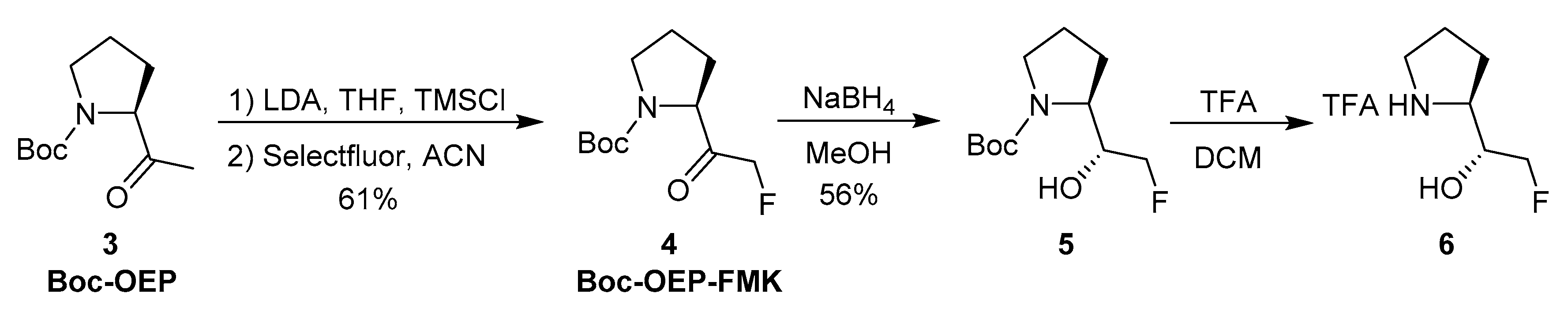
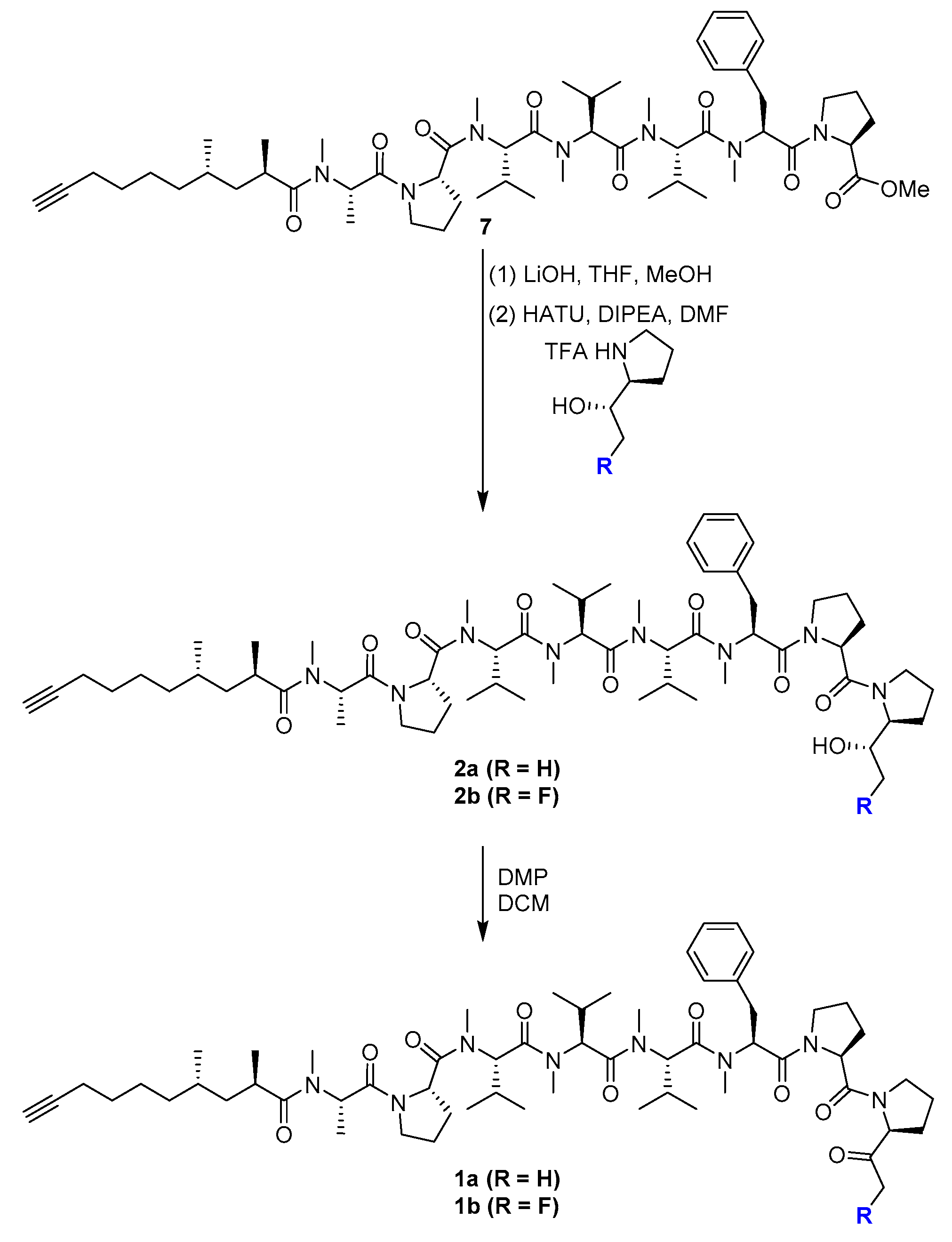
2.1. Chemistry
2.2. Biological Activity against Cancer Cells
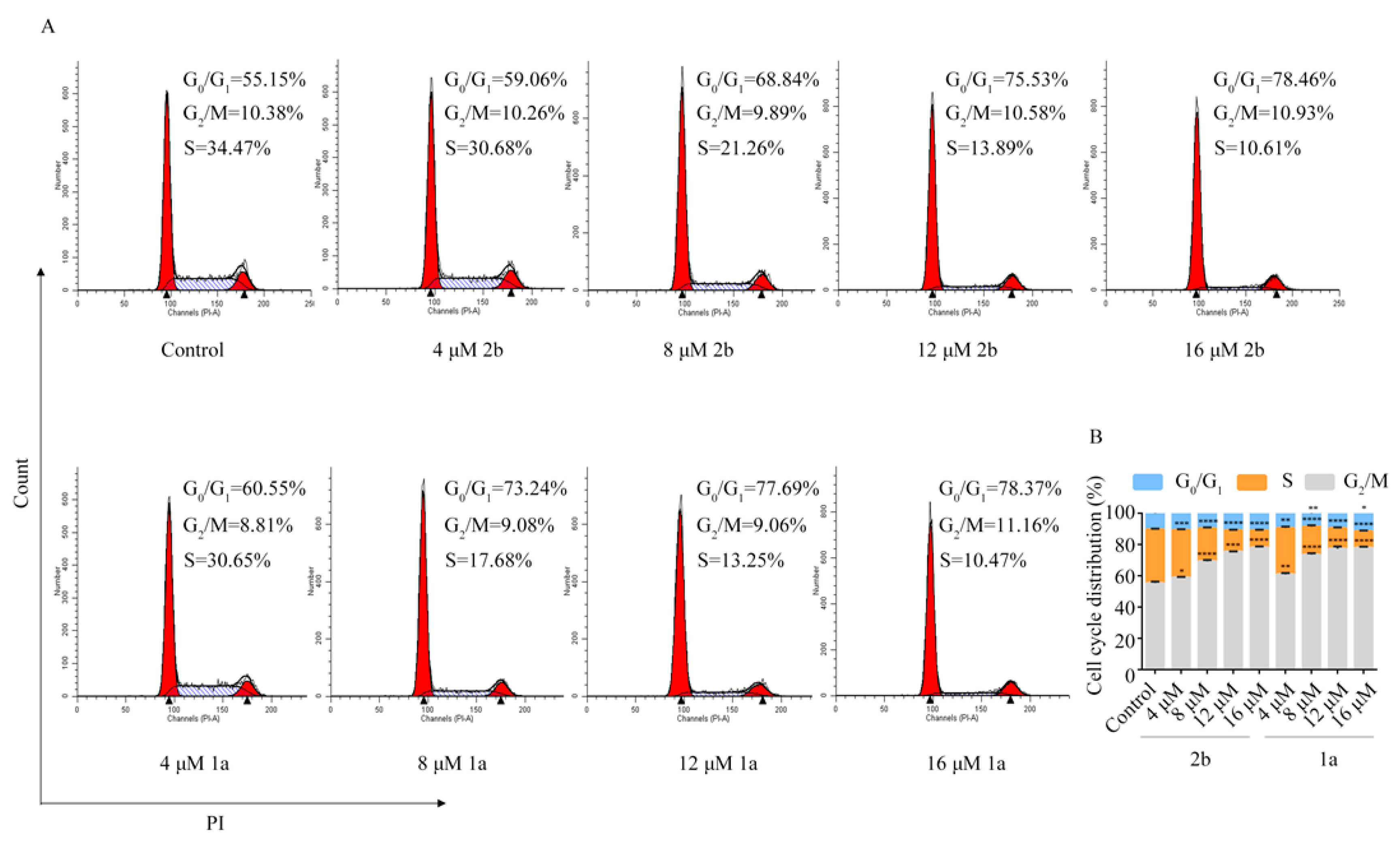
2.3. Compound 1a Induced G0/G1 Phase Arrest
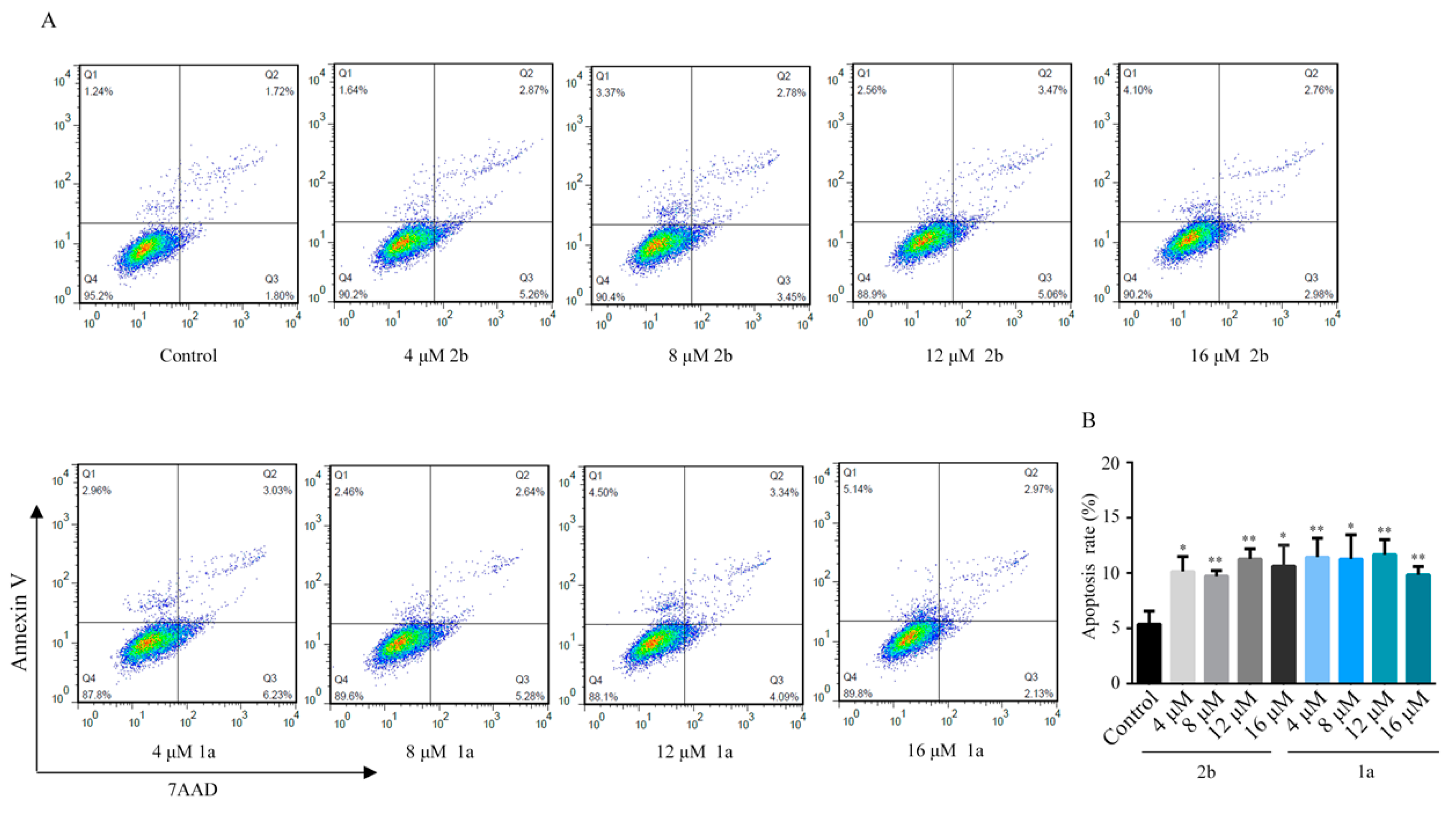
2.4. Cell Apoptosis Effects of Compound 2b and 1a
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.3. Cell Culture
3.4. CCK-8 Assay
3.5. Cell Cycle Assay
3.6. Apoptosis Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, Y.; Zhang, J.; He, S.; Yan, X. New peptides isolated from marine cyanobacteria, an overview over the past decade. Mar. Drugs 2017, 15, 132. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.; Garcia, M.; Costa-Rodrigues, J.; Costa, M.S.; Ribeiro, M.J.; Fernandes, M.H.; Barros, P.; Barreiro, A.; Vasconcelos, V.; Martins, R. Exploring bioactive properties of marine cyanobacteria isolated from the Portuguese coast: High potential as a source of anticancer compounds. Mar. Drugs 2014, 12, 98–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.; Wang, J.; Hao, Y.; Wang, Y. Recent advances in the discovery and development of marine microbial natural products. Mar. Drugs 2013, 11, 700–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.; Costa-Rodrigues, J.; Fernandes, M.H.; Barros, P.; Vasconcelos, V.; Martins, R. Marine cyanobacteria compounds with anticancer properties: A review on the implication of apoptosis. Mar. Drugs 2012, 10, 2181–2207. [Google Scholar] [CrossRef] [Green Version]
