Astaxanthin Modulates Apoptotic Molecules to Induce Death of SKBR3 Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
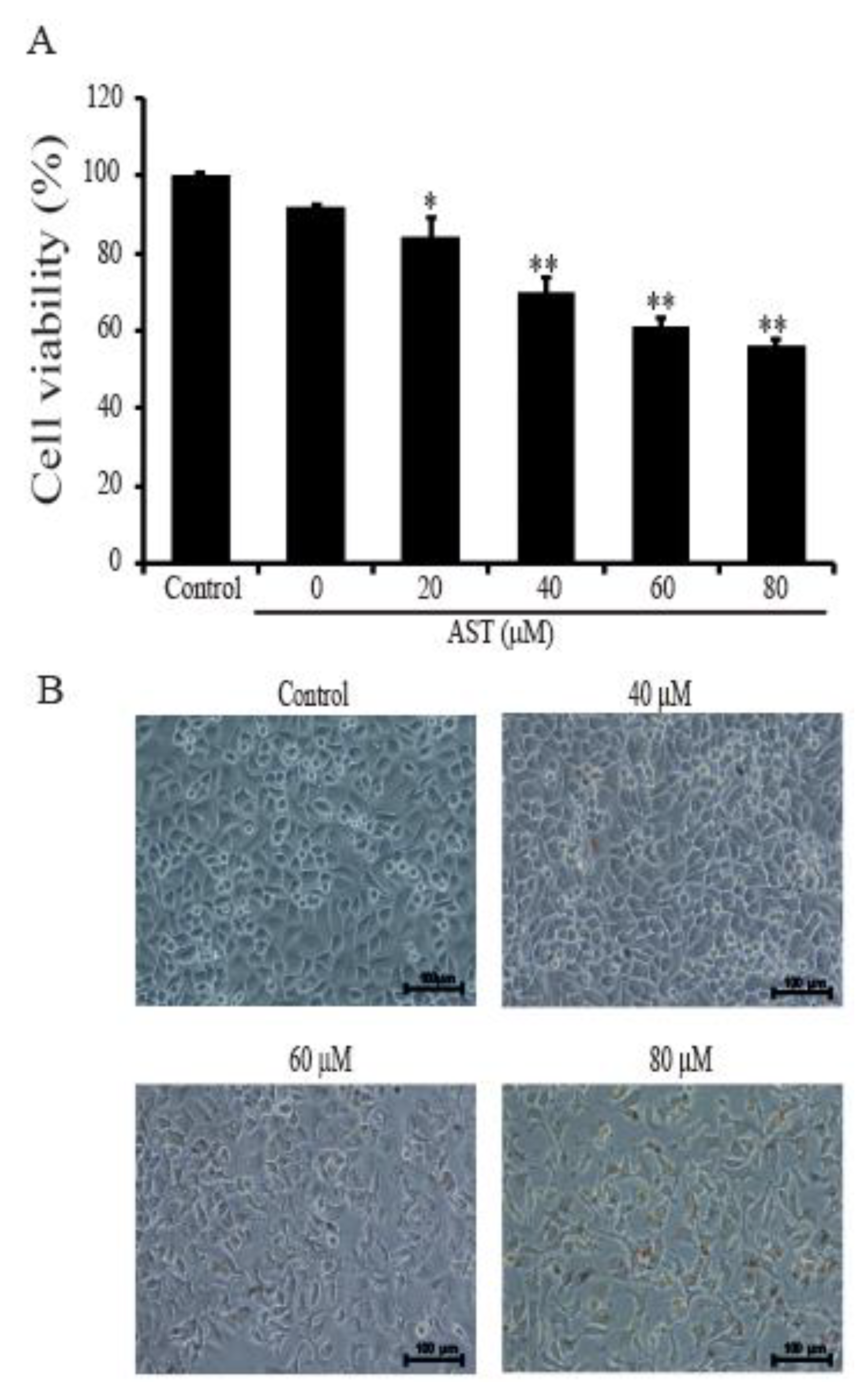
2.1. AST Inhibited Cell Proliferation and Changed Cellular Morphology of SKBR3 Cells
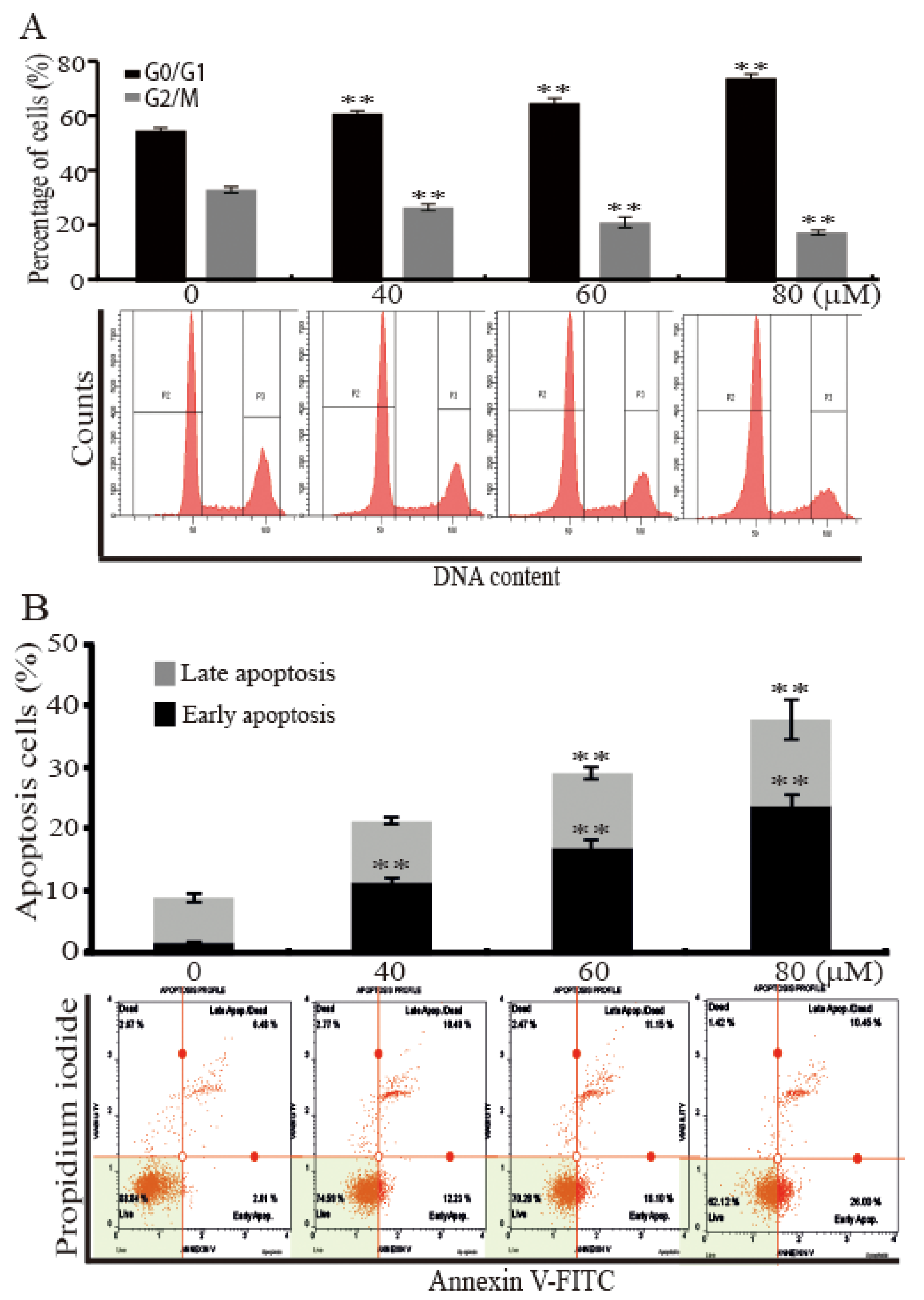
2.2. AST Induced Cell Cycle Arrest and Apoptosis of the SKBR3 Cells
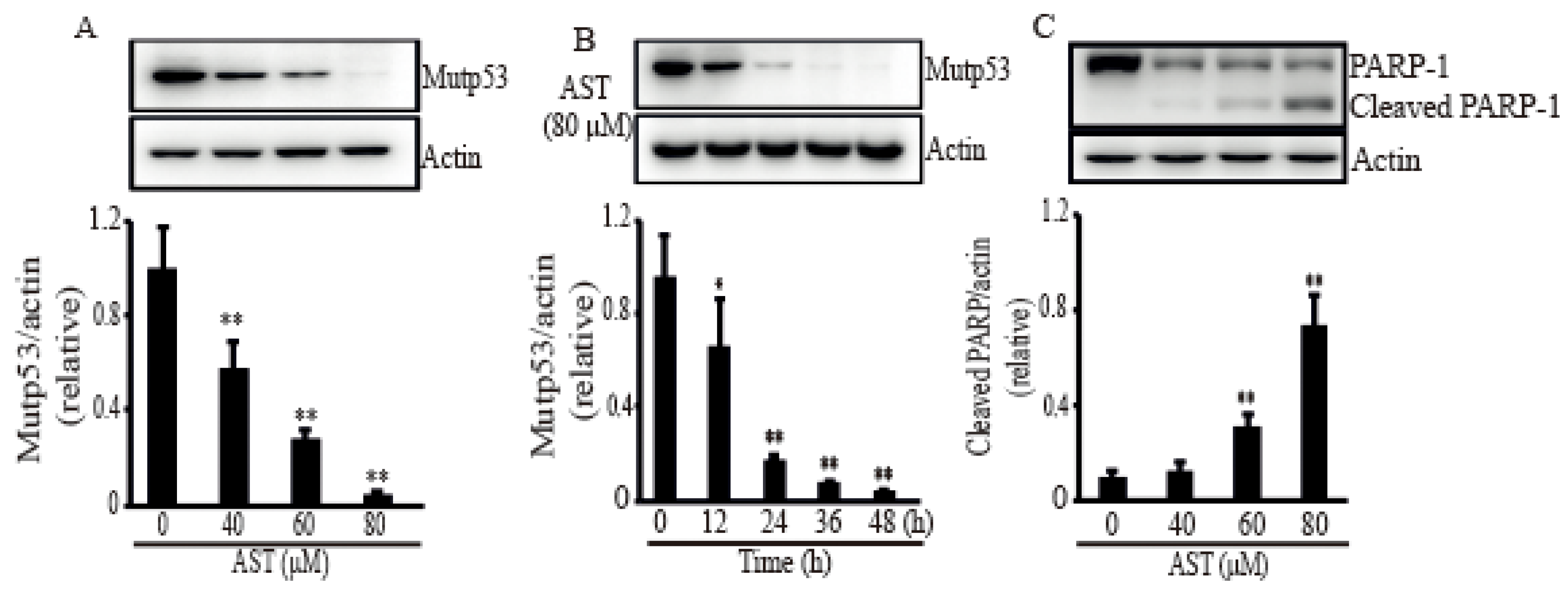
2.3. AST Reduced the Level of Mutp53 Expression and Generated a PARP-1 Fragment in the SKBR3 Cells
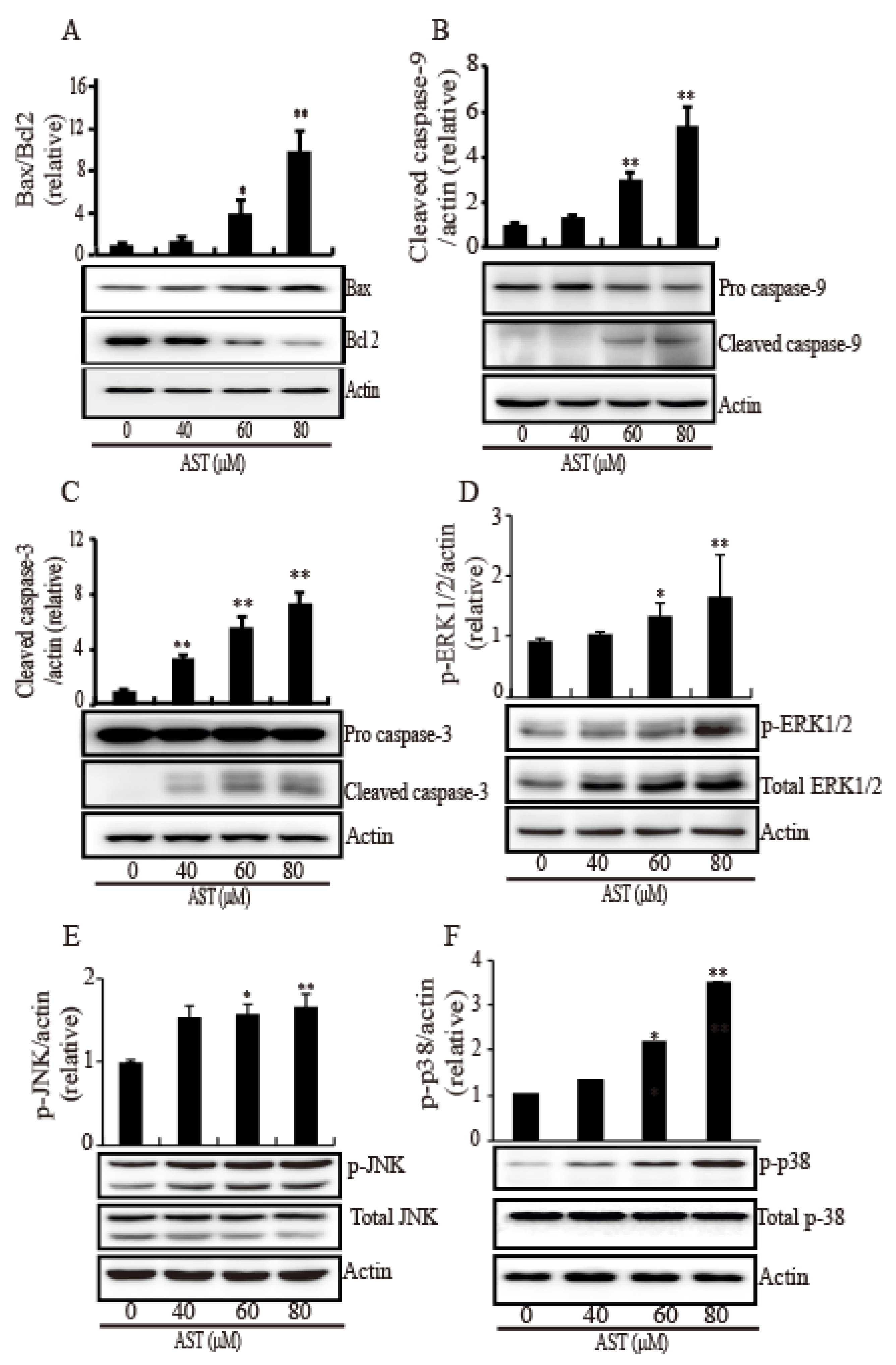
2.4. AST Induced Intrinsic Apoptosis Through Activation of the MAPKs in the SKBR3 Cells
2.5. AST Decreased Intracellular ROS Level and Modulated SOD1 and SOD2 Expressions in the SKBR3 Cells
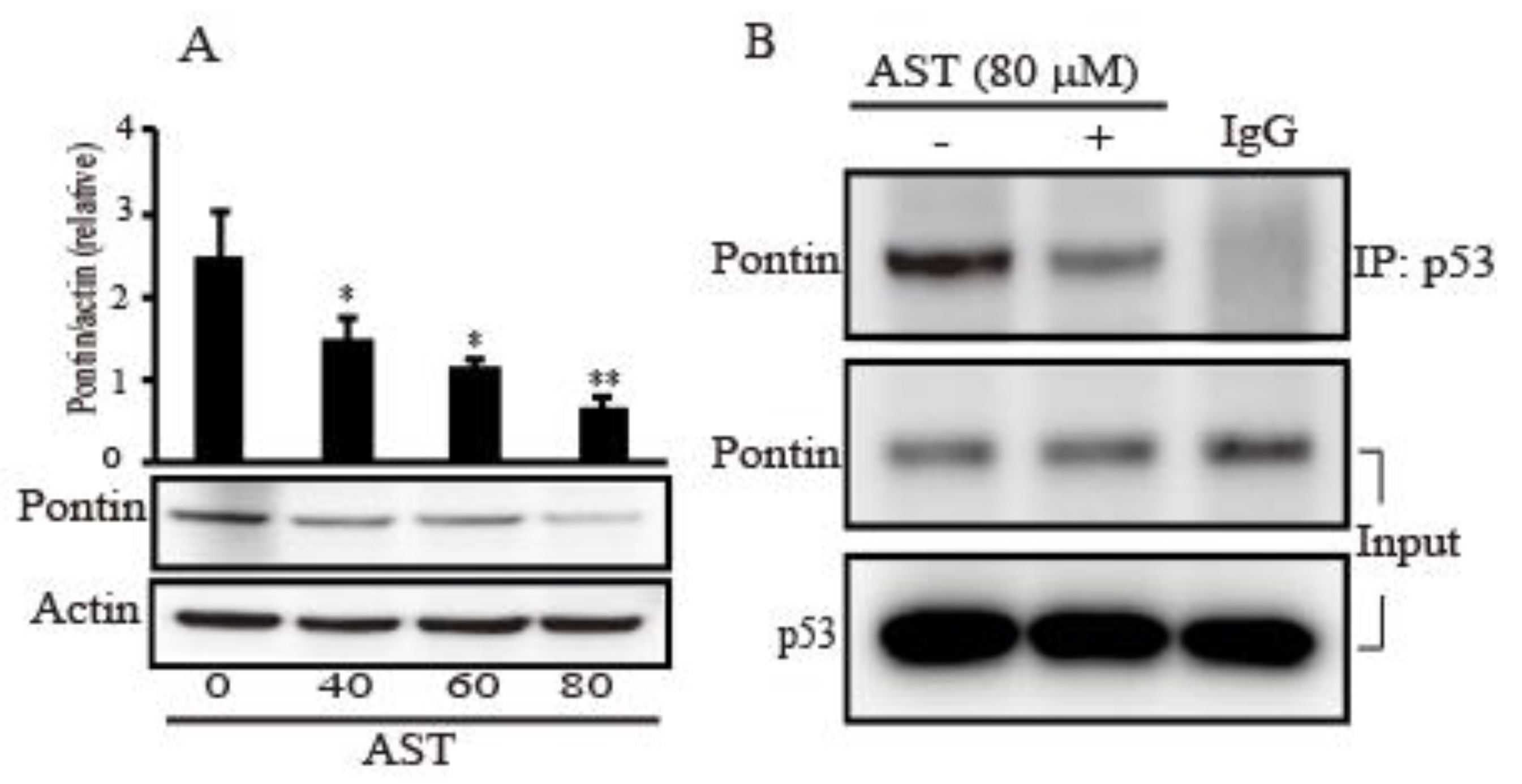
2.6. AST Decreased the Expression Level of Pontin and Reduced Association between Mutp53 and Pontin in the SKBR3 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Lines and Cell Culture
4.3. Cell Viability/Proliferation Assay
4.4. Cell Cycle Quantification and Apoptosis Assays
4.5. Cell Lysis and Western Blot Analysis
4.6. Co-Immunoprecipitation
4.7. ROS Determination
4.8. Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA. Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef] [PubMed]
- Sparreboom, A.; Scripture, C.D.; Trieu, V.; Williams, P.J.; De, T.; Yang, A.; Beals, B.; Figg, W.D.; Hawkins, M.; Desai, N. