A Metabolomics Exploration of the Sexual Phase in the Marine Diatom Pseudo-nitzschia multistriata

 and
and 
Abstract
:1. Introduction
2. Results
2.1. Effect of Sequential Solvent Extractions on the Endo-Metabolome
2.2. Identification of Known Metabolites
2.3. Mating Type and Harvesting Time Points
2.4. Differences between Sample Types in DCM Extracts
3. Discussion
4. Materials and Methods
4.1. Strains and Cultivation Procedure
4.2. Extraction of Endo-Metabolites
4.3. UPLC-ESI-QToF-MS/MS Untargeted Metabolomic Profiling
4.4. Data Processing and Metabolomics Analysis
4.4.1. Data Preprocessing
4.4.2. Statistical Analysis
4.4.3. Metabolite Annotation/Identification
4.4.4. Molecular Networking
4.5. Data Accessibility
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saha, M.; Berdalet, E.; Carotenuto, Y.; Fink, P.; Harder, T.; John, U.; Not, F.; Pohnert, G.; Potin, P.; Selander, E.; et al. Using chemical language to shape future marine health. Front. Ecol. Environ. 2019, 17, 530–537. [Google Scholar] [CrossRef]
- Reen, F.J.; Gutiérrez-Barranquero, J.A.; Dobson, A.D.W.; Adams, C.; O’Gara, F. Emerging concepts promising new horizons for marine biodiscovery and synthetic biology. Mar. Drugs 2015, 13, 2924–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarthou, G.; Timmermans, K.R.; Blain, S.; Tréguer, P. Growth physiology and fate of diatoms in the ocean: A review. J. Sea Res. 2005, 53, 25–42. [Google Scholar] [CrossRef]
- Huang, W.; Daboussi, F. Genetic and metabolic engineering in diatoms. Phil. Trans. R. Soc. B 2017, 372, 20160411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroth, P.G.; Bones, A.M.; Daboussi, F.; Ferrante, M.I.; Jaubert, M.; Kolot, M.; Nymark, M.; Río Bártulos, C.; Ritter, A.; Russo, M.T.; et al. Genome editing in diatoms: Achievements and goals. Plant Cell Rep. 2018, 37, 1401–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianora, A.; Miralto, A.; Poulet, S.A.; Carotenuto, Y.; Buttino, I.; Romano, G.; Casotti, R.; Pohnert, G.; Wichard, T.; Colucci-D’Amato, L.; et al. Aldehyde suppression of copepod recruitment in blooms of a ubiquitous planktonic diatom. Nature 2004, 429, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Vardi, A.; Formiggini, F.; Casotti, R.; De Martino, A.; Ribalet, F.; Miralto, A.; Bowler, C. A Stress Surveillance System Based on Calcium and Nitric Oxide in Marine Diatoms. PLoS Biol. 2006, 4, e60. [Google Scholar] [CrossRef] [Green Version]
- Moeys, S.; Frenkel, J.; Lembke, C.; Gillard, J.T.F.; Devos, V.; den Berge, K.V.; Bouillon, B.; Huysman, M.J.J.; Decker, S.D.; Scharf, J.; et al. A sex-inducing pheromone triggers cell cycle arrest and mate attraction in the diatom Seminavis robusta. Sci. Rep. 2016, 6, 19252. [Google Scholar] [CrossRef] [Green Version]
- Gillard, J.; Frenkel, J.; Devos, V.; Sabbe, K.; Paul, C.; Rempt, M.; Inzé, D.; Pohnert, G.; Vuylsteke, M.; Vyverman, W. Metabolomics Enables the Structure Elucidation of a Diatom Sex Pheromone. Angew. Chem. Int. Ed. 2013, 52, 854–857. [Google Scholar] [CrossRef]
- Le Manach, S.; Duval, C.; Marie, A.; Djediat, C.; Catherine, A.; Edery, M.; Bernard, C.; Marie, B. Global Metabolomic Characterizations of Microcystis spp. Highlights Clonal Diversity in Natural Bloom-Forming Populations and Expands Metabolite Structural Diversity. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Goulitquer, S.; Potin, P.; Tonon, T. Mass spectrometry-based metabolomics to elucidate functions in marine organisms and ecosystems. Mar. Drugs 2012, 10, 849–880. [Google Scholar] [CrossRef] [PubMed]