- Sarvesh, S.R.; Tabassum, K. Novel anti-inflammatory drugs from marine microbes. Nat. Prod. J. 2015, 5, 206–218. [Google Scholar]
- Jo, C.; Khan, F.F.; Khan, M.I.; Iqbal, J. Marine bioactive peptides: Types, structures, and physiological functions. Food Rev. Int. 2017, 33, 44–61. [Google Scholar] [CrossRef]
- Polakis, P. Antibody drug conjugates for cancer therapy. Pharmacol. Rev. 2016, 68, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, S.L.; Leal, R.M.; Tinley, T.L.; Luesch, H.; Moore, R.E.; Corbett, T.H. The molecular pharmacology of symplostatin 1: A new antimitotic dolastatin 10 analog. Int. J. Cancer 2003, 104, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.; Sammet, B.; Sewald, N. Recent approaches for the synthesis of modified 1241 cryptophycins. Nat. Prod. Rep. 2013, 30, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shan, G.; Zheng, Y.; Yu, X.; Ruan, Z.; Li, Y.; Lei, X. Synthesis and preliminary biological evaluation of two fluoroolefin analogs of largazole inspired by the structural similarity of the side chain unit in psammaplin A. Mar. Drugs 2019, 17, 333. [Google Scholar] [CrossRef] [Green Version]
- Mazard, S.; Penesyan, A.; Ostrowski, M.; Paulsen, I.T.; Egan, S. Tiny microbes with a big impact: The role of cyanobacteria and their metabolites in shaping our future. Mar. Drugs 2016, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- McPhail, K.L.; Correa, J.; Linington, R.G.; Gonzalez, J.; Ortega-Barria, E.; Capson, T.L.; Gerwick, W.H. Antimalarial linear lipopeptides