Interactions of the α3β2 Nicotinic Acetylcholine Receptor Interfaces with α-Conotoxin LsIA and its Carboxylated C-terminus Analogue: Molecular Dynamics Simulations



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
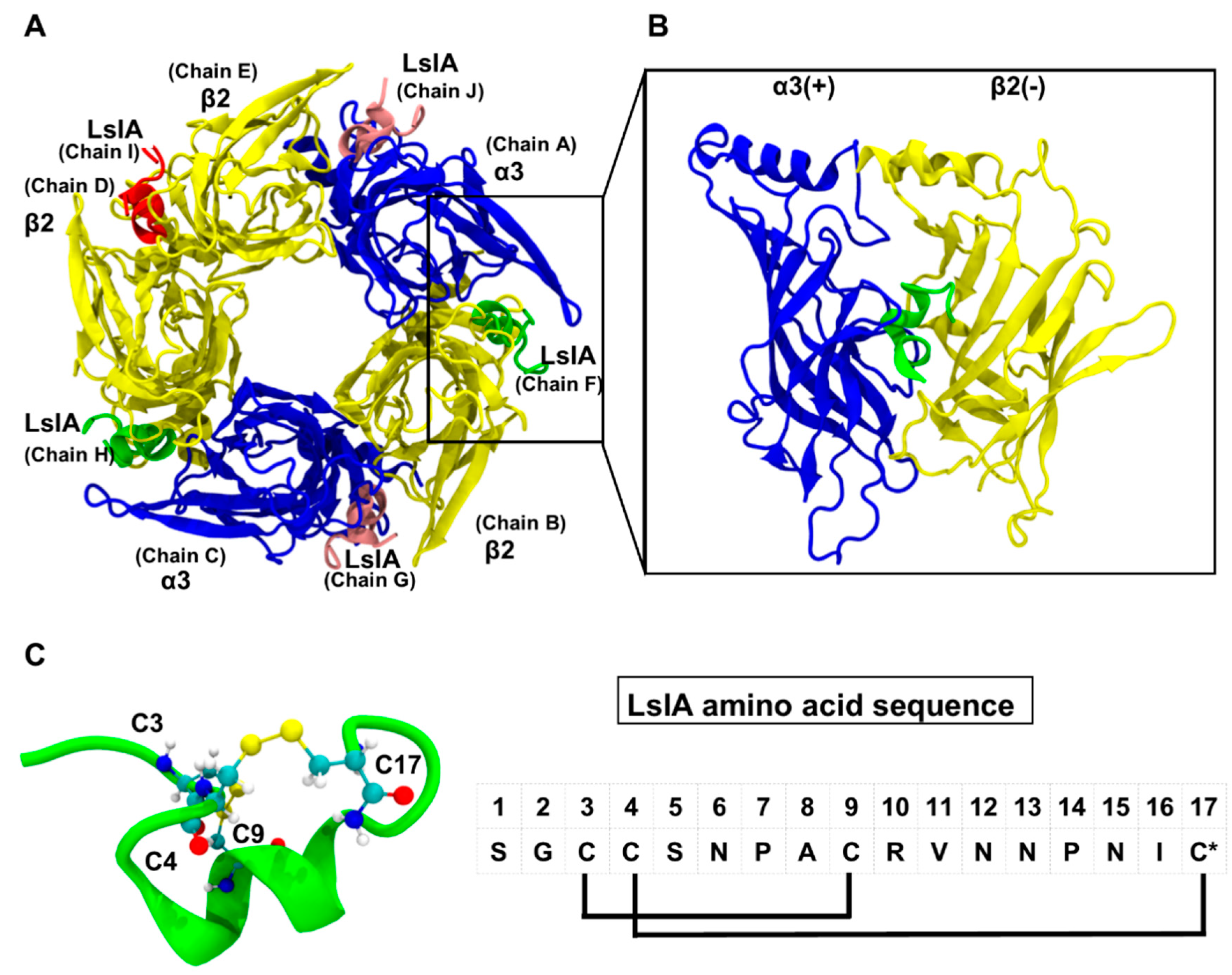
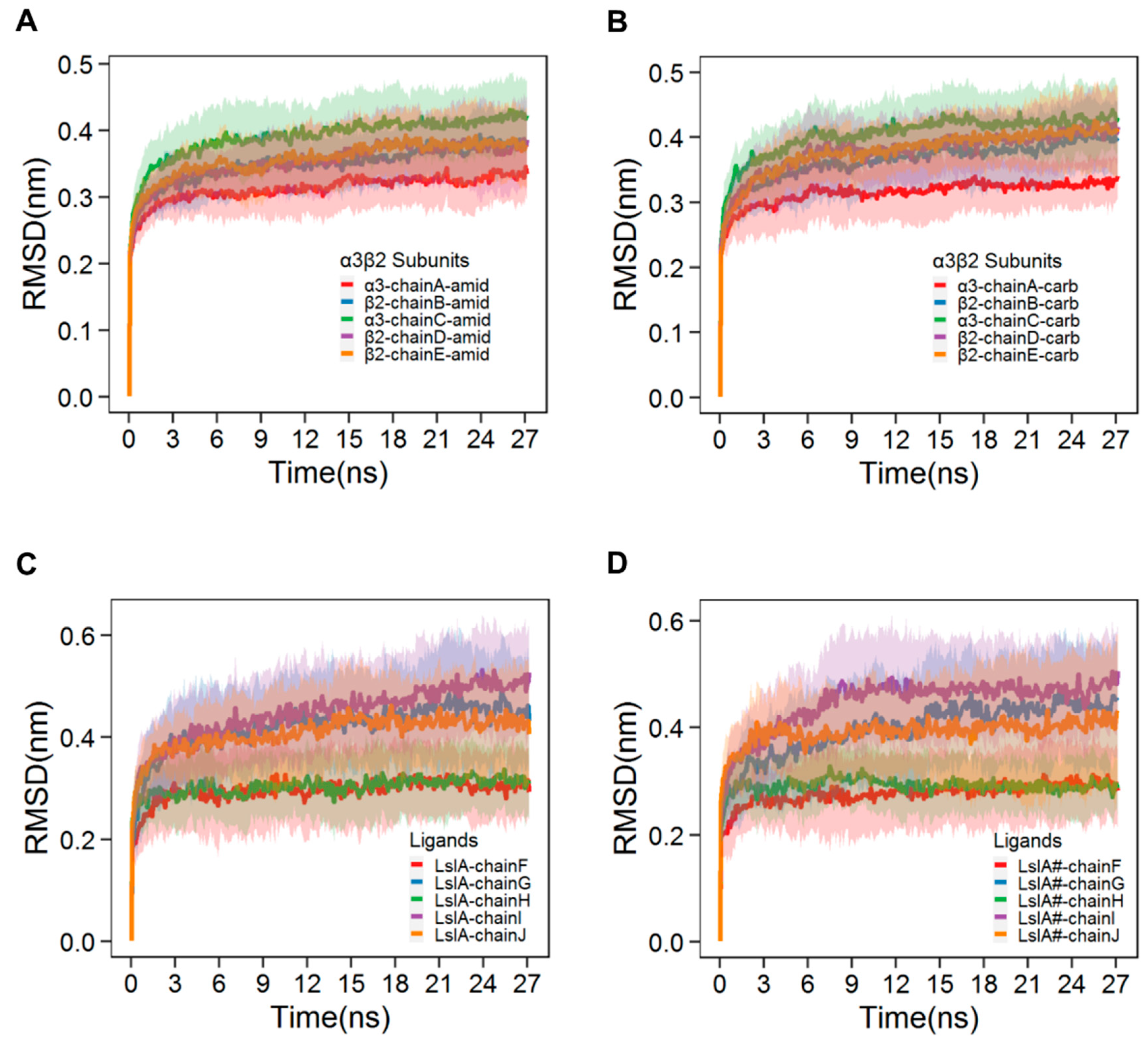
