Mussel Shell-Derived Macroporous 3D Scaffold: Characterization and Optimization Study of a Bioceramic from the Circular Economy

 ,
,  , ,
, , 
 ,
,  and
and
Abstract
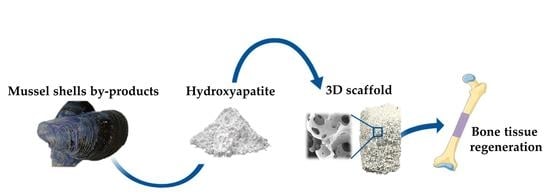
:
1. Introduction
2. Results and Discussion
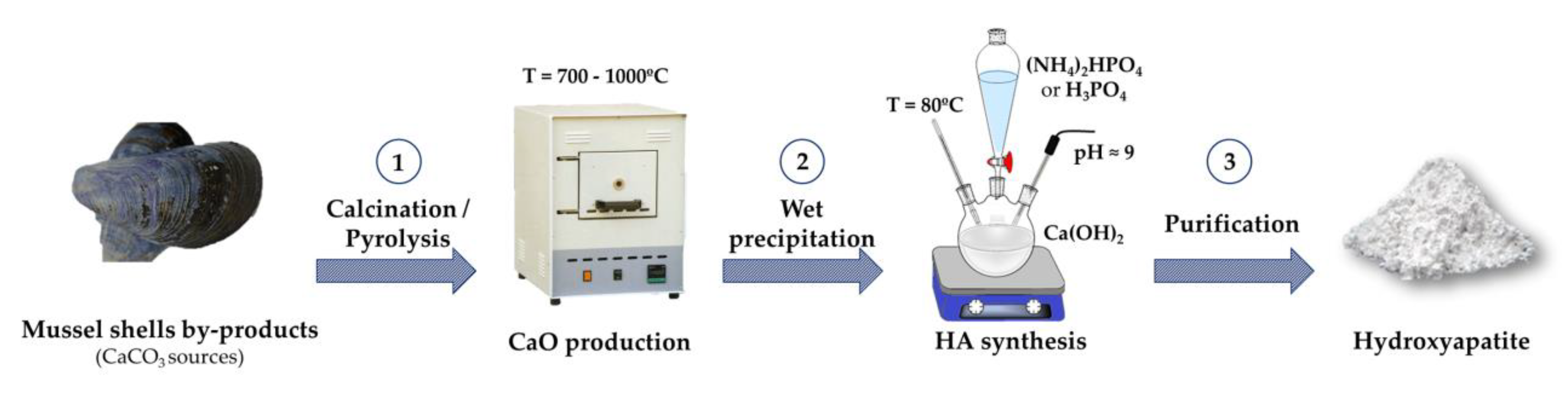
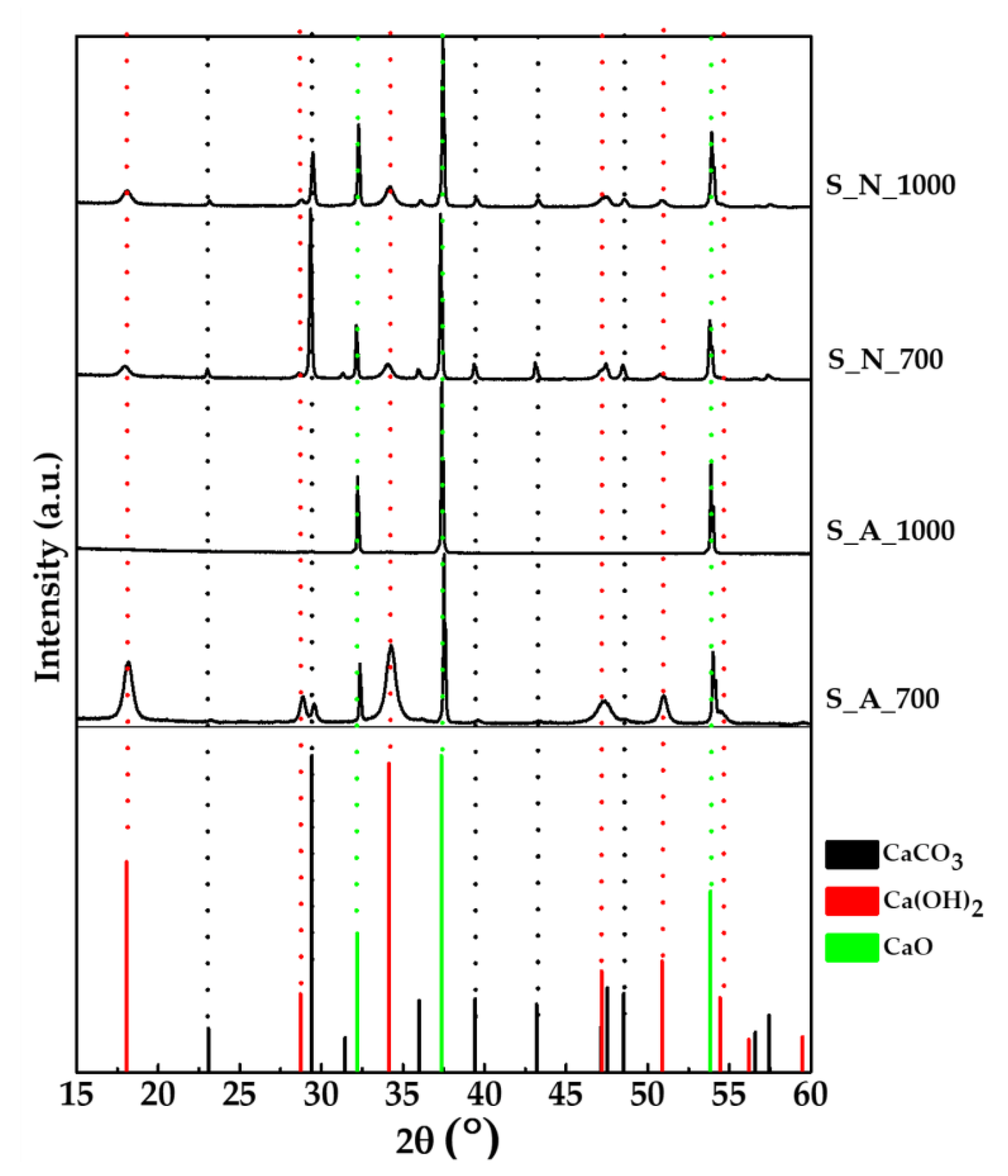
2.1. Preparation and Characterization of CaO Samples from Mussel Shells
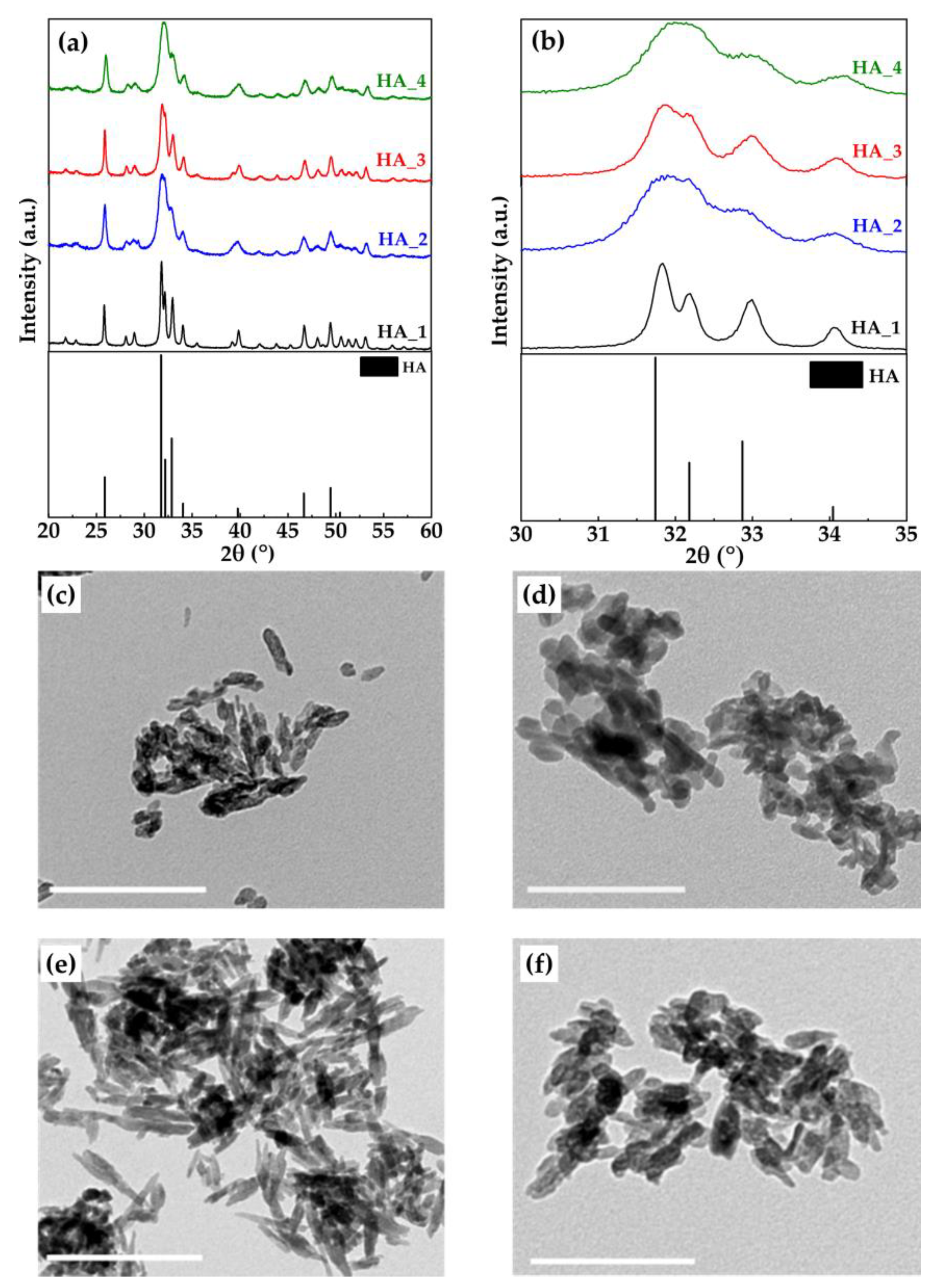
2.2. Preparation and Characterization of HA Samples
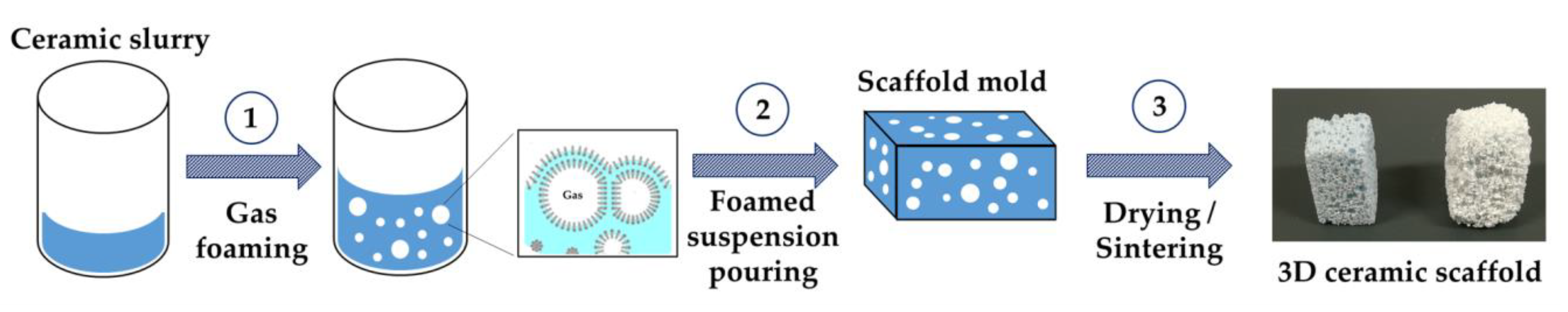
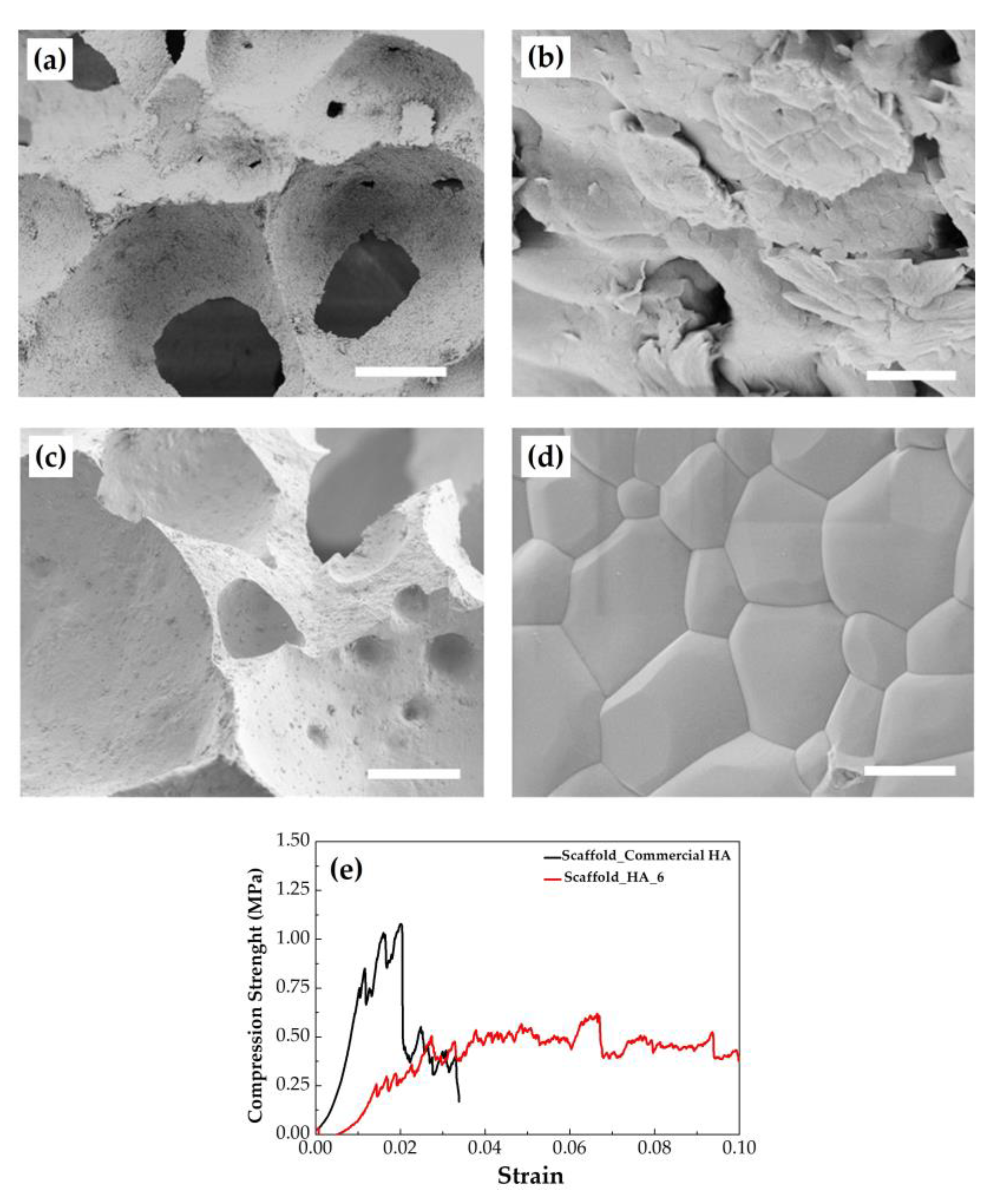
2.3. Preparation and Characterization of the 3D Scaffolds
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis of CaO Samples
3.3. Synthesis of Hydroxyapatite Samples
Scale-Up Synthesis of Hydroxyapatite Sample
3.4. Preparation of the 3D Scaffolds
3.5. Characterizations of the Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sammadar, P.; Ok, Y.S.; Kim, K.H.; Kwon, E.E.; Tsang, D.C.W. Synthesis of nanomaterials from various wastes and their new age applications. J. Clean. Prod. 2018, 197, 1190–1209. [Google Scholar] [CrossRef]
- Nayak, A.; Bhushan, B. An overview of the recent trends on the waste valorization techniques for food wastes. J. Environ. Manag. 2019, 233, 352–370. [Google Scholar] [CrossRef]
- Salinas, A.J.; Esbrit, P.; Vallet-Regí, M. A tissue engineering approach based on the use of bioceramics for bone repair. Biomater. Sci. 2012, 1, 40–51. [Google Scholar] [CrossRef]
- Rezwan, K.; Chen, Q.Z.; Blaker, J.J.; Boccaccini, A.R. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 3413–3431. [Google Scholar] [CrossRef]