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin. Cancer Res. 2005, 11, 4136–4143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb Perspect Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 10, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Robbins, D.; Zhao, Y. Manganese superoxide dismutase in cancer prevention. Antioxid. Redox Signal. 2014, 20, 1628–1645. [Google Scholar] [CrossRef] [Green Version]
- Somwar, R.; Erdjument-Bromage, H.; Larsson, E.; Shum, D.; Lockwood, W.W.; Yang, G.; Sander, C.; Ouerfelli, O.; Tempst, P.J.; Djaballah, H.; et al. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 16375–16380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, M.; Flores, E.R. The p53 family orchestrates the regulation of metabolism: Physiological regulation and implications for cancer therapy. Br. J. Cancer 2017, 116, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Iwakuma, T. Regulators of oncogenic mutant TP53 gain of function. Cancers 2019, 11, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, M.; Francis, P.; Bilke, S.; Li, X.L.; Hara, T.; Lu, X.; Jones, M.F.; Walker, R.L.; Zhu, Y.; Pineda, M.; et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene 2015, 34, 1094–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, C.; Yue, X.; Li, X.; Liu, J.; Yu, H.; Belyi, V.A.; Yang, Q.; Feng, Z.; Hu, W. Pontin, a new mutant p53-binding protein, promotes gain-of-function of mutant p53. Cell Death Differ. 2015, 22, 1824–1836. [Google Scholar] [CrossRef] [Green Version]
