Graph-Directed Approach for Downselecting Toxins for Experimental Structure Determination


Abstract
:1. Introduction
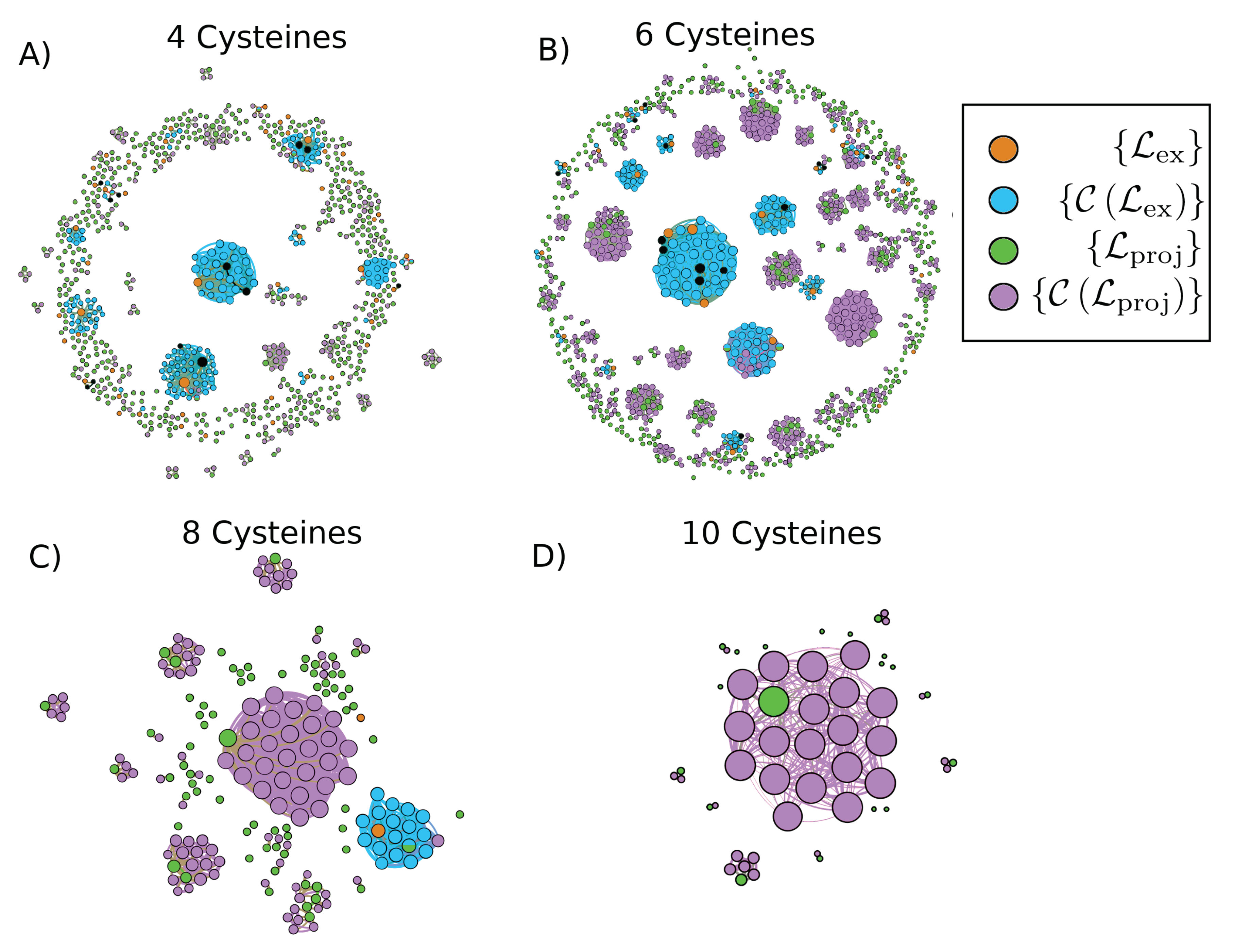
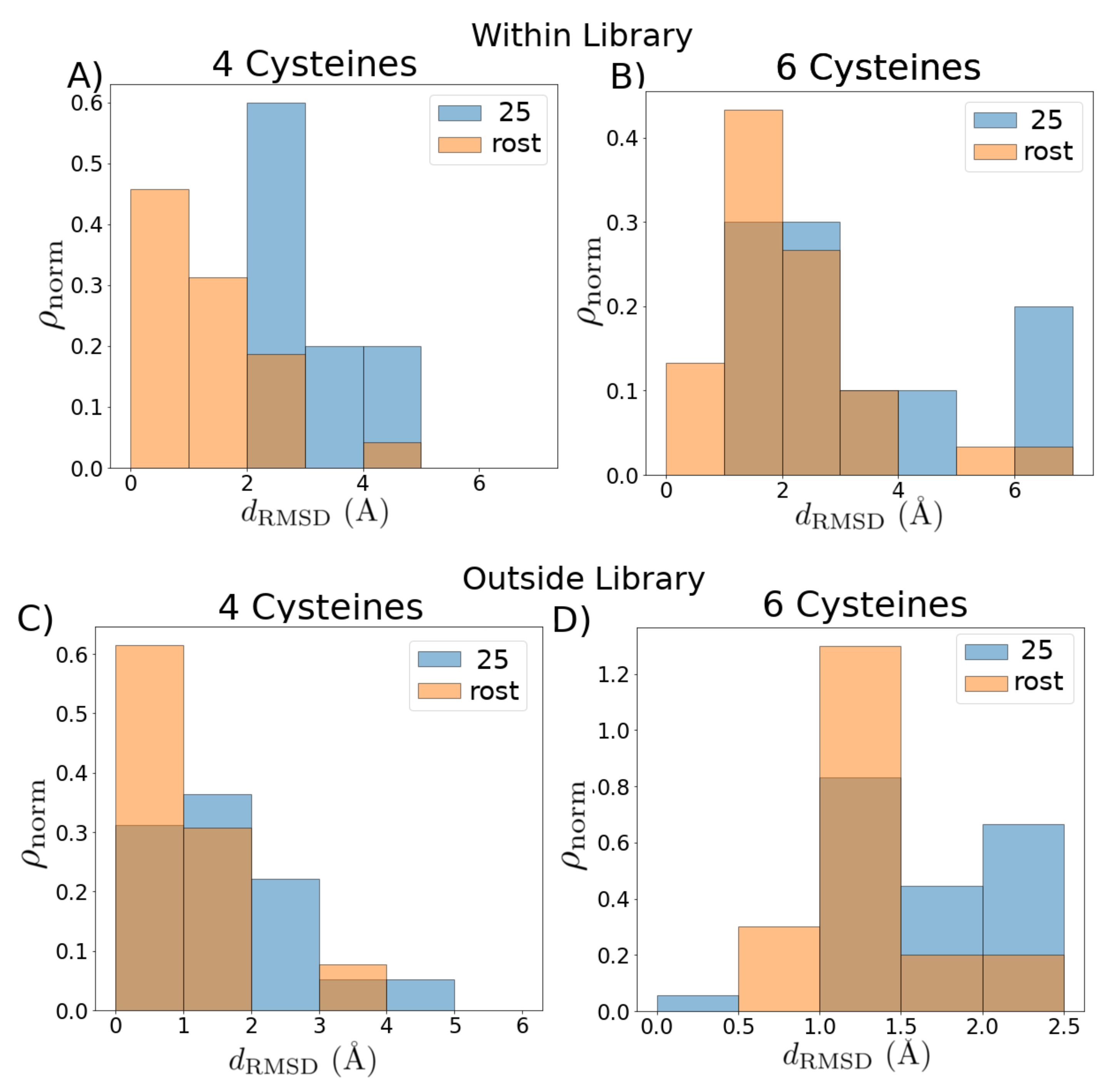
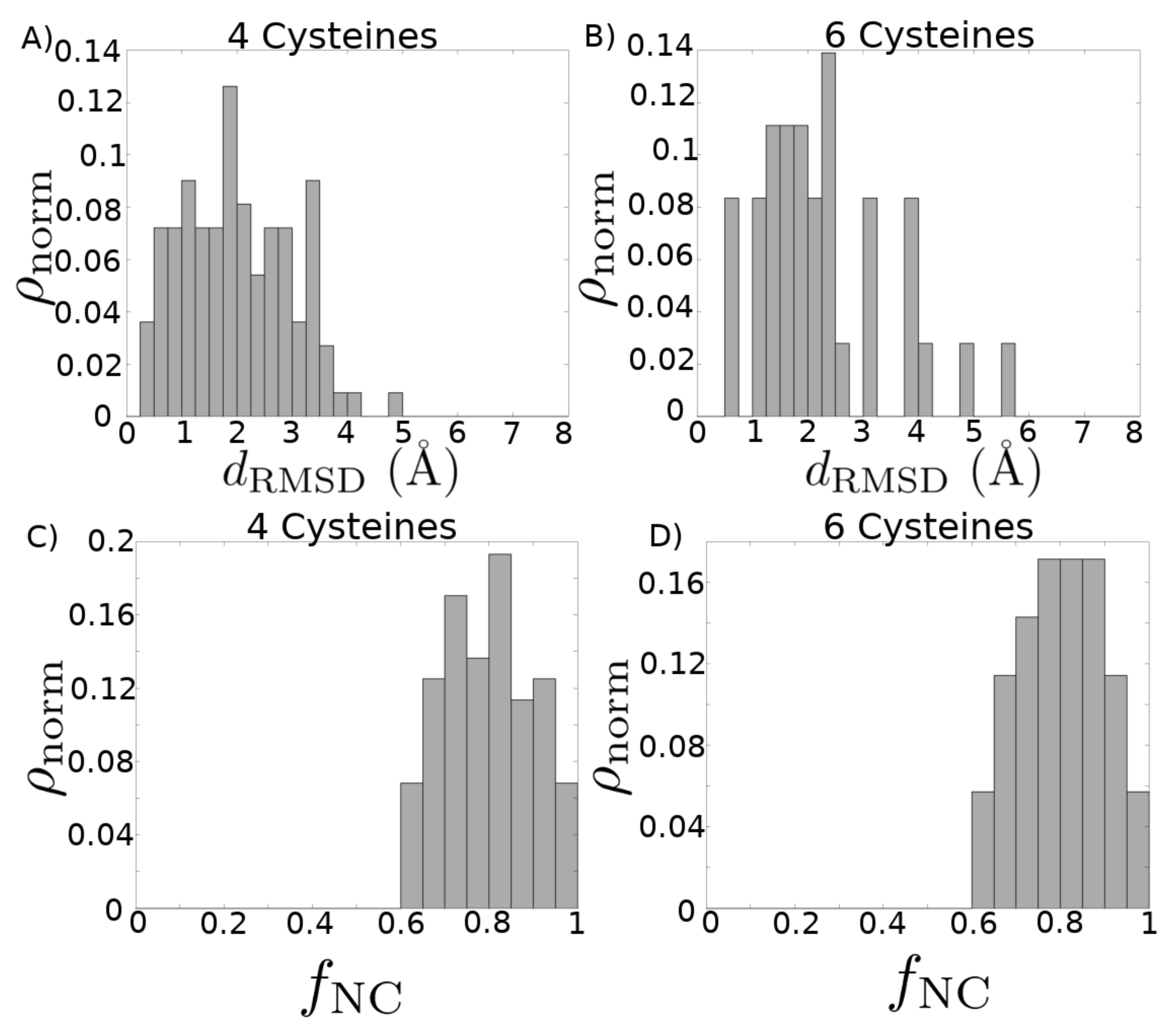
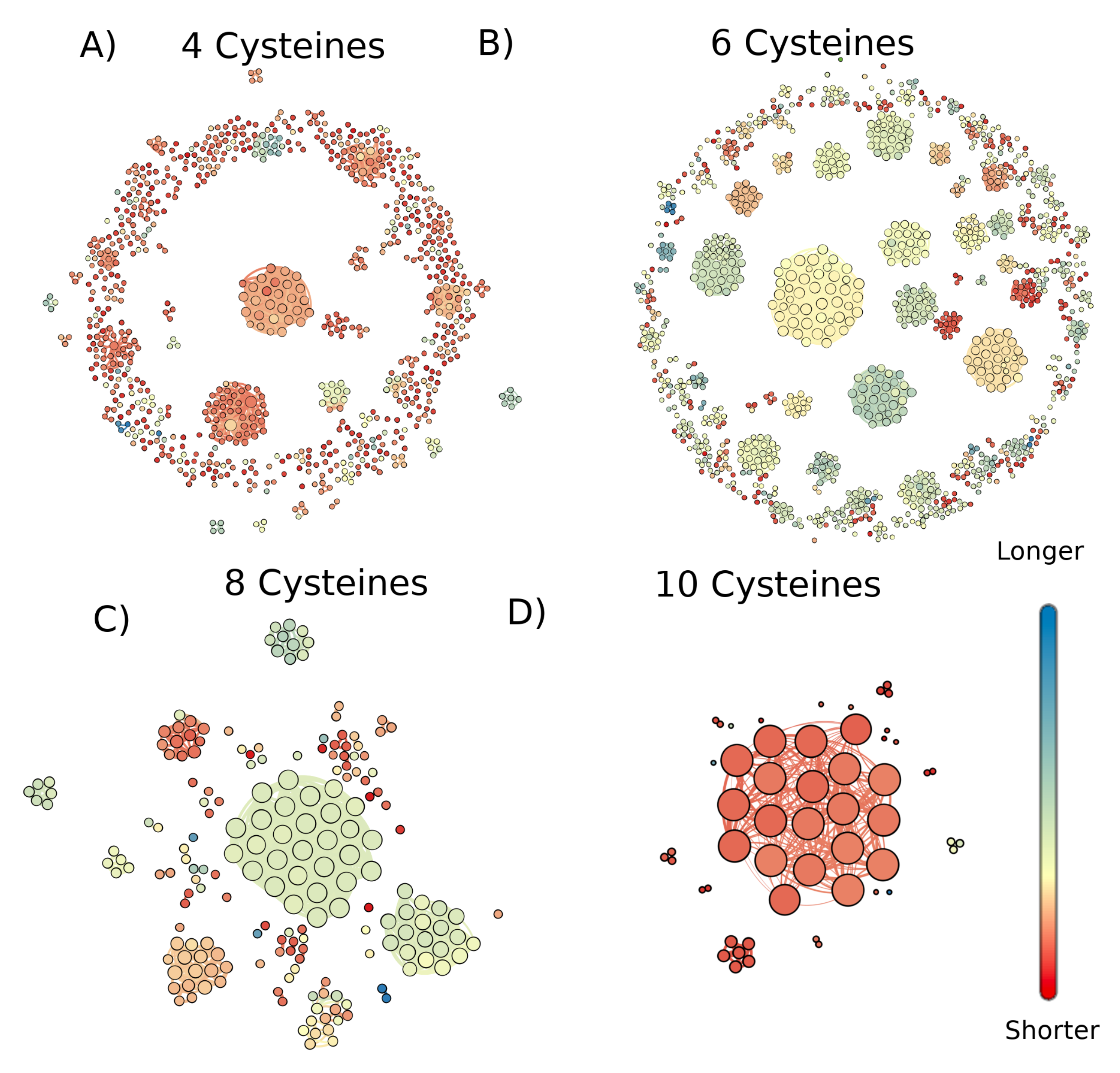
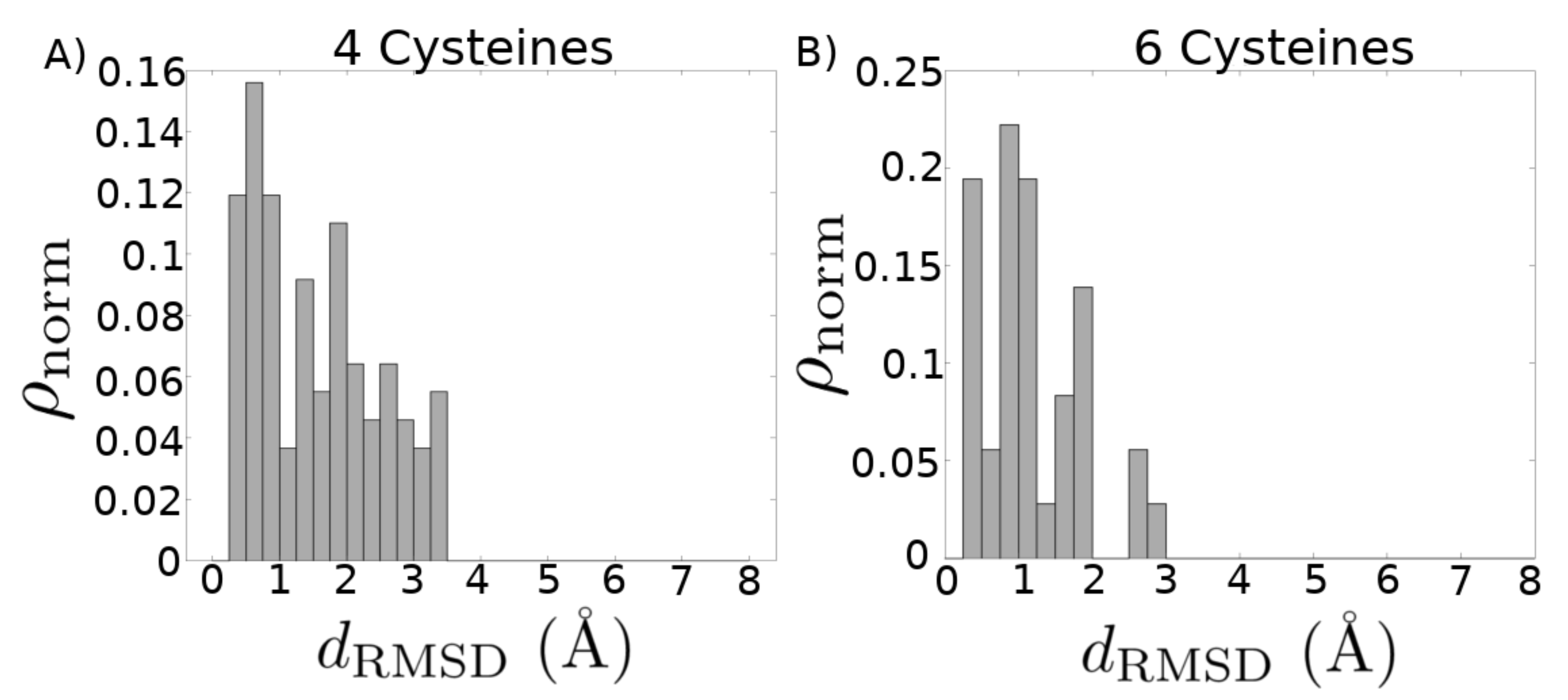
2. Results
3. Discussion
4. Materials and Methods
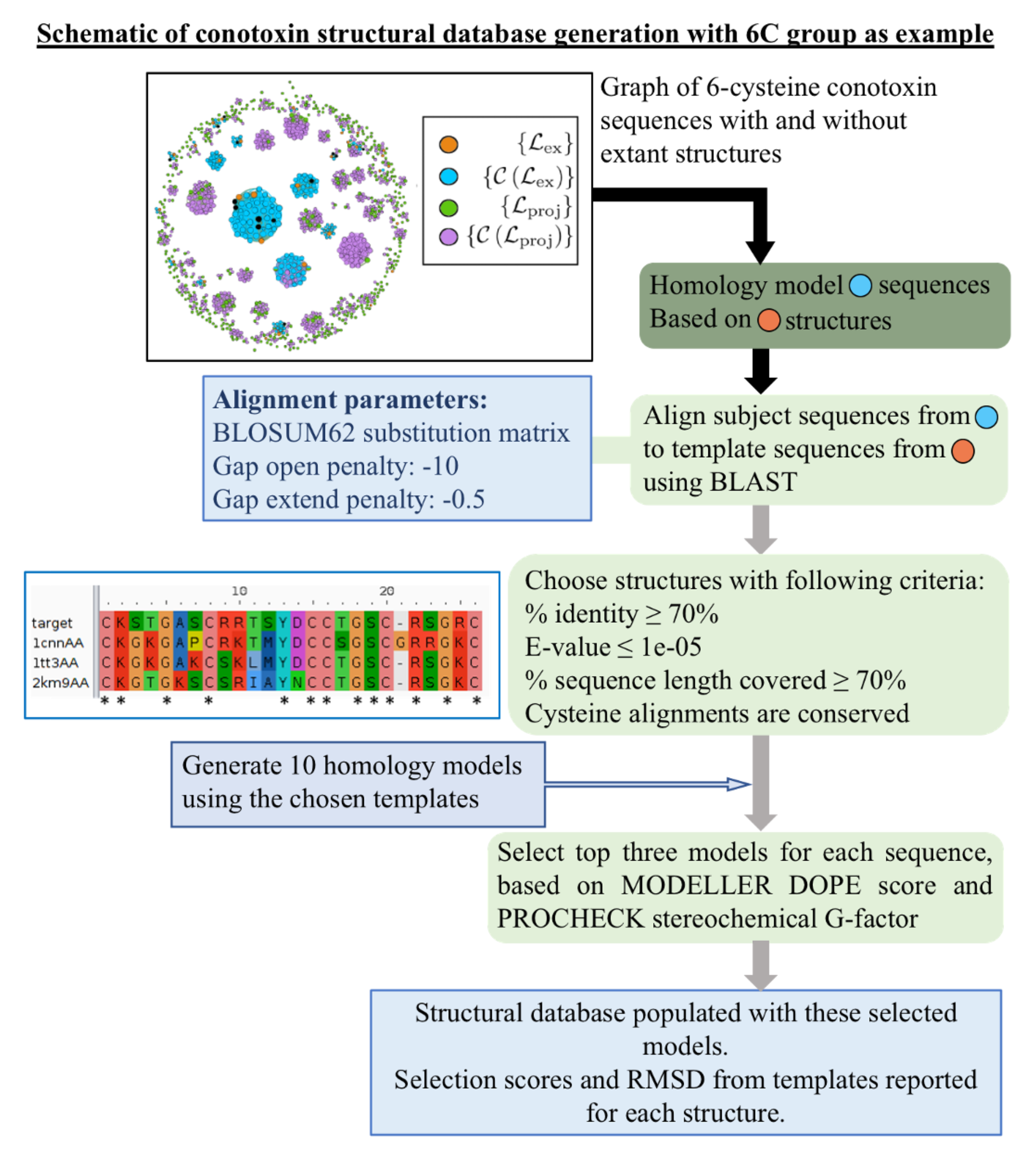
4.1. Data Acquisition and Curation
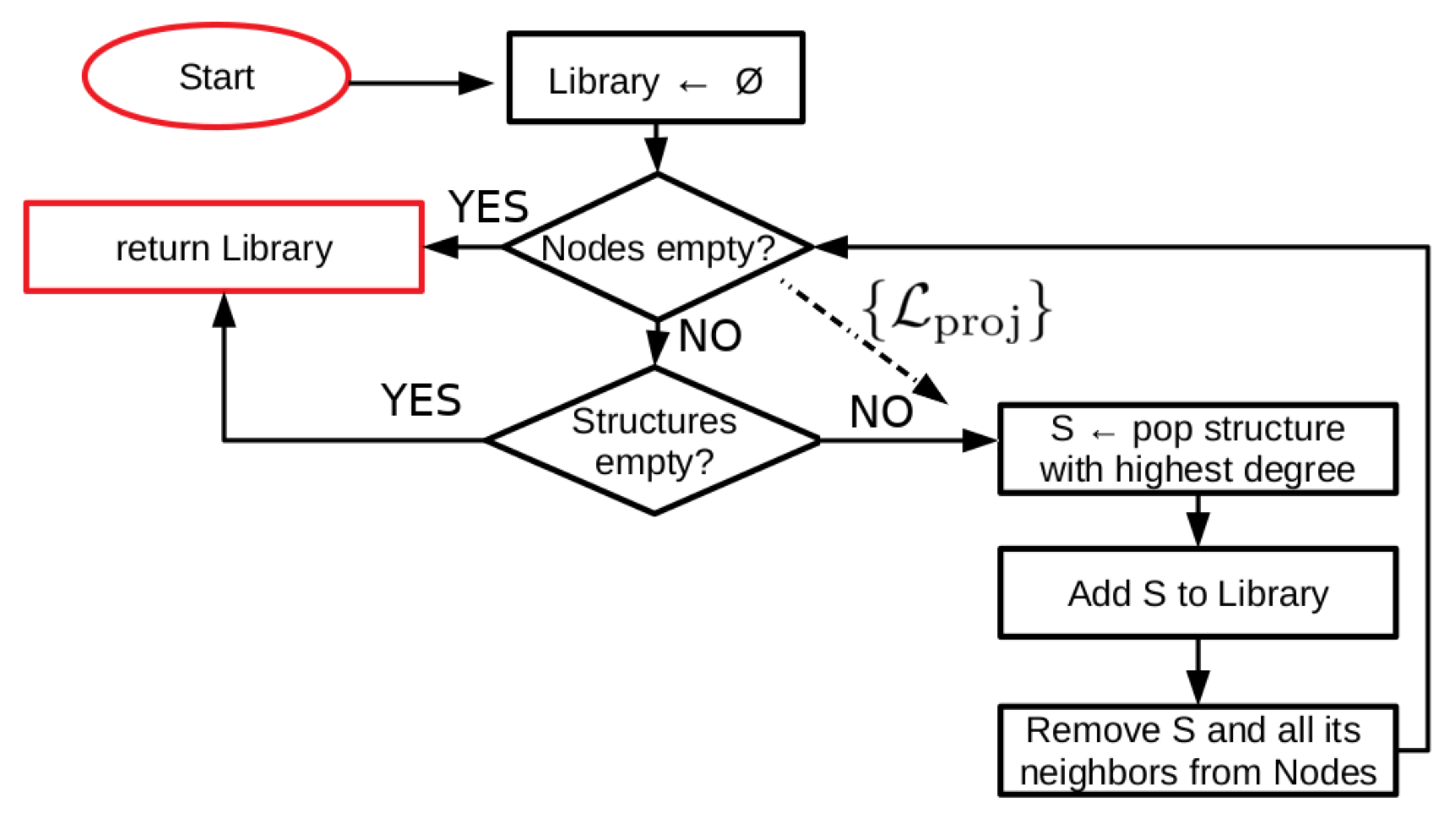
4.2. Details of Library Template Selection Procedure
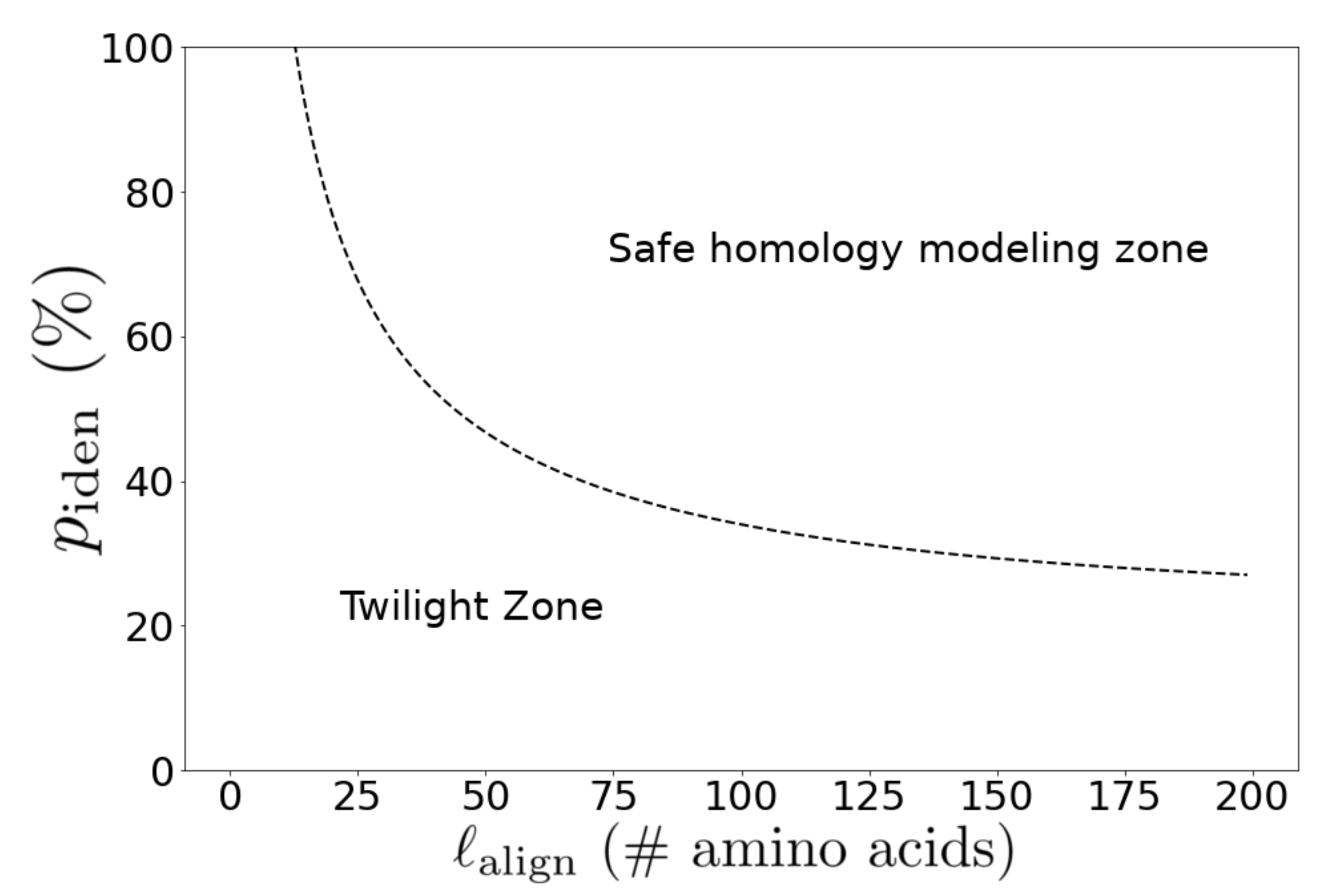
4.3. Homology Modeling Criteria
4.4. Quantification and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name(s) | PDB ID | Sequence |
|---|---|---|
| EpI [sTy15>Y], EpI | 1a0m | GCCSDPRCNMNNPDYC |
| PnIB, PnIB [sTy15Y] | 1akg | GCCSLPPCALSNPDYC |
| CnIA | 1b45 | GRCCHPACGKYYSC |
| AuIB, Ac-AuIB, AuIB [ribbon isoform] | 1mxp | GCCSYPPCFATNPDC |
| ImI [R11E] | 1e74 | GCCSDPRCAWEC |
| ImI [R7L] | 1e75 | GCCSDPLCAWRC |
| ImI [D5N] | 1e76 | GCCSNPRCAWRC |
| TIA | 2lr9 | FNWRCCLIPACRRNHKKFC |
| MrIB, MrIB C-term amidated | 1ieo | VGVCCGYKLCHPC |
| EI | 1k64 | RDPCCYHPTCNMSNPQIC |
| GID, GID*, GID*-NH2, GID*[O16P] | 1mtq | IRDECCSNPACRVNNPHVC |
| SI | 1hje | ICCNPACGPKYSC |
| TXIX | 1wct | ECCEDGWCCXAAP |
| GI | 1xga | ECCNPACGRHYSC |
| Conkunitzin-S1 | 1y62 | RPSLCDLPADSGSGTKAEKRIYYNSARKQ-CLRFDYTGQGGNENNFRRTYDCQRTCL |
| PIA, PIA [R1ADMA] | 1zlc | RDPCCSNPVCTVHNPQIC |
| cMII-6 | 2ajw | GCCSNPVCHLEHSNLCGGAAGG |
| PlXIVA | 2fqc | FPRPRICNLACRAGIGHKYPFCHCR |
| GI (SER12)-benzoylphenylalanine | 2fr9 | ECCNPACGRHYYC |
| GI (ASN4)-benzoylphenylalanine | 2frb | ECCYPACGRHYSC |
| OmIA | 2gcz | GCCSHPACNVNNPHICG |
| BuIA, BuIA[P6O], BuIA[P7O] | 2ns3 | GCCSTPPCAVLYC |
| ImI [P6A] | 2ifi | GCCSDARCAWRC |
| ImI [P6K], ImI [P6K] deamidated | 2ifj | GCCSDKRCAWRC |
| ImI, ImI [C2U,C8U], ImI [C2U,C3U,C8U,C12U], ImI deamidated, A c-ImI, ImI [A9S], ImI [C3U,C12U], ImI [P60], ImI [P6APro], ImI [P6A(S)Pro], ImI [P6guaPro], ImI [P6betPro], ImI [P6fluoPro], ImI [P6fluo(S)Pro], ImI [P6phiPro], ImI [P6phi(S)Pro], ImI [P6benzPro], ImI [P6naphPro], ImI [P6phi(3S)Pro], ImI [P6phi(5R)Pro] | 2bypF | GCCSDPRCAWRC |
| CMrVIA [K6P], CMrVIA [K6P] amidated | 2ih7 | VCCGYPLCHPC |
| CMrVIA, CMrVIA amidated | 2b5p | VCCGYKLCHPC |
| Cyclic MrIA | 2j15 | NGVCCGYKLCHPCAG |
| RgIA [P6V] | 2juq | GCCSDVRCRYRCR |
| RgIA [D5E] | 2jur | GCCSEPRCRYRCR |
| RgIA [Y10W] | 2jus | GCCSDPRCRWRCR |
| RgIA | 2jut | GCCSDPRCRYRCR |
| Pc16a | 2ler | SCSCKRNFLCC |
| Midi | 2lu6 | CNCSRWARDHSRCC |
| TxIB | 2lz5 | GCCSDPPCRNKHPDLC |
| Li1.12, TxID | 2m3i | GCCSHPVCSAMSPIC |
| Ar1248 | 2m62 | GVCCGVSFCYPC |
| Lo1a | 2md6 | EGCCSNPACRTNHPEVCD |
| LvIA | 5xgl | GCCSHPACNVDHPEIC |
| Exendin-4/conotoxin chimera (Ex-4[1-27]/pl14a) | 2naw | HGEGTFTSDLSKQMEEEAVRC-FIECLKGIGHKYPFCHCR |
| Bt1.8 | 2nay | GCCSNPACILNNPNQC |
| TXIA(A10L) | 2uz6 | GCCSRPPCILNNPDLC |
| CnVA | 3zkt | ECCHRQLLCCLRFV |
| Cyclic Vc1.1 | 4ttl | GCCSDPRCNYDHPEICGGAAGG |
| GIC | 1ul2 | GCCSHPACAGNNQHIC |
| PeIA, Bt1.4, PeIA[P6O], PeIA[P13O] | 5jmeF | GCCSHPACSVNHPELC |
| Pn10.1 | 5t6v | STCCGYRMCVPC |
| LsIA, LsIA# | 5t90F | SGCCSNPACRVNNPNIC |
| VilXIVA | 6efe | GGLGRCIYNCMNSGGGLSFIQCKTMCY |
| Name(s) | PDB ID | Sequence |
|---|---|---|
| conotoxin-GS | 1ag7 | ACSGRGSRCPPQCCMGLRCGRGNPQKCIGAHEDV |
| PIIIE, PIIIE [K9S], PIIIE [S17Y,S18N,S20L] | 1jlo | HPPCCLYGKCRRYPGCSSASCCQR |
| MVIIC, S6.6 | 1omn | CKGKGAPCRKTMYDCCSGSCGRRGKC |
| TVIIA | 1eyo | SCSGRDSRCPPVCCMGLMCSRGKCVSIYGE |
| TxVII | 1f3k | CKQADEPCDVFSLDCCTGICLGVCMW |
| TxVIA | 1fu3 | WCKQSGEMCNLLDQNCCDGYCIVLVCT |
| EVIA | 1g1z | DDCIKPYGFCSLPILKNGLCCSGACVGVCADL |
| GIIIB | 1gib | RDCCTPPRKCKDRRCKPMKCCA |
| GVIA | 1ttl | CKSPGSSCSPTSYNCCRSCNPYTKRCY |
| PIVA | 1p1p | GCCGSYPNAACHPCSCKDRPSYCGQ |
| EIVA | 1pqr | GCCGPYPNAACHPCGCKVGRPPYCDRPSGG |
| PIIIA | 1r9i | QRLCCGFPKSCRSRQCKPHRCC |
| MVIIA[R10K] | 1tt3 | CKGKGAKCSKLMYDCCTGSCRSGKC |
| Am2766 | 1yz2 | CKQAGESCDIFSQNCCVGTCAFICIE |
| MrIIIE | 2efz | VCCPFGGCHELCYCCD |
| FVIA | 2km9 | CKGTGKSCSRIAYNCCTGSCRSGKC |
| Im23a, Mr23a | 2lmz | IPYCGQTGAECYSWCIKQDLSKDWCCDFVKDIRMNPPADKCP |
| BuIIIB | 2lo9 | VGERCCKNGKRGCGRWCRDHSRCC |
| KIIIA, KIIA [W8dTrp] | 2lxg | CCNCSSKWCRDHSRCC |
| Ar1446 | 2m61 | CCRLACGLGCHPCC |
| cGm9a | 2mso | SCNNSCQSHSDCASHCICTFRGCGAVNGLP |
| cBru9a | 2msq | SCGGSCFGGCWPGCSCYARTCFRDGLP |