- Baino, F.; Novajra, G.; Vitale-Brovarone, C. Bioceramics and Scaffolds: A Winning Combination for Tissue Engineering. Front. Bioeng. Biotechnol. 2015, 3, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hench, L.L. Bioceramics. J. Am. Ceram. Soc. 2005, 81, 1705–1728. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, M.; Contessi Negrini, N.; Scialla, S.; Marelli, B.; Farè, S.; Danti, S.; Buehler, M.J. Additive Manufacturing Approaches for Hydroxyapatite-Reinforced Composites. Adv. Funct. Mater. 2019, 29, 1903055. [Google Scholar] [CrossRef] [Green Version]
- Novotna, L.; Kucera, L.; Hampl, A.; Drdlik, D.; Cihlar, J.; Cihlar, J. Biphasic calcium phosphate scaffolds with controlled pore size distribution prepared by in-situ foaming. Mater. Sci. Eng. C 2019, 9, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, M.; Deng, Y.; Chen, X.; Xiao, Y.; Zhang, X. Fabrication and properties of Ca-P bioceramic spherical granules with interconnected porous structure. ACS Biomater. Sci. Eng. 2017, 3, 1557–1566. [Google Scholar] [CrossRef]
- Scalera, F.; Gervaso, F.; Sanosh, K.P.; Sannino, A.; Licciulli, A. Influence of the calcination temperature on morphological and mechanical properties of highly porous hydroxyapatite scaffolds. Ceram. Int. 2013, 39, 4839–4846. [Google Scholar] [CrossRef]
- Colombro, P.; Vakifahmetoglu, C.; Costacurta, S. Fabrication of ceramic components with hierarchical porosity. J. Mater. Sci. 2010, 45, 5425–5455. [Google Scholar] [CrossRef]
- Ma, H.; Feng, C.; Chang, J.; Wu, C. 3D-printed bioceramic scaffolds: From bone tissue engineering to tumour therapy. Acta Biomater. 2018, 79, 37–59. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shao, H.; Lin, T. Effect of magnesium silicate on 3D gel-printing of hydroxyapatite ceramic composite scaffold. Int. J. Appl. Ceram. Technol. 2019, 16, 494–502. [Google Scholar] [CrossRef]