- Kuhlisch, C.; Pohnert, G. Metabolomics in chemical ecology. Nat. Prod. Rep. 2015, 32, 937–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennecke, P.; Ferrante, M.I.; Johnston, I.A.; Smith, D. A Collaborative European Approach to Accelerating Translational Marine Science. J. Mar. Sci. Eng. 2018, 6, 81. [Google Scholar] [CrossRef] [Green Version]
- Bromke, M.A.; Sabir, J.S.; Alfassi, F.A.; Hajarah, N.H.; Kabli, S.A.; Al-Malki, A.L.; Ashworth, M.P.; Méret, M.; Jansen, R.K.; Willmitzer, L. Metabolomic Profiling of 13 Diatom Cultures and Their Adaptation to Nitrate-Limited Growth Conditions. PLoS ONE 2015, 10, e0138965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, A.; Sabatino, V.; Nylund, G.M.; Bergkvist, J.; Basu, S.; Andersson, M.X.; Sanges, R.; Godhe, A.; Kiørboe, T.; Selander, E.; et al. Grazer-induced transcriptomic and metabolomic response of the chain-forming diatom Skeletonema marinoi. ISME J. 2018, 12, 1594–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, S.S.; Hubbard, K.A.; Lundholm, N.; Montresor, M.; Leaw, C.P. Pseudo-nitzschia, Nitzschia, and domoic acid: New research since 2011. Harmful Algae 2018, 79, 3–43. [Google Scholar] [CrossRef]
- Montresor, M.; Vitale, L.; D’Alelio, D.; Ferrante, M.I. Sex in marine planktonic diatoms: Insights and challenges. PiP 2016, 3, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.; Moeys, S.; von Dassow, P.; Huysman, M.J.J.; Mapleson, D.; De Veylder, L.; Sanges, R.; Vyverman, W.; Montresor, M.; Ferrante, M.I. Identification of the meiotic toolkit in diatoms and exploration of meiosis-specific SPO11 and RAD51 homologs in the sexual species Pseudo-nitzschia multistriata and Seminavis robusta. BMC Genom. 2015, 16, 930. [Google Scholar] [CrossRef] [Green Version]
- Di Dato, V.; Musacchia, F.; Petrosino, G.; Patil, S.; Montresor, M.; Sanges, R.; Ferrante, M.I. Transcriptome sequencing of three Pseudo-nitzschia species reveals comparable gene sets and the presence of Nitric Oxide Synthase genes in diatoms. Sci. Rep. 2015, 5, 12329. [Google Scholar] [CrossRef]
- Basu, S.; Patil, S.; Mapleson, D.; Russo, M.T.; Vitale, L.; Fevola, C.; Maumus, F.; Casotti, R.; Mock, T.; Caccamo, M.; et al. Finding a partner in the ocean: Molecular and evolutionary bases of the response to sexual cues in a planktonic diatom. New Phytol. 2017, 215, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, V.; Russo, M.T.; Patil, S.; d’Ippolito, G.; Fontana, A.; Ferrante, M.I. Establishment of Genetic Transformation in the Sexually Reproducing Diatoms Pseudo-nitzschia multistriata and Pseudo-nitzschia arenysensis and Inheritance of the Transgene. Mar. Biotechnol. 2015, 17, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.T.; Vitale, L.; Entrambasaguas, L.; Anestis, K.; Fattorini, N.; Romano, F.; Minucci, C.; Luca, P.D.; Biffali, E.; Vyverman, W.; et al. MRP3 is a sex determining gene in the diatom Pseudo-nitzschia multistriata. Nat. Commun. 2018, 9, 5050. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Computational strategies for metabolite identification in metabolomics. Bioanalysis 2009, 1, 1579–1596. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef]
- Diekmann, A.B.S.; Peck, M.A.; Holste, L.; St John, M.A.; Campbell, R.W. Variation in diatom biochemical composition during a simulated bloom and its effect on copepod production. J. Plankton Res. 2009, 31, 1391–1405. [Google Scholar] [CrossRef] [Green Version]