| Mo3964 | 2mw7 | DGECGDKDEPCCGRPDGAKVCNDPWVCILTSSRCENP |
| MfVIA | 2n7f | RDCQEKWEYCIVPILGFVYCCPGLICGPFVCV |
| cyclic PVIIA | 2n8e | CRIPNQKCFQHLDDCCSRKCNRFNKCVLPETGGG |
| conotoxin-muOxi-GVIIJ | 2n8h | GWCGDPGATCGKLRLYCCSGFCDSYTKTCKDKSSA |
| CnIIIC | 2yen | QGCCNGPKGCSSKWCRDHARCC |
| CcTx | 4b1qP | APWLVPSQITTCCGYNPGTMCPSCMCTNTC |
| Reg12i | 6bx9 | CCTALCSRYHCLPCC |
| MoVIB | 6ceg | CKPPGSKCSPSMRDCCTTCISYTKRCRKYY |
| Name(s) | PDB ID | Sequence |
|---|---|---|
| G11.1 | 6cei | CAVTHEKCSDDYDCCGSLCCVGICAKTIAPCK |
| RXIA, RXIA[Btr33>W] | 2p4l | GPSFCKADEKPCEYHADCCNCCLSGICAPSTNWILPGCSTSSFFKI |

References
- Zambelli, V.; Pasqualoto, K.; Picolo, G.; Chudzinski-Tavassi, A.; Cury, Y. Harnessing the knowledge of animal toxins to generate drugs. Pharmacol. Res. 2016, 112, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Verdes, A.; Anand, P.; Gorson, J.; Jannetti, S.; Kelly, P.; Leffler, A.; Simpson, D.; Ramrattan, G.; Holford, M.; Verdes, A.; et al. From Mollusks to Medicine: A Venomics Approach for the Discovery and Characterization of Therapeutics from Terebridae Peptide Toxins. Toxins 2016, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G. Ziconotide: Neuronal Calcium Channel Blocker for Treating Severe Chronic Pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Dang, B. Chemical synthesis and structure determination of venom toxins. Chin. Chem. Lett. 2019, 30, 1369–1373. [Google Scholar] [CrossRef]
- Romano, J.D.; Tatonetti, N.P. Informatics and Computational Methods in Natural Product Drug Discovery: A Review and Perspectives. Front. Genet. 2019, 10, 368. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.C.L.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [Green Version]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein–peptide docking: Opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef]
- Leffler, A.E.; Kuryatov, A.; Zebroski, H.A.; Powell, S.R.; Filipenko, P.; Hussein, A.K.; Gorson, J.; Heizmann, A.; Lyskov, S.; Tsien, R.W.; et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. Proc. Natl. Acad. Sci. USA 2017, 114, E8100–E8109. [Google Scholar] [CrossRef] [Green Version]
- Younis, S.; Rashid, S. Alpha conotoxin-BuIA globular isomer is a competitive antagonist for oleoyl-L-alpha-lysophosphatidic acid binding to LPAR6; A molecular dynamics study. PLoS ONE 2017, 12, e0189154. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Tatay, L.; Hernandez-Andreu, J.M. Biosafety and biosecurity in Synthetic Biology: A review. Crit. Rev. Env. Sci. Tec. 2019, 49, 1587–1621. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struc. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Mansbach, R.A.; Travers, T.; McMahon, B.H.; Fair, J.M.; Gnanakaran, S. Snails In Silico: A Review of Computational Studies on the Conopeptides. Mar. Drugs 2019, 17, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef]
- Pitera, J.W.; Swope, W. Understanding folding and design: Replica-exchange simulations of “Trp-cage” miniproteins. Proc. Natl. Acad. Sci. USA 2003, 100, 7587–7592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensign, D.L.; Kasson, P.M.; Pande, V.S. Heterogeneity Even at the Speed Limit of Folding: Large-scale Molecular Dynamics Study of a Fast-folding Variant of the Villin Headpiece. J. Mol. Biol. 2007, 374, 806–816. [Google Scholar] [CrossRef] [Green Version]
- Voelz, V.A.; Bowman, G.R.; Beauchamp, K.; Pande, V.S. Molecular Simulation of ab Initio Protein Folding for a Millisecond Folder. J. Am. Chem. Soc. 2010, 132, 1526–1528. [Google Scholar] [CrossRef] [Green Version]
- Sborgi, L.; Verma, A.; Piana, S.; Lindorff-Larsen, K.; Cerminara, M.; Santiveri, C.M.; Shaw, D.E.; de Alba, E.; Muñoz, V. Interaction Networks in Protein Folding via Atomic-Resolution Experiments and Long-Time-Scale Molecular Dynamics Simulations. J. Am. Chem. Soc. 2015, 137, 6506–6516. [Google Scholar] [CrossRef]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z. Advances in Homology Protein Structure Modeling. Curr. Protein Pept. Sci. 2006, 7, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Nabuurs, S.B.; Vriend, G. Homology modeling. Methods Biochem. Anal. 2003, 44, 509–524. [Google Scholar] [PubMed]