- Babaie, E.; Bhaduri, S.B. Fabrication aspects of porous biomaterials in orthopedic applications: A review. ACS Biomater. Sci. Eng. 2018, 4, 1–39. [Google Scholar] [CrossRef]
- Studart, A.R.; Gozenbach, U.T.; Tervoort, E.; Gauckler, L.J. Processing routes to macroporous ceramics: A review. J. Am. Ceram. Soc. 2006, 89, 1771–1789. [Google Scholar] [CrossRef]
- Gόmez-Morales, J.; Iafisco, M.; Delgado-Lόpez, J.M.; Sarda, S.; Drouet, C. Progress on the preparation of nanocrystalline apatites and surface characterization: Overview of fundamental and applied aspects. Prog. Cryst. Growth Charact. Mater. 2013, 59, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Salcedo, S.; Vila, M.; Diaz, A.; Acosta, C.; Barton, I.; Escobar, A.; Vallet-Regí, M. Synthesis of HA/β-TCP bioceramic foams from natural products. J. Sol Gel Sci. Technol. 2016, 79, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Piccirillo, C.; Pintado, M.M.; Castro, P.M.L. Hydroxyapatite and calcium phosphates from marine sources: Extraction and characterization. In Marine Biomaterials: Characterization, Isolation and Applications; Kim, S.K., Ed.; CRC Press: Boca Raton, FL, USA, 2013; ISBN 978113. [Google Scholar]
- Mohd Puad, N.A.S.; Koshy, P.; Abdullah, H.Z.; Idris, M.I.; Lee, T.C. Syntheses of hydroxyapatite from natural sources. Heliyon 2019, 5, e01588. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.S.; Girija, E.K.; Venkatesh, M.; Karunakaran, G.; Kolesnikov, E.; Kuznetsov, D. One step method to synthesize flower-like hydroxyapatite architecture using mussel shell bio-waste as a calcium source. Ceram. Int. 2017, 43, 3457–3461. [Google Scholar] [CrossRef]