- Heal, K.R.; Kellogg, N.A.; Carlson, L.T.; Lionheart, R.M.; Ingalls, A.E. Metabolic Consequences of Cobalamin Scarcity in the Diatom Thalassiosira pseudonana as Revealed Through Metabolomics. Protist 2019, 170, 328–348. [Google Scholar] [CrossRef]
- Allen, A.E.; LaRoche, J.; Maheswari, U.; Lommer, M.; Schauer, N.; Lopez, P.J.; Finazzi, G.; Fernie, A.R.; Bowler, C. Whole-cell response of the pennate diatom Phaeodactylum tricornutum to iron starvation. Proc. Natl. Acad. Sci. USA 2008, 105, 10438–10443. [Google Scholar] [CrossRef] [Green Version]
- Bromke, M.A.; Giavalisco, P.; Willmitzer, L.; Hesse, H. Metabolic Analysis of Adaptation to Short-Term Changes in Culture Conditions of the Marine Diatom Thalassiosira pseudonana. PLoS ONE 2013, 8, e67340. [Google Scholar] [CrossRef]
- Nappo, M.; Berkov, S.; Codina, C.; Avila, C.; Messina, P.; Zupo, V.; Bastida, J. Metabolite profiling of the benthic diatom Cocconeis scutellum by GC-MS. J. Appl. Phycol. 2008, 21, 295. [Google Scholar] [CrossRef]
- Vidoudez, C.; Pohnert, G. Comparative metabolomics of the diatom Skeletonema marinoi in different growth phases. Metabolomics 2012, 8, 654–669. [Google Scholar] [CrossRef]
- Azizan, A.; Ahamad Bustamam, M.S.; Maulidiani, M.; Shaari, K.; Ismail, I.S.; Nagao, N.; Abas, F. Metabolite Profiling of the Microalgal Diatom Chaetoceros Calcitrans and Correlation with Antioxidant and Nitric Oxide Inhibitory Activities via 1H NMR-Based Metabolomics. Mar. Drugs 2018, 16, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hano, T.; Tomaru, Y. Metabolomics-based approach to explore growth phase-dependent markers in cultured diatom Chaetoceros tenuissimus. J. Chromatogr. B 2019, 1128, 121779. [Google Scholar] [CrossRef] [PubMed]
- Scalco, E.; Stec, K.; Iudicone, D.; Ferrante, M.I.; Montresor, M. The dynamics of sexual phase in the marine diatom Pseudo-nitzschia multistriata (Bacillariophyceae). J. Phycol. 2014, 50, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Hiley, C.R.; Hoi, P.M. Oleamide: A fatty acid amide signaling molecule in the cardiovascular system? Cardiovasc. Drug Rev. 2007, 25, 46. [Google Scholar] [CrossRef]
- Getachew, P.; Getachew, M.; Joo, J.; Choi, Y.S.; Hwang, D.S.; Hong, Y.-K. The slip agents oleamide and erucamide reduce biofouling by marine benthic organisms (diatoms, biofilms and abalones). Toxicol. Environ. Health Sci. 2016, 8, 341–348. [Google Scholar] [CrossRef]
- Scalco, E.; Amato, A.; Ferrante, M.I.; Montresor, M. The sexual phase of the diatom Pseudo-nitzschia multistriata: Cytological and time-lapse cinematography characterization. Protoplasma 2016, 253, 1421–1431. [Google Scholar] [CrossRef]
- Fenizia, S.; Thume, K.; Wirgenings, M.; Pohnert, G. Ectoine from Bacterial and Algal Origin Is a Compatible Solute in Microalgae. Mar. Drugs 2020, 18, 42. [Google Scholar] [CrossRef] [Green Version]
- Jaramillo-Madrid, A.C.; Ashworth, J.; Fabris, M.; Ralph, P.J. Phytosterol biosynthesis and production by diatoms (Bacillariophyceae). Phytochemistry 2019, 163, 46–57. [Google Scholar] [CrossRef]
- Borgonuovo, C. Exploring the Sexual Phase of the Diatom Pseudo-Nitzschia Multistriata: From Genes to Metabolites. Ph.D. Thesis, The Open University, Milton Keynes, UK, 2019. [Google Scholar]
- R Development Core Team, R: A language and environment for statistical computing; R Foundation for Statistical Computing: Vienna, Austria, 2013.