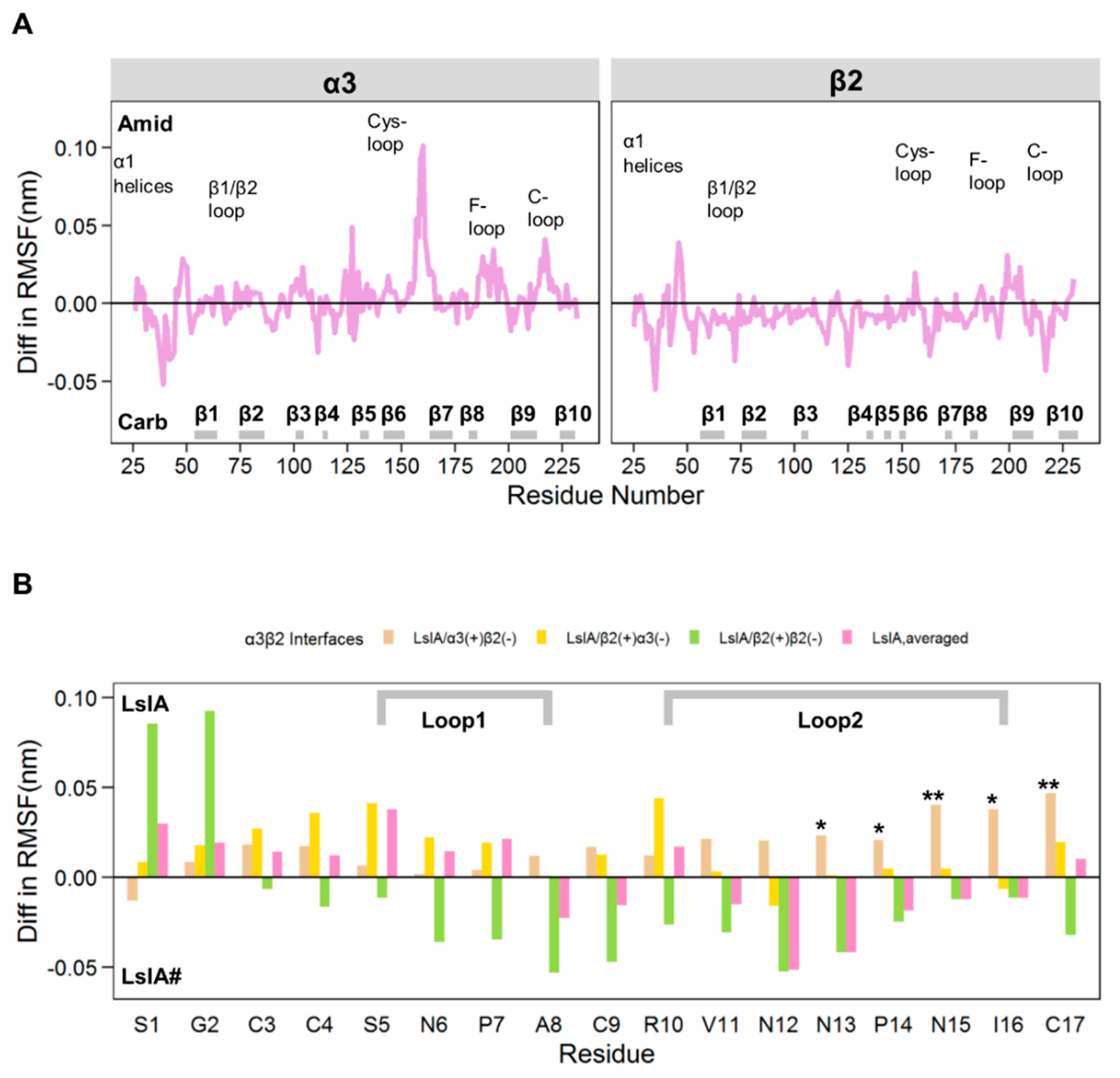
2.1. Stability and Flexibility of the LsIA-α3β2 Complexes