- Miculescu, F.; Mocanu, A.C.; Dascălu, C.A.; Maidaniuc, A.; Batalu, D.; Berbecaru, A.; Voicu, S.I.; Miculescu, M.; Thakur, V.K.; Ciocan, L.T. Facile synthesis and characterization of hydroxyapatite particles for high value nanocomposites and biomaterials. Vacuum 2017, 146, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Maity, S.; Chabri, S.; Bera, S.; Chowdhury, A.R.; Das, M.; Sinha, A. Mechanochemical synthesis of nanocrystalline hydroxyapatite from Mercenaria clam shells and phosphoric acid. Biomed. Phys. Eng. Express 2017, 3, 015010. [Google Scholar] [CrossRef]
- Shavandi, A.; Bekhit, A.E.D.A.; Ali, A.; Sun, Z. Synthesis of nano-hydroxyapatite (nHA) from waste mussel shells using a rapid microwave method. Mater. Chem. Phys. 2015, 149, 607–616. [Google Scholar] [CrossRef]
- Dapporto, M.; Sprio, S.; Fabbi, C.; Figallo, E.; Tampieri, A. A novel route for the synthesis of macroporous bioceramics for bone regeneration. J. Eur. Ceram. Soc. 2016, 36, 2383–2388. [Google Scholar] [CrossRef]
- Hou, Y.; Shavandi, A.; Carne, A.; Bekhit, A.A.; Ng, T.B.; Cheung, R.C.F.; Bekhit, A.E.A. Marine shells: Potential opportunities for extraction of functional and health-promoting materials. Crit. Rev. Environ. Sci. Technol. 2016, 46, 1047–1116. [Google Scholar] [CrossRef]
- Kwon, H.B.; Lee, C.W.; Jun, B.S.; Yun, J.D.; Weon, S.Y.; Koopman, B. Recycling waste oyster shells for eutrophication control. Res. Conserv. Recycl. 2004, 41, 75–82. [Google Scholar] [CrossRef]
- Piccirillo, C.; Moreira, I.S.; Novais, R.M.; Fernades, A.J.S.; Pullar, R.C.; Castro, P.M.L. Biphasic apatite-carbon materials derived from pyrolysed fish bones for effective adsorption of persistent pollutants and heavy metals. J. Environ. Chem. Eng. 2017, 5, 4884–4894. [Google Scholar] [CrossRef]