- Lauscher, J.C.; Elezkurtaj, S.; Dullat, S.; Lipka, S.; Gröne, J.; Buhr, H.J.; Huber, O.; Kruschewski, M. Increased Pontin expression is a potential predictor for outcome in sporadic colorectal carcinoma. Oncol. Rep. 2012, 28, 1619–1624. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Mao, Y.; Brandt-Rauf, P.W.; Williams, A.C.; Fine, R.L. Selective induction of apoptosis in mutant p53 premalignant and malignant cancer cells by PRIMA-1 through the c-Jun-NH2-kinase pathway. Mol. Cancer Ther. 2005, 4, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Fassett, R.G.; Coombes, J.S. Astaxanthin: A potential therapeutic agent in cardiovascular disease. Mar. Drugs 2011, 9, 447–465. [Google Scholar] [CrossRef] [Green Version]
- Jyonouchi, H.; Sun, S.; Iijima, K.; Gross, M.D. Antitumor activity of astaxanthin and its mode of action. Nutr. Cancer 2000, 36, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Kowshik, J.; Baba, A.B.; Giri, H. Astaxanthin Inhibits JAK/STAT-3 Signaling to Abrogate Cell Proliferation, Invasion and Angiogenesis in a Hamster Model of Oral Cancer. PLoS ONE 2014, 9, e109114. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, K.; Kowshik, J.; Kishore, T.K.; Baba, A.B.; Nagini, S. Astaxanthin inhibits NF-κB and Wnt/β-catenin signaling pathways via inactivation of Erk/MAPK and PI3 K/Akt to induce intrinsic apoptosis in a hamster model of oral cancer. Biochem. Biophys. Acta 2013, 1830, 4433–4444. [Google Scholar] [CrossRef] [PubMed]