- R. Team. RStudio: Integrated development for R; RStudio, Inc.: Boston, MA, USA, 2014. [Google Scholar]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- De Livera, A.M.; Bowne, J.B. Analysis of Metabolomics Data, R package, version 0.1.4. 2014. Available online: https://cran.r-project.org/web/packages/metabolomics/index.html (accessed on 12 May 2020).
- Gaude, E.; Chignola, F.; Spiliotopoulos, D.; Spitaleri, A.; Ghitti, M.; Garcia-Manteiga, J.M.; Mari, S.; Musco, G. muma, An R Package for Metabolomics Univariate and Multivariate Statistical Analysis. Curr. Metabolomics 2013, 1, 180. [Google Scholar] [CrossRef]
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes. R package, version 1.1-2. 2014. Available online: https://cran.r-project.org/web/packages/RColorBrewer/index.html (accessed on 12 May 2020).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer, Chem: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Wickham, H.; François, R.; Lionel, H.; Müller, K. dplyr: A Grammar of Data Manipulation. R package version 0.8.0.1. 2019. Available online: https://dplyr.tidyverse.org/ (accessed on 12 May 2020).
- Thevenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L. Exact and approximate area-proportional circular Venn and Euler diagrams. IEEE Trans. Vis. Comput. Graph. 2012, 18, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Böcker, S.; Letzel, M.C.; Lipták, Z.; Pervukhin, A. SIRIUS: Decomposing isotope patterns for metabolite identification†. Bioinformatics 2009, 25, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
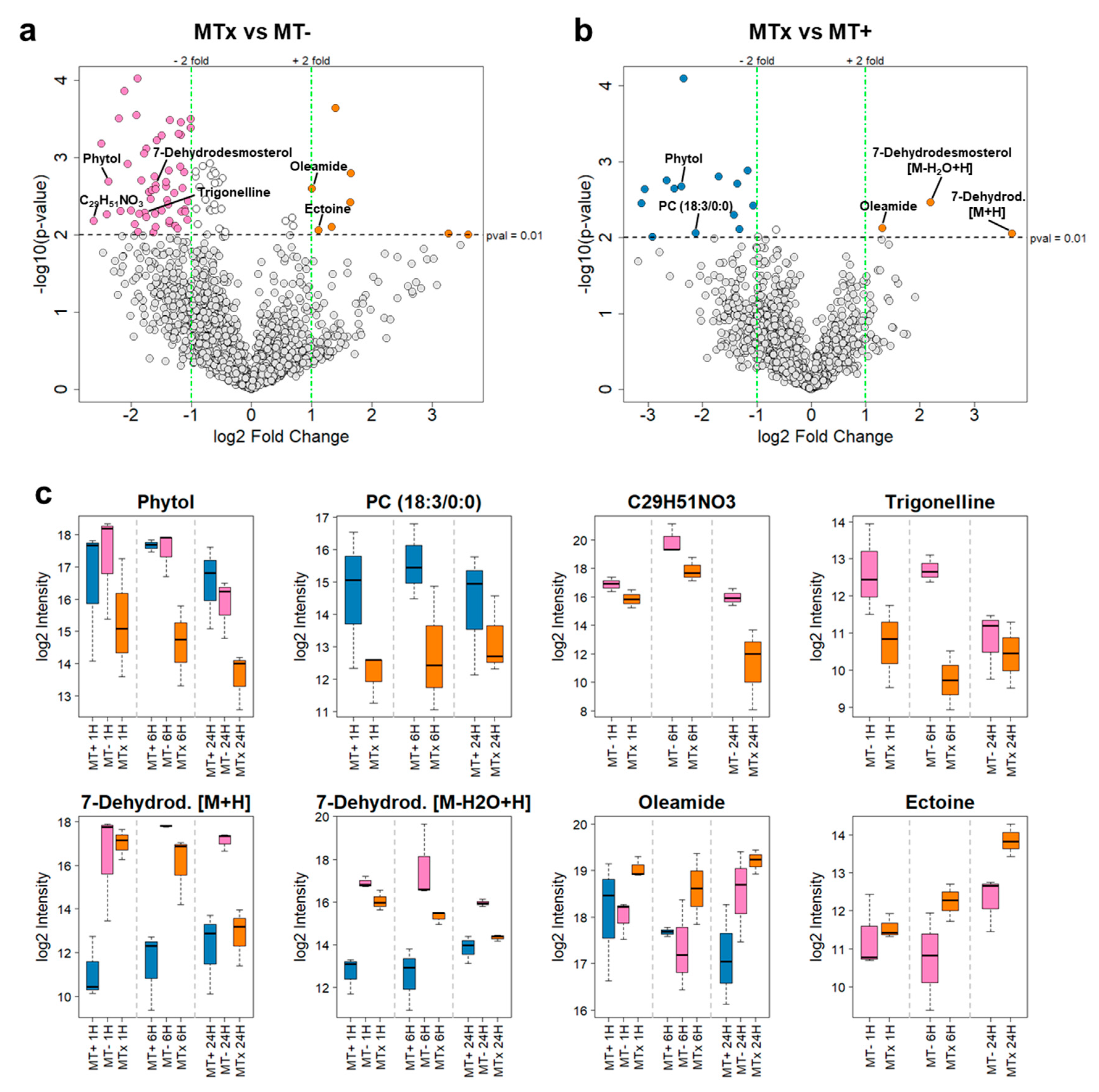