- Soisuwan, S.; Phommachant, J.; Wisaijorn, W.; Praserthdam, P. The characteristics of green calcium oxide derived from aquatic materials. Procedia Chem. 2014, 9, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Oldfield, T.L.; Patsios, S.I.; Holden, N.M. Hydrid life cycle assessment of agro-industrial wastewater valorisation. Water Res. 2020, 170, 115275. [Google Scholar] [CrossRef]
- Xia, X.; Shen, J.; Cao, F.; Wang, C.; Tang, M.; Zhang, Q.; Wei, S. A facile synthesis of hydroxyapatite for effective removal strontium ion. J. Hazard. Mater. 2019, 368, 326–355. [Google Scholar] [CrossRef]
- Iafisco, M.; Margiotta, N. Silica xerogels and hydroxyapatite nanocrystals for the local delivery of platinum–bisphosphonate complexes in the treatment of bone tumors: A mini-review. J. Inorg. Biochem. 2012, 117, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Bastan, F.E.; Erdogan, G.; Ustel, F. Role of strontium in spray drying of hydroxyapatite: A comparative study on physical properties. Appl. Ceram. Technol. 2020, 17, 1155–1166. [Google Scholar] [CrossRef]
- Belpassi, A.; Dolcini, L.; Martinetti, R. A Process for the Production of Porous Calcium Phosphate Articles for Bone Regeneration and as a Support for Cells and the Porous Articles Themselves. European Patent Office EP1411035, 15 October 2002. [Google Scholar]
- Tan, C.Y.; Singh, R.; The, Y.C.; Tan, Y.M.; Yap, B.K. The effects of calcium-to-phosphorus ratio on the densification and mechanical properties of hydroxyapatite ceramic. Int. J. Appl. Ceram. Technol. 2015, 12, 223–227. [Google Scholar] [CrossRef]