- Nagendraprabhu, P.; Sudhandiran, G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expressions of ERK-2, NFkB and COX-2. Invest. New Drugs 2011, 29, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Maraldi, T.; Prata, C.; Fiorentini, D.; Zambonin, L.; Landi, L.; Hakim, G. Induction of apoptosis in a human leukemic cell line via reactive oxygen species modulation by antioxidants. Free Radic. Biol. Med. 2009, 46, 244–252. [Google Scholar] [CrossRef]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H. Multiple Mechanisms of Anti-Cancer Effects Exerted by Astaxanthin. Mar. Drugs 2015, 13, 4310–4330. [Google Scholar] [CrossRef] [Green Version]
- McCall, B.; McPartland, C.K.; Moore, R.; Frank-Kamenetskii, A.; Booth, B.W. Effects of Astaxanthin on the Proliferation and Migration of Breast Cancer Cells In Vitro. Antioxidants (Basel) 2018, 7, 135. [Google Scholar] [CrossRef] [Green Version]
- Sowmya, P.R.; Arathi, B.P.; Vijay, K.; Baskaran, V.; Lakshminarayana, R. Astaxanthin from shrimp efficiently modulates oxidative stress and allied cell death progression in MCF-7 cells treated synergistically with β-carotene and lutein from greens. Food Chem. Toxicol. 2017, 106, 58–69. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, J.J.; Lee, B.J.; Joo, M.K.; Chun, H.J.; Lee, S.W.; Bak, Y.T. Astaxanthin inhibits proliferation of human gastric cancer cell lines by interrupting cell cycle progression. Gut Liver 2016, 10, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Palozza, P.; Torelli, C.; Boninsegna, A.; Simone, R.; Catalano, A.; Mele, M.C.; Picci, N. Growth-inhibitory effects of the astaxanthin-rich alga Haematococcus pluvialis in human colon cancer cells. Cancer Lett. 2009, 283, 108–117. [Google Scholar] [CrossRef]