- Kong, L.; Lee, B.T.K.; Tong, J.C.; Tan, T.W.; Ranganathan, S. SDPMOD: An automated comparative modeling server for small disulfide-bonded proteins. Nucleic Acids Res. 2004, 32, W356–W359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. Des. Sel. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Van Steen, M. Graph Theory and Complex Networks—An Introduction; van Steen, Maarten: Lexington, KY, USA, 2010; Volume 144. [Google Scholar]
- Green, S.; Şerban, M.; Scholl, R.; Jones, N.; Brigandt, I.; Bechtel, W. Network analyses in systems biology: New strategies for dealing with biological complexity. Synthese 2018, 195, 1751–1777. [Google Scholar] [CrossRef] [Green Version]
- Pavlopoulos, G.A.; Secrier, M.; Moschopoulos, C.N.; Soldatos, T.G.; Kossida, S.; Aerts, J.; Schneider, R.; Bagos, P.G. Using graph theory to analyze biological networks. BioData Min. 2011, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Morel, P.A.; Lee, R.E.; Faeder, J.R. Demystifying the cytokine network: Mathematical models point the way. Cytokine 2017, 98, 115–123. [Google Scholar] [CrossRef]
- Aburatani, S.; Kokabu, Y.; Teshima, R.; Ogawa, T.; Araki, M.; Shiarai, T. Application of Graph Theory to Evaluate Chemical Reactions in Cells. J. Phys. Conf. Ser. 2019, 1391, 012047. [Google Scholar] [CrossRef]
- Sethi, A.; Tian, J.; Derdeyn, C.A.; Korber, B.; Gnanakaran, S. A mechanistic understanding of allosteric immune escape pathways in the HIV-1 envelope glycoprotein. PLoS Comput. Biol. 2013, 9, e1003046. [Google Scholar] [CrossRef]
- Chakraborty, S.; Berndsen, Z.T.; Hengartner, N.W.; Korber, B.T.; Ward, A.B.; Gnanakaran, S. A Network-based approach for Quantifying the Resilience and Vulnerability of HIV-1 Native Glycan Shield. bioRxiv Preprint 2019. bioRxiv:10.1101/856071. [Google Scholar]
- Long, A.W.; Ferguson, A.L. Rational design of patchy colloids via landscape engineering. Mol. Syst. Des. Eng. 2018, 3, 49–65. [Google Scholar] [CrossRef]
- Santiago, C.; Pereira, V.; Digiampietri, L. Homology Detection Using Multilayer Maximum Clustering Coefficient. J. Comput. Biol. 2018, 25, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Bolten, E.; Schliep, A.; Schneckener, S.; Schomburg, D.; Schrader, R. Clustering protein sequences–structure prediction by transitive homology. Bioinformatics 2001, 17, 935–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipenbacher, P.; Schliep, A.; Schneckener, S.; Schonhuth, A.; Schomburg, D.; Schrader, R. ProClust: Improved clustering of protein sequences with an extended graph-based approach. Bioinformatics 2002, 18, S182–S191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhang, S.; Wu, F.X. Applications of graph theory in protein structure identification. Proteome Sci. 2011, 9, S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abascal, F.; Valencia, A. Clustering of proximal sequence space for the identification of protein families. Bioinformatics 2002, 18, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Enright, A.J.; Ouzounis, C.A. GeneRAGE: A robust algorithm for sequence clustering and domain detection. Bioinformatics 2000, 16, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Karp, R.M. Reducibility among Combinatorial Problems. In Complexity of Computer Computations: Proceedings of a Symposium on the Complexity of Computer Computations, Held March 20–22, 1972, at the IBM Thomas J. Watson Research Center, Yorktown Heights, New York, and Sponsored by the Office of Naval Research, Mathematics Program, IBM World Trade Corporation, and the IBM Research Mathematical Sciences Department; Miller, R.E., Thatcher, J.W., Bohlinger, J.D., Eds.; Springer: Boston, MA, USA, 1972; pp. 85–103. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. Experimental accuracy in protein structure refinement via molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2018, 115, 13276–13281. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large data sets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Undheim, E.A.; Mobli, M.; King, G.F. Toxin structures as evolutionary tools: Using conserved 3D folds to study the evolution of rapidly evolving peptides. BioEssays 2016, 38, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araujo, A.D.; Mobli, M.; Castro, J.; Harrington, A.M.; Vetter, I.; Dekan, Z.; Muttenthaler, M.; Wan, J.; Lewis, R.J.; King, G.F.; et al. Selenoether oxytocin analogues have analgesic properties in a mouse model of chronic abdominal pain. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanujam, V.; Shen, Y.; Ying, J.; Mobli, M. Residual dipolar couplings for resolving cysteine bridges in disulfide-rich peptides. Front. Chem. 2020, 7, 889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, M.P. Peptide structure determination by NMR. In Spectroscopic Methods and Analyses; Humana Press: Totowa, NJ, USA, 1993; pp. 69–85. [Google Scholar]