from a panamanian strain of the marine cyanobacterium lyngbya majuscula. J. Nat. Prod. 2007, 70, 984–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, A.; Ohno, O.; Sumimoto, S.; Suda, S.; Suenaga, K. Kurahyne, an acetylene-containing lipopeptide from a marine cyanobacterial assemblage of Lyngbya sp. RSC Adv. 2014, 4, 12840–12843. [Google Scholar] [CrossRef] [Green Version]
- Simmons, T.L.; Engene, N.; Ureña, L.D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, L.; Gerwick, W.H. Viridamides A and B, lipodepsipeptides with anti-protozoal activity from the marine cyanobacterium Oscillatoria nigro-viridis. J. Nat. Prod. 2008, 71, 1544–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, G.J.; Orjala, J.; Schatzman, R.C.; Gerwick, W.H. Carmabins A and B, new lipopeptides from the caribbean cyanobacterium Lyngbya majuscula. J. Nat. Prod. 1998, 61, 529–533. [Google Scholar] [CrossRef]
- Sanchez, L.M.; Lopez, D.; Vesely, B.A.; Togna, G.D.; Gerwick, W.H.; Kyle, D.E.; Linington, R.G. Almiramides A−C: Discovery and development of a new class of leishmaniasis lead compounds. J. Med. Chem. 2010, 53, 4187–4197. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Ohno, O.; Sumimoto, S.; Ogawa, H.; Nguyen, K.A.; Suenaga, K. Jahanyne, an apoptosis-inducing lipopeptide from the marine cyanobacterium Lyngbya sp. Org. Lett. 2015, 17, 652–655. [Google Scholar] [CrossRef]