- Kuno, T.; Tsukamoto, T.; Hara, A.; Tanaka, T. Cancer chemoprevention through the induction of apoptosis by natural compounds. J. Biophys. Chem. 2012, 3, 156–173. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Rennie, P.S.; Dragowska, V.; Nelson, C.C.; Jia, W. A mutant P53 can activate apoptosis through a mechanism distinct from those induced by wild type P53. FEBS Lett. 2002, 517, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.Y.; Vidnovic, N.; Ellisen, L.W.; Leong, C.O. Mutant p53 mediates survival of breast cancer cells. Br. J. Cancer 2009, 101, 1606–1612. [Google Scholar] [CrossRef]
- Haiming, D.; Meng, X.W.; Kaufmann, S.H. BCL2 Family, mitochondrial apoptosis, and beyond. Cancer Transl. Med. 2016, 2, 7–20. [Google Scholar]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef] [Green Version]
- Dantzer, F.; Amé, J.C.; Schreiber, V.; Nakamura, J.; Ménissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 2006, 409, 493–510. [Google Scholar]
- Hormozi, M.; Ghoreishi, S.; Baharvand, P. Astaxanthin induces apoptosis and increases activity of antioxidant enzymes in LS-180 cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 891–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, D.N.; Johnson, G.L. MAPKs: Function, regulation, role in cancer and therapeutic targeting. Oncogene 2017, 26, 3097–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Song, M.; Gao, Z.; Cai, X.; Dixon, W.; Chen, X.; Cao, Y.; Xiao, H. Stereoisomers of Astaxanthin Inhibit Human Colon Cancer Cell Growth by Inducing G2/M Cell Cycle Arrest and Apoptosis. J. Agric. Food Chem. 2016, 64, 7750–7759. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bdana, A.K.; Malla, R. Reactive Oxygen Species: A key constituent in cancer survival. Biomark. Insights 2018, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer. A double-edged sword with therapeutic potential. Oxid. Med. Cell Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Papa, L.; Manfredi, G.; Germain, D. SOD1, an unexpected novel target for cancer therapy. Genes Cancer 2014, 5, 15–21. [Google Scholar]
- Papa, L.; Hahn, M.; Marsh, E.L.; Evans, B.S.; Germain, D. SOD2 to SOD1 switch in breast cancer. J. Biol. Chem. 2014, 289, 5412–5416. [Google Scholar] [CrossRef] [Green Version]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.Z.; Ni, X.F.; Yu, H.N.; Wang, S.S.; Shen, S.R. Effects of astaxanthin on oxidative stress induced by Cu2+ in prostate cells. J. Zhejiang Univ. Sci. B 2017, 18, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Matias, P.M.; Baek, S.H.; Bandeiras, T.M.; Dutta, A.; Houry, W.A.; Llorca, O.; Rosenbaum, J. The AAA+ proteins Pontin and Reptin enter adult age: From understanding their basic biology to the identification of selective inhibitors. Front Mol. Biosci. 2015, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugan, K.A.; Wood, M.A.; Cole, M.D. TIP49, but not TRRAP, modulates c-Myc and E2F1 dependent apoptosis. Oncogene 2002, 21, 5835–5843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Huang, X.; Wu, C.; Xue, L. Pontin/Tip49 negatively regulates JNK-mediated cell death in Drosophila. Cell Death Discov. 2018, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Ménard, L.; Taras, D.; Grigoletto, A.; Haurie, V.; Nicou, A.; Dugot-Senant, N.; Costet, P.; Rousseau, B.; Rosenbaum, J. In vivo silencing of Reptin blocks the progression of human hepatocellular carcinoma in xenografts and is associated with replicative senescence. J. Hepatol. 2010, 52, 681–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.S.; Ahn, Y.T.; Lee, C.W.; Kim, H.; An, W.G. Astaxanthin Modulates Apoptotic Molecules to Induce Death of SKBR3 Breast Cancer Cells. Mar. Drugs 2020, 18, 266. https://doi.org/10.3390/md18050266
Kim MS, Ahn YT, Lee CW, Kim H, An WG. Astaxanthin Modulates Apoptotic Molecules to Induce Death of SKBR3 Breast Cancer Cells. Marine Drugs. 2020; 18(5):266. https://doi.org/10.3390/md18050266
Chicago/Turabian StyleKim, Min Sung, Yong Tae Ahn, Chul Won Lee, Hyungwoo Kim, and Won Gun An. 2020. "Astaxanthin Modulates Apoptotic Molecules to Induce Death of SKBR3 Breast Cancer Cells" Marine Drugs 18, no. 5: 266. https://doi.org/10.3390/md18050266
APA StyleKim, M. S., Ahn, Y. T., Lee, C. W., Kim, H., & An, W. G. (2020). Astaxanthin Modulates Apoptotic Molecules to Induce Death of SKBR3 Breast Cancer Cells. Marine Drugs, 18(5), 266. https://doi.org/10.3390/md18050266