- Combelles, C.; Gracy, J.; Heitz, A.; Craik, D.J.; Chiche, L. Structure and folding of disulfide-rich miniproteins: Insights from molecular dynamics simulations and MM-PBSA free energy calculations. Proteins 2008, 73, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Paul George, A.A.; Heimer, P.; Maaß, A.; Hamaekers, J.; Hofmann-Apitius, M.; Biswas, A.; Imhof, D. Insights into the Folding of Disulfide-Rich μ-Conotoxins. ACS Omega 2018, 3, 12330–12340. [Google Scholar] [CrossRef] [Green Version]
- Dutton, J.L.; Bansal, P.S.; Hogg, R.C.; Adams, D.J.; Alewood, P.F.; Craik, D.J. A new level of conotoxin diversity, a non-native disulfide bond connectivity in alpha-conotoxin AuIB reduces structural definition but increases biological activity. J. Biol. Chem. 2002, 277, 48849–48857. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, S.; Belgi, A.; Bartels, P.; van Lierop, B.J.; Robinson, S.D.; Kompella, S.N.; Hung, A.; Callaghan, B.P.; Adams, D.J.; Robinson, A.J.; et al. Dicarba Analogues of α-Conotoxin RgIA. Structure, Stability, and Activity at Potential Pain Targets. J. Med. Chem. 2014, 57, 9933–9944. [Google Scholar] [CrossRef]
- Steiner, A.M.; Bulaj, G. Optimization of oxidative folding methods for cysteine-rich peptides: A study of conotoxins containing three disulfide bridges. J. Pept. Sci. 2011, 17, 1–7. [Google Scholar] [CrossRef]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, Synthesis, and Structure–Activity Relationships of Conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef]
- Kasheverov, I.E.; Chugunov, A.O.; Kudryavtsev, D.S.; Ivanov, I.A.; Zhmak, M.N.; Shelukhina, I.V.; Spirova, E.N.; Tabakmakher, V.M.; Zelepuga, E.A.; Efremov, R.G.; et al. High-Affinity α-Conotoxin PnIA Analogs Designed on the Basis of the Protein Surface Topography Method. Sci. Rep. 2016, 6, 36848. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; Volume 54, pp. 1–5. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.8; Technical Report; Schrödinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [Green Version]
- Braun, W.; Go, N. Calculation of protein conformations by proton-proton distance constraints: A new efficient algorithm. J. Mol. Biol. 1985, 186, 611–626. [Google Scholar] [CrossRef]
- Shen, M.y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M.; IUCr. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- John, B.; Sali, A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003, 31, 3982–3992. [Google Scholar] [CrossRef]
- Oliphant, T.E. Python for Scientific Computing. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar] [CrossRef] [Green Version]





| Sequence | Name(s) | Degree | Pharm. Fam. |
|---|---|---|---|
| AAKVKYSNTPEECCSNPPCFATHSEICG | Li1.28 | 10 | Unknown |
| GCCSDPRCAYDHPEIC | Vc1.1[N9A] | 10 | alpha |
| GCCSNPVCHLEHSNAC | MII [L15A] | 8 | Unknown |
| AALEDADMKTEKGFLSSIVGNLGTVGNLV– | |||
| GSVCCQITNSCCPED | Pu5.7 | 7 | Unknown |
| RAALEDADMKTEKGVLNAIFSNLGDLGNL– | |||
| VSSVCCKATTSCCPED | Pu5.9 | 6 | Unknown |
| AGLTDADLKTEKGFLSGLLNVAGSVCCKVDTSCCSNQ | Lt5g | 6 | Unknown |
| GCCSNPVCALEHSNLC | MII [H9A] | 6 | Unknown |
| VPAEQMMEELCPDMCNRGEGEIICTCVLRRHVVSPSIR | Lt14.4 | 5 | Unknown |
| TNEGPGRDPAPCCQHPIETCC | Cal5b | 5 | Unknown |
| RPECCTHPACHVSNPELCS | Mr1.8 | 4 | Unknown |
| GCCSRPPCIANNPDLC | TxIA | 4 | alpha |
| SPGSTICKMACRTGNGHKYPFCNCR | Fe14.1 | 4 | Unknown |
| GCCSLPPCALNNPDYC | PnIA [A10L,sTy15Y] | 4 | Unknown |
| YAAVVNRASALMAQAVLRDCCSNPPCAHNIHCA | Ec1.7 | 4 | Unknown |
| NGRCCHPACGKHFSC | Ac1.1b, CnIH, R1.1, Bt1.6, Mn1.2, C4.3 | 4 | Unknown |
| NGRCCHPACGKYFSC | Mn1.5 | 3 | Unknown |
| GCCSRAACAGIHQELC | LtIA [A4S] | 3 | Unknown |
| GCCSNPVCHLAHSNAC | MII [E11A,L15A] | 3 | Unknown |
| GCCSHPACSGNNREYCRES | O1.3 | 3 | Unknown |
| GGCCSHPVCYFNNPQMCR | Cr1.6 | 3 | Unknown |
| GGGCCSHPACAANNQDYC | Gly-AnIB | 3 | Unknown |
| DGCCSSPSCSVNNPDICGG | Eb1.1, Qc1.18 | 3 | Unknown |
| LDPCCREPPCASTHTDICT | Li1.4, Sa1.12 | 3 | Unknown |