| Metabolite | m/z [Da] | r.t. med [min] | log2 fc MTx vs. MT+ | log2 fc MTx vs. MT− |
|---|---|---|---|---|
| Phytol | 319.2974 | 22 | −2.39 | −2.37 |
| PC (18:3/0:0) | 518.3244 | 16 | −2.13 | Not significant (p > 0.01) |
| C29H51NO3 | 462.3944 | 15 | Not significant (p > 0.01) | −2.61 |
| Trigonelline | 138.0551 | 1 | Not significant (p > 0.01) | −1.77 |
| 7-Dehydrodesmosterol [M + H] | 383.3309 | 21 | 3.70 | Not significant (p > 0.01) |
| 7-Dehydrodesmosterol [M − H2O + H] | 365.3204 | 21 | 2.20 | −1.59 |
| Oleamide | 282.2794 | 19 | 1.31 | 1.00 |
| Ectoine | 143.0817 | 1 | Not significant (p > 0.01) | 1.12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorini, F.; Borgonuovo, C.; Ferrante, M.I.; Brönstrup, M. A Metabolomics Exploration of the Sexual Phase in the Marine Diatom Pseudo-nitzschia multistriata. Mar. Drugs 2020, 18, 313. https://doi.org/10.3390/md18060313
Fiorini F, Borgonuovo C, Ferrante MI, Brönstrup M. A Metabolomics Exploration of the Sexual Phase in the Marine Diatom Pseudo-nitzschia multistriata. Marine Drugs. 2020; 18(6):313. https://doi.org/10.3390/md18060313
Chicago/Turabian StyleFiorini, Federica, Camilla Borgonuovo, Maria Immacolata Ferrante, and Mark Brönstrup. 2020. "A Metabolomics Exploration of the Sexual Phase in the Marine Diatom Pseudo-nitzschia multistriata" Marine Drugs 18, no. 6: 313. https://doi.org/10.3390/md18060313
APA StyleFiorini, F., Borgonuovo, C., Ferrante, M. I., & Brönstrup, M. (2020). A Metabolomics Exploration of the Sexual Phase in the Marine Diatom Pseudo-nitzschia multistriata. Marine Drugs, 18(6), 313. https://doi.org/10.3390/md18060313