- Kallepu, S.; Kavitha, M.; Yeeravalli, R.; Manupati, K.; Jadav, S.S.; Das, A.; Mainkar, P.S.; Chandrasekhar, S. Total synthesis of desmethyl jahanyne and its lipo-tetrapeptide conjugates derived from parent skeleton as bcl-2-mediated apoptosis-inducing agents. ACS Omega 2018, 3, 63–75. [Google Scholar] [CrossRef]
- Siow, A.; Opiyo, G.; Kavianinia, I.; Li, F.F.; Furkert, D.P.; Harris, P.W.R.; Brimble, M.A. Total synthesis of the highly N-methylated acetylene-containing anticancer peptide jahanyne. Org. Lett. 2018, 20, 788–791. [Google Scholar] [CrossRef]
- Ye, B.; Jiang, P.; Zhang, T.; Ding, Y.; Sun, Y.; Hao, X.; Li, L.; Wang, L.; Chen, Y. Total synthesis of the highly N-methylated peptide jahanyne. J. Org. Chem. 2018, 83, 6741–6747. [Google Scholar] [CrossRef]
- Iwasaki, A.; Fujimura, H.; Okamoto, S.; Kudo, T.; Hoshina, S.; Sumimoto, S.; Teruya, T.; Suenaga, K. Isolation of jahanene and jahanane, and total synthesis of the jahanyne family. J. Org. Chem. 2018, 83, 9592–9603. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isanbor, C.; O’Hagan, D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluorine Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Cohen, M.S.; Zhang, C.; Shokat, K.M.; Taunton, J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005, 308, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angliker, H.; Wikstrom, P.; Rauber, P.; Shaw, E. The synthesis of lysylfluoromethanes and their properties as inhibitors of trypsin, plasmin and cathepsin B. Biochem. J. 1987, 241, 871–875. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) a | ||||
|---|---|---|---|---|---|