| NECCDNPPCKSSNPDLCDWRS | Qc1.1b, LiC22 | 3 | Unknown |
| DECCSNPSCAQTHPEVC | Li1.24, Sa1.6 | 3 | Unknown |
| GCCSHPACAGNNPHICS | Li1.11 | 3 | Unknown |
| SFRFIPGGIKEIACHRYCAKGIASAFCNCPDKRDVVSPRI | G14.1 | 3 | Unknown |
| VPPEPILEIICPGMCDEGVGKEPFCHCTKKRDAVSSRI | Vc14.4 | 3 | Unknown |
| GCCSYPPCNVSYPEICG | Su1.6 | 2 | Unknown |
| AANDKASVQIALTVQECCADSACSLTNPLIC | Dd1.7, Li1.21 | 2 | Unknown |
| TAFGLRLCCKRHHGCHPCGRT | Cal1b | 2 | Unknown |
| AANAKLFDVGQSCCSAPLCALLYMVIC | Sa1.7 | 2 | Unknown |
| TVRDACCSDPRCSGKHQDLC | Li1.16, Sa1.3 | 2 | Unknown |
| NLQILCCKHTPACCT | S5.3, Eb5.5 | 2 | Unknown |
| ECPPWCPTSHCNAGTC | Cl14c | 2 | Unknown |
| GIWCDPPCPKGETCRGGECSDEFNSDV | Cal14.1a | 2 | Unknown |
| GMWDECCDDPPCRQNNMEHCPAS | Lp1.7 | 2 | Unknown |
| GRCCHPACGGKYFKC | CnIJ | 2 | Unknown |
| IALIATRECCANPQCWSKNC | Co1.3 | 2 | Unknown |
| GCCSHPVCHARHPELC | PeIA[A7V, S9H,V10A,N11R] | 2 | Unknown |
| DGCCSDPACSVNHPDICGG | Qc1.7 | 2 | Unknown |
| PPGCCNNPACVKHRCG | Bu1.2 | 2 | Unknown |
| LINTRCCPGQPCCRM | Vc5.11 | 2 | Unknown |
| NAAANDKASDVIPLALQGCCSNPVCHVDHPELCL | Cn1.6 | 2 | Unknown |
| GCCSHPVCHARHPALC | PeIA[A7V, S9H,V10A,N11R,E14A] | 2 | Unknown |
| WDVNDCIHFCLIGVVGRSYTECHTMCT | FlfXIVB | 2 | Unknown |
| NGRCCHPACAKYFSC | Mn1.4b | 2 | Unknown |
| NGRCCHPACGGKYVKC | Ac1.2 | 2 | Unknown |
| GCCSYPPCFATNSDYC | AuIA | 2 | alpha |
| DECCAIPLCAKIFPGRCP | Pc1b | 1 | Unknown |
| AANLMALLQESLCPPGCYPSCTNCRYMFP | Pu14.6 | 1 | Unknown |
| GCCAIRECRLQNAAYCGGIY | Ca1.2 | 1 | Unknown |
| FLTQQSPRDFAKSVMQLLHYNWIDCCNYGVSDCCI | Lv5.7 | 1 | Unknown |
| APAELILETICPHMCGTGIGEPFCNCRNKRDVVSSRII | Bt14.3 | 1 | Unknown |
| EIVNIIDSISDVAKQICCEITVQCCVLDEE | Vn5.5 | 1 | Unknown |
| ECCEDGWCCTAAPLTAP | Vc5.7 | 1 | Unknown |
| CCPGWELCCEWDDWW | Mr5.7 | 1 | Unknown |
| GCCSFPACRKYRPEMCG | Su1.2 | 1 | Unknown |
| DDCCPDPACRQNHPELCST | PuSG1.1 | 1 | Unknown |
| APNVKDSKASGSCCDNPSCAVNNSHC | Li1.32 | 1 | Unknown |
| YHECCKNPPCRNKHPDLC | Sa1.16 | 1 | Unknown |
| GCCSNPACAGSNAHIC | Li1.14 | 1 | Unknown |
| GCCVYPPCAVNHPDICRG | Qc1.9 | 1 | Unknown |
| VMQLRYYNWIDCCFDGDCCN | Qc5.3 | 1 | Unknown |
| TGCCEYPYCAENNPELCG | Co1.4 | 1 | Unknown |
| SVEGVISTIKDFAVKVCCSVSLKFCCPTA | Ts5.5 | 1 | Unknown |
| SCCSDSDCNANHPDMCS | Leo-A1 | 1 | Unknown |
| SCCPQEFLCCLYLVK | Lp5.1 | 1 | Unknown |
| RCCHPACGKNYSC | MI[del1G] | 1 | Unknown |
| QTPGCCWNPACVKNRC | EIIA | 1 | Unknown |
| QGCCSYPACAVSNPDICGG | Qc1.12 | 1 | Unknown |
| PECCSDPRCNSTHPELCG | Ai1.2 | 1 | Unknown |
| NIQIICCKHTPKCCT | Tx5.5 | 1 | Unknown |
| NAWLTPEECCAAPACREMILEFCLAGEAFAAAL– | |||
| DGFRRLPYR | Pu1.5 | 1 | Unknown |
| KVYCCLGVRDDWCCAGQIQI | Lt5i | 1 | Unknown |
| IINWCCLIFYQCCL | Sr5.7 | 1 | Unknown |
| YCCHPACGKNFDC | SIA | 1 | alpha |
| GILELAKTVCCSATGISICC | Tx5.13, Tr5.3, Vr5.1 | 1 | Unknown |
| GGCCSRPPCILKHPEIC | Qc1.13 | 1 | Unknown |
| GCPADCPNTCDSSNKCSPGFP | Cal14a | 1 | Unknown |
| GIRGNCCMFHTCPIDYSRFYCP | Vt1.24 | 1 | Unknown |
| Sequence | Name(s) | Degree | Pharm. Fam. |
|---|---|---|---|
| LPPCCSLNLRLCPAPACKYKPCCKS | RIIIJ 6-11 | 29 | Unknown |
| QKGLVPSVITTCCGYDPGTMCPPCRCTNSCPKKPKKP | S4.4 | 28 | Unknown |
| QPWLVPSKITNCCGYNTMEMCPTCMCTYSCRPKKKKP | Mn4.2 | 27 | Unknown |
| DDECEPPGDFCGFFKIGPPCCSGWCFLWCA | MaIr137, G6.2 | 20 | Unknown |
| DCVAGGHFCGFPKIGGPCCSGWCFFVCA | Vn6.8 | 20 | Unknown |
| VCREKGQGCTNTALCCPGLECEGQSQGGLCVDN | Mi010 | 20 | Unknown |
| ECREQSQGCTNTSPPCCSGLRCSGQSQGGVCISN | CaHr91 | 17 | Unknown |
| TVDEACNEYCEERNKNCCGRTDGEPVCAQACL | Vi6.7 | 15 | Unknown |
| ECTRSGGACYSHNQCCDDFCSTATSTCV | Eb6.22 | 15 | Unknown |
| GCTPPGGACGGHAHCCSQSCNILASTCNA | ABVIC | 15 | Unknown |
| TVGEECNEYCEQRNKNCCGKTNGEPVCAQACL | Tr7.4 | 15 | Unknown |
| TATEECEEYCEDEEKTCCGEEDGEPVCARFCL | Ar6.24 | 14 | Unknown |
| EACYNAGTFCGIKPGLCCSAICLSFVCISFDLIDVFSSP | M6.2 | 13 | Unknown |
| TTEECHEYCEDQNKNCCGLTDGEPRCAGMCL | Tr7.3 | 13 | Unknown |
| MTMGCTHPGGACGGHYHCCSQSCNTAANSCN | MIL3-b (partial) | 12 | Unknown |
| VPEECEESCEEEEKTCCGLENGQPFCSRICW | Ar6.28 | 12 | Unknown |
| DECYPPGTFCGIKPGLCCSERCFPFVCLSLEF | Ac6.2 | 12 | Unknown |
| CLDAGEVCDIFFPTCCGYCILLFCA | TxO1 | 11 | omega |
| DCTPPDGACGFHYHCCSKFCITISSTCN | MIL2-a | 11 | Unknown |
| CIDGGEICDIFFPNCCSGWCIILVCA | Mr6.8 | 11 | Unknown |
| TTAESWWEGECLGWSNGCTHPSDCCSNYCKGIYCDL | Mr6.16 | 11 | Unknown |
| GCTHPGGACGGHHHCCSLFCNTAANACN | MIL3-f | 11 | Unknown |
| CLGSGETCWLDSSCCSFSCTNNVCF | Vn6.15 | 11 | Unknown |