- Li, W.X.; Gao, M.; Song, Y.; Wang, F.P. Thermal stability and mechanical properties of zirconia-hydroxyapatite composites. Mater. Res. Innov. 2014, 18, S4487–S4489. [Google Scholar] [CrossRef]
- Yang, L.; Ning, X.; Bai, Y.; Jia, W. A scalable synthesis of non-agglomerated and low-aspect ratio hydroxyapatite nanocrystals using gelatinized starch matrix. Mater. Lett. 2013, 113, 142–145. [Google Scholar] [CrossRef]
- Fahami, A.; Beall, G.W.; Betancourt, T. Synthesis, bioactivity and zeta potential investigations of chlorine and fluorine substituted hydroxyapatite. Mater. Sci. Eng. C 2016, 59, 78–85. [Google Scholar] [CrossRef]
- Predoi, D.; Iconaru, S.L.; Predoi, M.V.; Motelica-Heino, M.; Guegan, R.; Buton, N. Evaluation of antibacterial activity of zinc-doped hydroxyapatite colloids and dispersion stability using ultrasound. Nanomaterials 2019, 9, 515. [Google Scholar] [CrossRef] [Green Version]
- Hadagalli, K.; Panda, A.K.; Mandal, S.; Basu, B. Faster biomineralization and tailored mechanical properties of marine resources-derived hydroxyapatite scaffolds with tunable interconnected porous architecture. ACS Appl. Bio Mater. 2019, 2, 2171–2184. [Google Scholar] [CrossRef]
- He, L.H.; Standard, O.C.; Huang, T.T.; Latella, B.A.; Swain, M.V. Mechanical behaviour of porous hydroxyapatite. Acta Biomater. 2008, 4, 577–586. [Google Scholar] [CrossRef]
- Walsh, P.J.; Buchanan, J.; Dring, M.; Maggs, C.; Bell, S.; Walker, G.M. Low-pressure synthesis and characterisation of hydroxyapatite derived from mineralised red algae. Chem. Eng. J. 2008, 137, 173–179. [Google Scholar] [CrossRef]
- Santhosh, S.; Prabu, B. Thermal stability of nanohydroxyapatite synthesized from sea shells through wet chemical synthesis. Mater. Lett. 2013, 97, 121–124. [Google Scholar] [CrossRef]