| HL60 | H1688 | H1299 | H820 | A549 | |
| 2a | 19.53 ± 4.73 | 17.73 ± 2.80 | 16.73 ± 3.03 | 9.01 ± 1.75 | 16.68 ± 1.05 |
| 2b | 29.68 ± 9.41 | 13.47 ± 3.10 | 10.98 ± 1.31 | 7.64 ± 1.70 | 14.65 ± 2.06 |
| 1a (jahanyne) | 13.98 ± 1.84 | 21.90 ± 3.26 | 18.70 ± 1.55 | 11.48 ± 1.87 | 17.11 ± 2.79 |
| 1b | 14.70 ± 0.56 | 29.35 ± 4.37 | 28.56 ± 9.15 | 19.38 ± 1.87 | 13.83 ± 2.21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, B.; Gong, J.; Li, Q.; Bao, S.; Zhang, X.; Chen, J.; Meng, Q.; Chen, B.; Jiang, P.; Wang, L.; et al. Design, Synthesis and Biological Evaluation of Jahanyne Analogs as Cell Cycle Arrest Inducers. Mar. Drugs 2020, 18, 176. https://doi.org/10.3390/md18030176
Ye B, Gong J, Li Q, Bao S, Zhang X, Chen J, Meng Q, Chen B, Jiang P, Wang L, et al. Design, Synthesis and Biological Evaluation of Jahanyne Analogs as Cell Cycle Arrest Inducers. Marine Drugs. 2020; 18(3):176. https://doi.org/10.3390/md18030176
Chicago/Turabian StyleYe, Baijun, Jianmiao Gong, Qiuying Li, Shiqi Bao, Xuemei Zhang, Jing Chen, Qing Meng, Bolin Chen, Peng Jiang, Liang Wang, and et al. 2020. "Design, Synthesis and Biological Evaluation of Jahanyne Analogs as Cell Cycle Arrest Inducers" Marine Drugs 18, no. 3: 176. https://doi.org/10.3390/md18030176
APA StyleYe, B., Gong, J., Li, Q., Bao, S., Zhang, X., Chen, J., Meng, Q., Chen, B., Jiang, P., Wang, L., & Chen, Y. (2020). Design, Synthesis and Biological Evaluation of Jahanyne Analogs as Cell Cycle Arrest Inducers. Marine Drugs, 18(3), 176. https://doi.org/10.3390/md18030176