| CLDAGEMCDLFNSKCCSGWCIILFCA | Mr6.1 | 10 | Unknown |
| SCGEEGEGCYTRPCCPGLKCIGTAHGGLCREE | Pu6.7 | 10 | Unknown |
| GCLEVDYFCGIPFVNNGLCCSGNCVFVCTPQ | Pn6.7 | 10 | Unknown |
| SIAGRTTTEECDEYCEDLNKNCCGLSNGEPVCATACL | Ts6.7 | 10 | Unknown |
| DGCYNAGTFCGIRPGLCCSEFCFLWCITFVDS | MVIA, Cn6.1 | 10 | delta |
| NCCNGGCSSKWCRDHARCC | SIIIA[del1] | 9 | Unknown |
| KTTAESWWEGECYGWWTSCSSPEQC– | |||
| CSLNCENIYCRAW | TsMEKL-03 | 8 | Unknown |
| RHGCCKGPKGCSSRECRPQHCC | TIIIA | 8 | mu |
| DCGEQGQGCYTRPCCPGLHCAAGATGGGSCQP | Conotoxin-1 | 8 | Unknown |
| CLAGSAPCEFHRGYTCCSGHCLIWVCA | Cal6.1d | 8 | Unknown |
| KTTAESWWEGECRTWYAPCNFPSQC– | |||
| CSEVCSSKTGRCLTW | Vn6.5 | 7 | Unknown |
| CRPPGMVCGFPKPGPYCCSGWCFAVCLPV | MaIr193 | 7 | Unknown |
| CTPGGEACDATTNCCFLTCNLATNKCRSPNFP | ABVIL | 7 | Unknown |
| WWEGECRGWSNGCTTNSDCCSNNCDGTFCKLW | Vn6.3 | 7 | Unknown |
| WWWGGCTWWFGRCSTDSECCSNSCDQTYC– | |||
| ELYRFPSRY | Vc6.26 | 7 | Unknown |
| YECYSTGTFCGINGGLCCSNLCLFFVCLTFS | CnVIA, St6.2 | 7 | delta |
| CCSRDCWVCIPCCPNGSA | Lv3-IP01 | 7 | Unknown |
| VCVDGGTFCGFPKIGGPCCSGWCIFVCL | Ar6.2 | 6 | Unknown |
| ECIEGSEPCEVFRPYTCCSGHCIIFVCA | Cal6.1h | 6 | Unknown |
| CCSQDCWVCIPCCPN | Eu3.2 | 6 | Unknown |
| CCSQDCSVCIPCCPN | Co3-IP02, Ts3-IP07, Vr3-IP08, Rt3-IP03, Ca3-IP02, Ec3-IP03 | 6 | Unknown |
| CYDSGTSCNTGNQCCSGWCIFVSCL | Tx6.3 | 6 | Unknown |
| CTVDSDFCDPDNHDCCSGRCIDEGGSGVCAIVPVLN | Ar6.19 | 6 | Unknown |
| FPCNPGGCACRPLDSYSYTCQSPSSSTANCEGNECVS– | |||
| EADW | Cl9.4 | 6 | Unknown |
| GPPCCLYGSCRPFPGCSSASCCRK | PIIIF [Y17S,N18S,L20S] | 5 | Unknown |
| DCQEKWDYCPVPFLGSRYCCDGFICPSFFCA | Da6.6, Tx6.6 | 5 | Unknown |
| CCGVPNAACHPCVCNNTC | OIVA [K15N] | 5 | Unknown |
| CTPRNGYCYYRYFCCSRACNLTIKRCL | Ml6.2 | 5 | Unknown |
| CCSQDCRVCIPCCPN | Ts3.1 | 5 | Unknown |
| QCTPVGGSCSRHYHCCSLYCNKNIGQCLATSYP | Ar6.17 | 5 | Unknown |
| CLNDGDDCDTGDDCCSGLCIFDEYFSYCDDSDP– | |||
| YYDDYDEYYY | Mi029 | 5 | Unknown |
| SCGNLHESCSAHRCCPGLKCIGTAHGGLCRE | Pu6.15 (partial) | 5 | Unknown |
| VKPCSEEGQLCDPLSQNCCRGWHCVLVSCV | Da6.2 | 5 | Unknown |
| FAVIFTCTPPGSHCTGHSDCCSDFCSTMSDVCQ | Co6.1 | 4 | Unknown |
| WWDGECRLWSNGCRKHKECCSNHCKGIYCDIW | VeG52 | 4 | Unknown |
| CTPCGPDLCCEPGTTCDTVLHHTRFGEPSCSY | Fla6.16 | 4 | Unknown |
| CCGKPNAACHPCVCNGSCS | G4.1 | 4 | Unknown |
| MGYILPALSQQTCCVRPWCDGACDCCVDS | Co3-D01 | 4 | Unknown |
| MKLMLSALRQQECCKPSTCDGGCYHCC | Lv3-YH04 | 4 | Unknown |
| SCGNLHESCSAHRCCPGLMCFTLPTPICIW | Pu6.17 | 4 | Unknown |
| CIPQFDPCDMVRHTCCKGLCVLIACSKTA | Pn6.3 | 4 | Unknown |
| STSCMEAGSYCGSTTRICCGYCAYFGKKCIDYPSN | SO5 | 4 | omega |
| GGCTPCGPNLCCSEEFRCGTSTHHQTYGEPACLSY | Ca6.2 | 4 | Unknown |
| CLGFGEACLMLYSDCCSYCVALVCL | Ep6.1 | 4 | Unknown |
| CIEQFDPCEMIRHTCCVGVCFLMACI | King-Kong 1 | 4 | Unknown |
| TCSPAGEVCTSKSPCCTGFLCTHIGGMCHH | LvVIA 2 | 4 | Unknown |
| CTPSGGACYVASTCCSNACNLNSNKCV | M1 | 4 | Unknown |
| CCGVPNAACHPCVCTGKC | PeIVA | 4 | alpha |
| ATDCIEAGNYCGPTVMKICCGFCSPFSKICMNYPQN | Ac6.5 | 4 | Unknown |
| GCTPRNGACGYHSHCCSNFCHTWANVCL | LvVID | 3 | Unknown |
| DVCELPFEEGPCFAAIRVYAYNAKTGDCEQLTY– | |||
| GGCEGNGNRFATLEDCDNACARY | Cal9.1d | 3 | Unknown |
| KFCCDSNWCHISDCECCY | Tx3h | 3 | Unknown |
| GCGYLGEPCCVAPKRAYCHGDLECNSVAMCVN | Mr2 | 3 | Unknown |
| ECTPPEGACNHPSHCCEDFCDRGRNRCM | At6.7 | 3 | Unknown |
| WWEGDCTDWLGSCSSPSECCYDNCETYCTLW | Lt7b | 3 | Unknown |
| CRSSGSPCGVTSICCGRCYRGKCT | SVIA | 3 | omega |
| CKAESEACNIITQNCCDGKCLFFCIQIPE | Pn6.5 | 3 | Unknown |
| CKSPGTPCSRTMRDCCTSCLSYSKKCR | G6.12 | 3 | Unknown |
| CTPPSGYCYHPYYCCSRACNLTRKRCL | At6.2 | 3 | Unknown |
| PCKTPGRKCFPHQKDCCGRACIITICP | P2a | 3 | Unknown |
| CVPYEGPCNWLTQNCCDELCVFFCL | Gm6.3 | 3 | Unknown |
| SKQCCHLPACRFGCTPCCW | Mr3.4 | 3 | Unknown |
| CCKYGWTCWLGCSPCGC | PnIVB | 2 | mu |
| EIILHALGTRCCSWDVCDHPSCTCC | Vr3-T05 | 2 | Unknown |
| CNNRGGGCSQHPHCCSGTCNKTFGVCL | VxVIA, MgJ42 | 2 | Unknown |
| CAGIGSFCGLPGLVDCCSGRCFIVCLP | Bt6.4, ErVIA | 2 | Unknown |
| CCHWNWCDHLCSCCGS | Mr3.8 | 2 | Unknown |
| CCQAACSPWLCLPCC | Eu3.3, Bt3.3 | 2 | Unknown |
| CIPFLHPCTFFFPDCCNSICAQFICL | VcVIC | 2 | Unknown |
| CTQSSEFCDVIDPDCCSGVCMAFFCI | Vc6.40 | 2 | Unknown |
| CTVNGVVCDPGNHNCCSGSCLDDEDTPVCGIHV– | |||
| EIQHVHMLS | Pu6.23 | 2 | Unknown |
| CCDDSECDYSCWPCCMF | Gm3-WP04 | 2 | Unknown |
| DAINVAPGTSITRTETDQECIDTCKQEDKKCCG– | |||
| RSNGVPTCAKICL | Di6.11 | 2 | Unknown |
| CLAPQRWCSMHDDSLHDDNCCKTCIILWCS | Pu6.20 | 2 | Unknown |