- Coelho, A. Topas Academic v4.1; Coelho Software: Brisbane, Australia, 2007. [Google Scholar]
- Klung, H.P.; Alexander, L.E. X-ray Diffraction Procedures for Polycrystalline and Amorphous Materials; Wiles: New York, NY, USA, 1974. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Treatment | Temperature (°C) | Phase Composition (wt%) | Specific Surface Area (SSA) (m2/g) |
|---|---|---|---|---|
| S_A_700 | Air | 700 | CaO (19.0 ± 0.3); Ca(OH)2 (70.1 ± 0.4); Calcite (10.9 ± 0.4) | 5.6 |
| S_A_1000 | Air | 1000 | CaO (100%) | 3.1 |
| S_N_700 | N2 | 700 | CaO (32.0 ± 0.3); Ca(OH)2 (17.8 ± 0.4); Calcite (50.2 ± 0.4) | 2.5 |
| S_N_1000 | N2 | 1000 | CaO (43.9 ± 0.3); Ca(OH)2 (27.6 ± 0.3); Calcite (28.6 ± 0.4) | 6.6 |
| Sample Name | Ca Containing Reagent | P Containing Reagent | Phase Composition (wt%) | Ca/P Ratio (mol/mol) | Specific Surface Area (m2/g) |
|---|---|---|---|---|---|
| HA_1 | S_A_700 | (NH4)2HPO4 | HA (100%) | 1.88 ± 0.01 | 49.6 |
| HA_2 | S_A_700 | H3PO4 | HA (99.0 ± 0.1); Calcite (1.0 ± 0.1) | 1.73 ± 0.01 | 100.9 |
| HA_3 | S_A_1000 | (NH4)2HPO4 | HA (100%) | 1.84 ± 0.01 | 83.5 |
| HA_4 | S_A_1000 | H3PO4 | HA (100%) | 1.73 ± 0.01 | 93.1 |
| Sample Name | D(002) (nm) | D(310) (nm) | D(002)/D(310) |
|---|---|---|---|
| HA_1 | 108.7 ± 1.5 | 55.2 ± 0.6 | 2.0 |
| HA_2 | 33.8 ± 0.5 | 11.9 ± 0.1 | 2.8 |
| HA_3 | 70.3 ± 0.6 | 21.0 ± 0.2 | 3.4 |
| HA_4 | 30.2 ± 0.5 | 12.0 ± 0.2 | 2.5 |
| HA_5 | 80.7 ± 0.7 | 30.6 ± 0.1 | 2.6 |
| Sample Name | Phase Composition (wt%) | Specific Surface Area (m2/g) |
|---|---|---|
| HA_6 | HA (96.0 ± 0.1); CaO (4.0 ± 0.2) | 4.5 |
| Commercial HA | HA (100%) | 5.1 |