| CIVGTPCHVCRSQSKSCNGWLGKQRYCGYC | Im9.11 | 2 | Unknown |
| CCDRPCSIGCVPCCLP | Ca3-VP01, Cp3-VP05 | 2 | Unknown |
| YWTECCGRIGPHCSRCICPGVVCPKR | Bu25 | 2 | Unknown |
| WFGHEECTYWLGPCEVDDTCCSASCESKFCGLW | RVIIA | 2 | Unknown |
| QCEDVWMPCTSSHWECCSLDCEMYCTQI | Mr6.29 | 2 | Unknown |
| QCPYCVVHCCPPSYCQASGCRPP | Vc7.4 | 2 | Unknown |
| QGCCNVPNGCSGRWCRDHAQCC | MIIIA | 2 | mu |
| TCSSSSDCPTGQECCPDKLDEPEGSCANECIIT | Pu6.37 | 2 | Unknown |
| SCSDDWQYCEYPHDCCSWSCDVVCS | Vc6.12 | 2 | Unknown |
| TCNTPTRYCTLHRHCCSLHCHKTIHACA | Pu6.30 | 2 | Unknown |
| TTSTRKCKGPLVFCPENHECCSKFCDFIDIPLRYCSTP | Br7.9 | 2 | Unknown |
| MTKHCTPPEVGCLFAYECCSKICWRPRCYPS | ABVIE | 2 | Unknown |
| VCCPFGGCHELCLCCD | MrIIIF | 2 | Unknown |
| RCCISPACHDDCICCIT | S3-I05 | 2 | Unknown |
| RCCISPACHEECYCCQ | S3-Y01 | 2 | Unknown |
| VSIWFCASRTCSTPADCNPCTCESGVCVDWL | Lt9a variant 2 | 2 | Unknown |
| QCLPPLSLCTMDDDECCDDCILFLCLVTS | Ar6.5 | 2 | Unknown |
| STDDCSTAGCKNVPCCEGLVCTGPSQGPVCQPLA | Vn6.18 | 2 | Unknown |
| GCCDPQWCDAGCYDGCC | Qc3-YDG01 | 2 | Unknown |
| GCWLCLGPNACCRGSVCHDYCPS | Cal6.4c | 2 | Unknown |
| GCSDFGSDCVPATHNCCSGECFGFEDFGLCT | Pu6.25 | 2 | Unknown |
| STDCNGVPCQFGCCVTINGNDECRELDC | Mr6.23 | 2 | Unknown |
| RCCTWQECDGNCHCCQ | Cp3-H02 | 2 | Unknown |
| RCCVHPACHDDCICCIT | Bt3-I03, Vx3-I03 | 2 | Unknown |
| WWGENDCSWTGPCTVNAECCLGVCDETC | Tx7.31 | 2 | Unknown |
| GCCHPSTCHVRKGCSRCCS | Tx3g, Vt3-SR01 | 2 | Unknown |
| SSDEECVGLSGYCGPWNNPPCCSWWECEVYCAVPGPSF | Mi034 | 2 | Unknown |
| SCCNAGFCRFGCTPCCY | Tx3e, Vt3-TP01, Ec3-TP01-2 | 2 | Unknown |
| TCDPYYCNDGKVCCPEYPTCGDSTGKLICVRVTD | Im6.7 | 1 | Unknown |
| TCLEIGEFCGKPMMVGSLCCSPGWCFFICVG | Pc6b | 1 | Unknown |
| CGGYSTYCEVDSECCSDNCVRSYCTLF | TxVIIA | 1 | gamma |
| GCCCNPACGPNYGCGTSCSRPSEP | S1.7 | 1 | Unknown |
| TRGCKSKGSFCWNGIECCGGNCFFACVY | Cl6.6b | 1 | Unknown |
| CFESWVACESPKRCCSHVCLFVCT | Pn6.6 | 1 | Unknown |
| WREGSCTSWLATCTDASQCCTGVCYKRAYCALWE | TxMEKL-022/TxMEKL-021 | 1 | Unknown |
| YCSDSGGWCGLDPELCCNSSCFVLC | Cl6.8 | 1 | Unknown |
| YCSDDWQPCSHFYDCCKWSCNNGYCP | Vc6.25 | 1 | Unknown |
| CCDDSECSYSCWPCCY | TxMMSK-02, Cp3-WP03, Vr3-WP04, S3-WP01, Rt3-WP01 | 1 | Unknown |
| WRVDSECISFWGSCTVDADCCFNSCDETYGYC | Tx7.30 | 1 | Unknown |
| CCDWPCTIGCVPCCLP | TsMMSK-021 | 1 | Unknown |
| CCFWPMCRGCDCCYL | Lv3-D02 | 1 | Unknown |
| CCGPTACLAGCKPCCY | Tx3-KP03 | 1 | Unknown |
| CESYGKPCGIYNDCCNACDPAKKTCT | Conotoxin-3 | 1 | Unknown |
| VQPSECKLPAAKGPCKGKYRKVYFNNFKKQCRM– | |||
| FTYGGCGGNGNKFRNAKECYHKCAYGV | conkunitzin-G1 | 1 | Unknown |
| VCCSFGSCDSLCQCCD | Mr3.16 | 1 | Unknown |
| CCLWPECGGCVCCYL | Lv3-V02 | 1 | Unknown |
| TRGCKTKGTWCWASRECCLKDCLFVCVY | Cl6.10 | 1 | Unknown |
| CCSVSICQSPPVCECCA | S3-E03 | 1 | Unknown |
| CCVVCNAGCSGNCCS | Ts3-SGN01 | 1 | Unknown |
| SCSGSGYGCKNTPCCAGLTCRGPRQGPICL | Vn6.16 | 1 | Unknown |
| RCCIWPECGSCVCCL | Cp3-V08 | 1 | Unknown |
| SCGNLHEMCNYHLPCCRPWRCRASRTGTR– | |||
| CLNKPRYRPV | Pu6.13 | 1 | Unknown |
| RDCRPVGQYCGIPYEHNWRCCSQLCAIICVS | PuIA | 1 | omega |
| GCCGSFACRFGCVPCCV | MrIIIA | 1 | Unknown |
| GCCHLLACRMGCTPCCW | Tx3-TP01 | 1 | Unknown |
| GCCIEPLCYQYDCDCCRYL | Cp3-D03 | 1 | Unknown |
| ECSSPDESCTYHYNCCQLYCNKEENVCLENSPEV | LtVIB | 1 | Unknown |
| ECRGYNAPCSAGAPCCSWWTCSTQTSRCF | Vc6.10 | 1 | Unknown |
| GCCPIGPCMQSVCSPCCP | Vr3-SP01 | 1 | Unknown |
| GMWGKCKDGLTTCLAPSECCSGNCEQNCKMW | TxMEKL-011, LeD51 | 1 | Unknown |
| GVWSECSDWLAGCSSPSECCSEKCDTFCRLW | G6.8 | 1 | Unknown |
| GWDTPAPCRYCQWNGPQCCVYYCSSCNYEEARE– | |||
| EGHYVSSHLLERQ | Cal6.3a | 1 | Unknown |
| DECCEPQWCDGACDCCS | LtIIIA | 1 | iota |
| KFILHALGQWQCCTMQWCDKACYCCE | Vc3.4 | 1 | Unknown |
| DDCTTYCYGVHCCPPAFKCAASPSCKQT | Cal6.5a | 1 | Unknown |
| KTCQRRWDFCPGSLVGVITCCGGLICFLFFCV | Om6.6 | 1 | Unknown |
| LCPDYTEPCSHAHECCSWNCYNGHCTG | Gla(3)-TxVI | 1 | Unknown |
| MQGKISSEQHPMFDPIEGCCTQSCTTCFPCCLI | Lt3.6 | 1 | Unknown |
| DCCSMSACVPPPACECC | Mi3-E04 | 1 | Unknown |
| DCCPLPACPFGCNPCCGWPALLSGPHQVMNNE | Mr020 | 1 | Unknown |
| DCCGVKLEMCHPCLCDNSCKNYGK | PIVE | 1 | kappa |
| DAMQKSKGSGSCAYISEPCDILPCCPGLKCNEDFVPICL | LtVIA | 1 | Unknown |
| NPKLSKLTKTCDPPGDSCSRWYNHCCSKLCTSR– | |||
| NSGPTCSRP | LiCr95 | 1 | Unknown |
| QCADLGEECYTRFCCPGLRCKDLQVPTCLLA | Ar6.10 | 1 | Unknown |
| QCCDSNSCEYPKCLCCN | Tx3-L02, Vr3-L01, Vt3-L01, S3-L02 | 1 | Unknown |
| CVEDGDFCGPGYEECCSGFCLYVCI | Pu6.2 | 1 | Unknown |
| QKCCGKGMTCPRYFRDNFICGCC | CnIIIG | 1 | Unknown |
| QQCCPPVACNMGCEPCC | TxMMSK-04, Vt3-EP01 | 1 | Unknown |
| RCCGEGASCPVYSRDRLICSCC | CnIIIE | 1 | Unknown |
| RCCISPACNDTCYCCQD | Vr3-Y02, Vt3-Y01, Ts3-Y01 | 1 | Unknown |
| CPNTGELCDVVEQNCCYTYCFIVVCPI | Mr6.2 | 1 | Unknown |
| RCCTGKKGSCSGRACKNLKCCA | SxIIIA | 1 | mu |
| APWTVVTATTNCCGITGPGCLPCRCTQTC | A4.4 | 1 | Unknown |
| Sequence | Name(s) | Degree | Pharm. Fam. |
|---|---|---|---|
| TDVCKKSPGKCIHNGCFCEQDKPQGNCCDSGGC– | |||
| TVKWWCPGTKGD | Cal12.1p2 | 28 | Unknown |
| GHVPCGKDGRKCGYHADCCNCCLSGICKPSTSW– | |||
| TGCSTSTVQLTR | R11.10 | 18 | Unknown |
| QCTPKNQICEEDGECCPNLECKCFTRPDCQSGYKCRP | Vr15b | 14 | Unknown |
| CFPPGVYCTRHLPCCRGRCCSGWCRPRCFPRY | Cp1.1 | 10 | Unknown |
| QCTQQGYGCDETEECCSNLSCKCSGSPLCTSSYCRP | Cap15a | 9 | Unknown |
| SCDSEFSSEFCEQPEERICSCSTHVCCHLSSSK– | |||
| RDQCMTWNRCLSAQTGN | Gla-MrII, Eu12.4 | 9 | Unknown |
| SRCFPPGIYCTPYLPCCWGICCGTCRNVCHLRF | Em11.8 | 8 | Unknown |
| DKWGTCSLLGKGCRHHSDCCWDLCCTGKTCVMT– | |||
| VLPCLFLSLIVRWT | Mr11.1 | 6 | Unknown |
| TCSLPGDGCIRDFHCCGHMCCQGNKCVVTVRRCFNFPY | Pu11.5 | 6 | Unknown |
| YDAPYCSQEEVRECQDDCSGNAVRDSCLCAYDPAGSP– | |||
| ACECRCVEPW | Cal22d | 5 | Unknown |
| GTCSGRGQECKHDSDCCGHLCCAGITCQFTYIPCK | Tx11.3 | 5 | Unknown |
| GTCSYLGEGCKRDSDCCGHFCCGGKTCVITARPCKV | Vc11.4 | 5 | Unknown |
| RGVCSTPEGSCVHNGCICQNAPCCHPSGCNWANVCPG– | |||
| YLWDKN | Cal12.2c | 4 | Unknown |
| TCSDLGQACVHESDCCAQMCCLNKKCAMTMPPCNFY | Vc11.1 | 3 | Unknown |
| CLSEGSPCSMSGSCCHKSCCRSTCTFPCLIP | Ep11.12 | 2 | Unknown |
| TCSNKGQQCGDDSDCCWHLCCVNNKCAHLILLCNL | M11.2 | 2 | Unknown |
| RCSDDTGATCSNRFDCCESMCCIGGHCVISTVGCP | Im11.14 | 1 | Unknown |
| CRLEGSSCRRSYQCCHKSCCIRECKFPCRWV | Vi11.5 | 1 | Unknown |
| TRSFADLPDDWGMCSDIGEGCGQDYDCCGDMCCDGQI– | |||
| CAMTFMACMF | Vc11.6 | 1 | Unknown |
| CLRDGQSCGYDSDCCRYSCCWGYCDLTCLIN | Im11.1 | 1 | Unknown |
| CNGRGEWCSTHRSCCDSGDVCCITTPVGPICTRGCSG– | |||
| RIIPQRRGAQLRHFF | Pu11.9 | 1 | Unknown |
| CRAEGTYCENDSQCCLNECCWGGCGHPCRHP | BtX, Sx11.2 | 1 | kappa |
| CTSEGYSCSSDSNCCKNVCCWNVCESHCRHPGKR | Lt11.3 | 1 | Unknown |
| CRSGKTCPRVGPDVCCERSDCFCKLVPARPFWRYRCICL | Mr15.2 | 1 | Unknown |
| DCPTSCPTTCANGWECCKGYPCVRQHCSGCNH | De13b | 1 | Unknown |
| EGGYVREDCGSDCMPCGGECCCEPNSCIDGTCHHESSPN | Mi045 | 1 | Unknown |
| SCRNEGAMCSFGFQCCKKKCCMSHCTDFCRNP | Vt11.3 | 1 | Unknown |
| WPRLYDSDCVRGRNMHITCFKDQTCGLTVKRNGRLNC– | |||
| SLTCSCRRGESCLHGEYIDWDSRGLKVHICPKPWF | Mr22.1 | 1 | Unknown |
| MCLSLGQRCGRHSNCCGYLCCFYDKCVVTAIGCGHY | Bt11.4 | 1 | Unknown |
| ASICYGTGGRCTKDKHCCGWLCCGGPSVGCVVSVAPC | Ca11.3 | 1 | Unknown |
| Sequence | Name(s) | Degree | Pharm. Fam. |
|---|---|---|---|
| DRDVQDCQVSTPGSKWGRCCLNRVCGPMCCPAS– | |||
| HCYCVYHRGRGHGCSC | Cp20.1 | 19 | Unknown |
| LHCYEISDLTPWILCSPEPLCGGKGCCAQEVCD– | |||
| CSGPACTCPPCL | Lt15.6 | 5 | Unknown |
| YNRQCCIDKTYDCLKKYRGRENTFASVCQQEAA– | |||
| VYCGAWDEAEGCCYGYSHCMSMYAQQSGLDVA– | |||
| HNGCKDRKCDNP | Vc21.1 | 2 | Unknown |
| QCTLVNNCDRNGERACNGDCSCEGQICKCGYRV– | |||
| SPGKSGCACTCRNA | Ac8.1 | 2 | Unknown |
| GCSGTCRRHRDGKCRGTCECSGYSYCRCGDAHH– | |||
| FYRGCTCTC | Ca8c | 2 | Unknown |
| TCDPTPDCRTTVCETDTGPCCCPHGYNCQTTNS– | |||
| GRRACVLVCPHNCPP | Pu19.1 | 1 | Unknown |
| SGSTCTCFTSTNCQGSCECLSPPGCYCSNNGIR– | |||
| QRGCSCTCPGT | G8.3 | 1 | Unknown |
| GCTRTCGGPKCTGTCTCTNSSKCGCRYNVHPSG– | |||
| WGCGCACS | GVIIIA | 1 | sigma |
| GCTISCGYEDNRCQGECHCPGKTNCYCTSGHHN– | |||
| KGCGCAC | Tx8.1 | 1 | Unknown |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansbach, R.A.; Chakraborty, S.; Travers, T.; Gnanakaran, S. Graph-Directed Approach for Downselecting Toxins for Experimental Structure Determination. Mar. Drugs 2020, 18, 256. https://doi.org/10.3390/md18050256
Mansbach RA, Chakraborty S, Travers T, Gnanakaran S. Graph-Directed Approach for Downselecting Toxins for Experimental Structure Determination. Marine Drugs. 2020; 18(5):256. https://doi.org/10.3390/md18050256
Chicago/Turabian StyleMansbach, Rachael A., Srirupa Chakraborty, Timothy Travers, and S. Gnanakaran. 2020. "Graph-Directed Approach for Downselecting Toxins for Experimental Structure Determination" Marine Drugs 18, no. 5: 256. https://doi.org/10.3390/md18050256
APA StyleMansbach, R. A., Chakraborty, S., Travers, T., & Gnanakaran, S. (2020). Graph-Directed Approach for Downselecting Toxins for Experimental Structure Determination. Marine Drugs, 18(5), 256. https://doi.org/10.3390/md18050256