| Sample Name | Porosity (%) | Compressive Strength (MPa) | Young’s Modulus (MPa) | Work of Fracture (mJ/m3) |
|---|---|---|---|---|
| Scaffold HA_6 | 87.8 ± 0.1 | 0.51 ± 0.14 | 36 ± 12 | 34 ± 7 |
| Scaffold Commercial HA | 85.4 ± 0.7 | 1.06 ± 0.26 | 68 ± 26 | 16 ± 6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scialla, S.; Carella, F.; Dapporto, M.; Sprio, S.; Piancastelli, A.; Palazzo, B.; Adamiano, A.; Degli Esposti, L.; Iafisco, M.; Piccirillo, C. Mussel Shell-Derived Macroporous 3D Scaffold: Characterization and Optimization Study of a Bioceramic from the Circular Economy. Mar. Drugs 2020, 18, 309. https://doi.org/10.3390/md18060309
Scialla S, Carella F, Dapporto M, Sprio S, Piancastelli A, Palazzo B, Adamiano A, Degli Esposti L, Iafisco M, Piccirillo C. Mussel Shell-Derived Macroporous 3D Scaffold: Characterization and Optimization Study of a Bioceramic from the Circular Economy. Marine Drugs. 2020; 18(6):309. https://doi.org/10.3390/md18060309
Chicago/Turabian StyleScialla, Stefania, Francesca Carella, Massimiliano Dapporto, Simone Sprio, Andreana Piancastelli, Barbara Palazzo, Alessio Adamiano, Lorenzo Degli Esposti, Michele Iafisco, and Clara Piccirillo. 2020. "Mussel Shell-Derived Macroporous 3D Scaffold: Characterization and Optimization Study of a Bioceramic from the Circular Economy" Marine Drugs 18, no. 6: 309. https://doi.org/10.3390/md18060309
APA StyleScialla, S., Carella, F., Dapporto, M., Sprio, S., Piancastelli, A., Palazzo, B., Adamiano, A., Degli Esposti, L., Iafisco, M., & Piccirillo, C. (2020). Mussel Shell-Derived Macroporous 3D Scaffold: Characterization and Optimization Study of a Bioceramic from the Circular Economy. Marine Drugs, 18(6), 309. https://doi.org/10.3390/md18060309