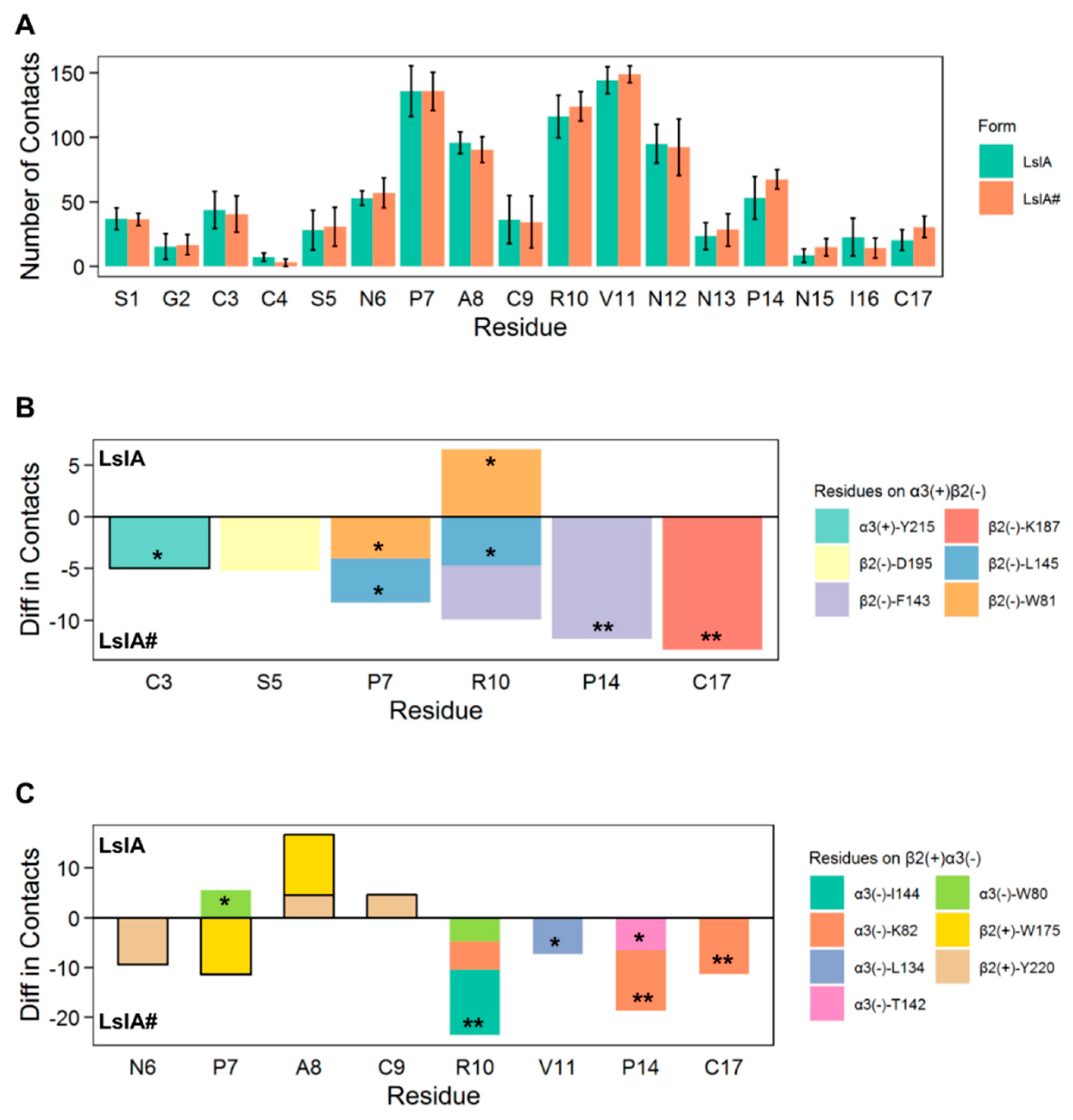
2.2. LsIA and LsIA# Toxin-Receptor Contacts
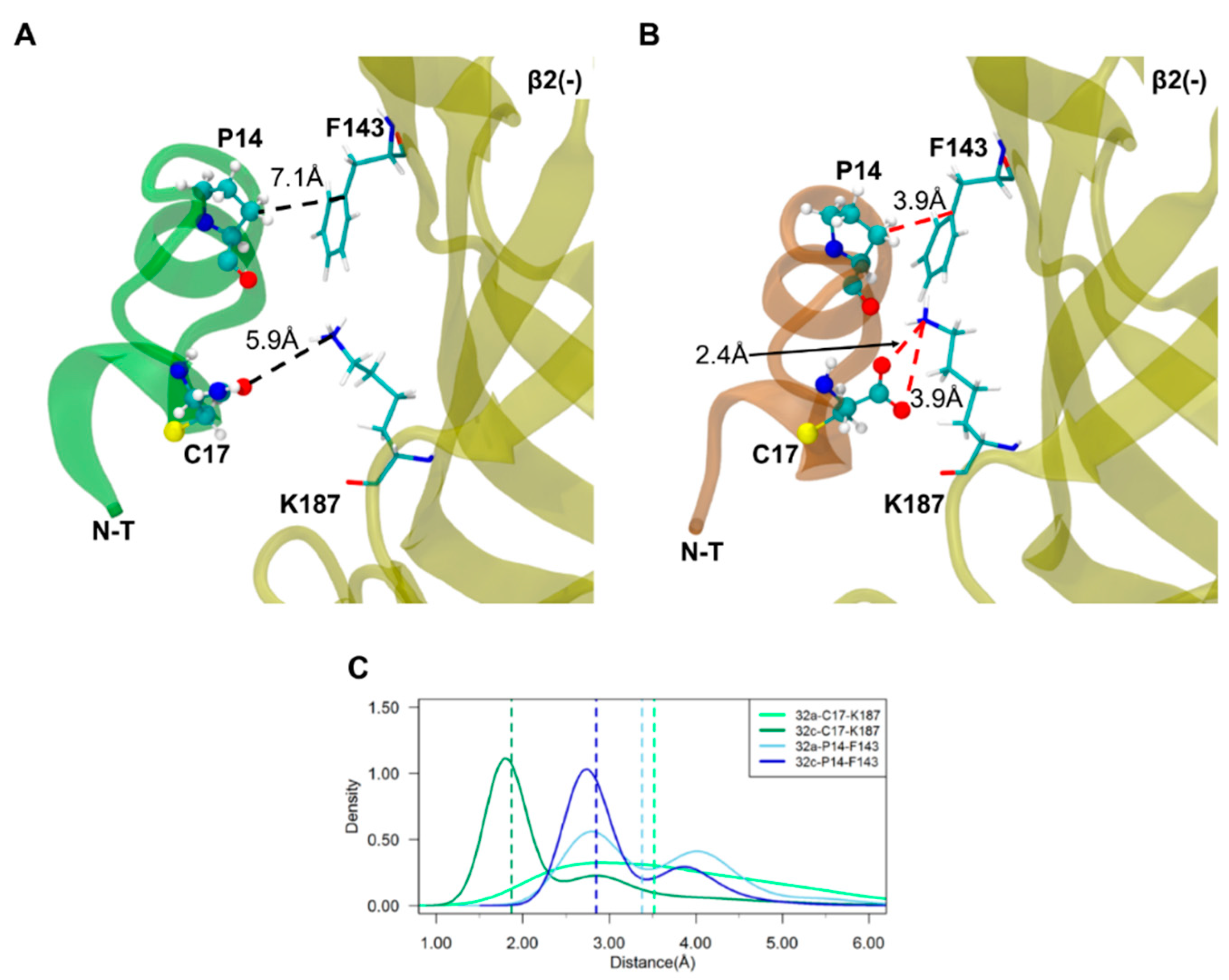
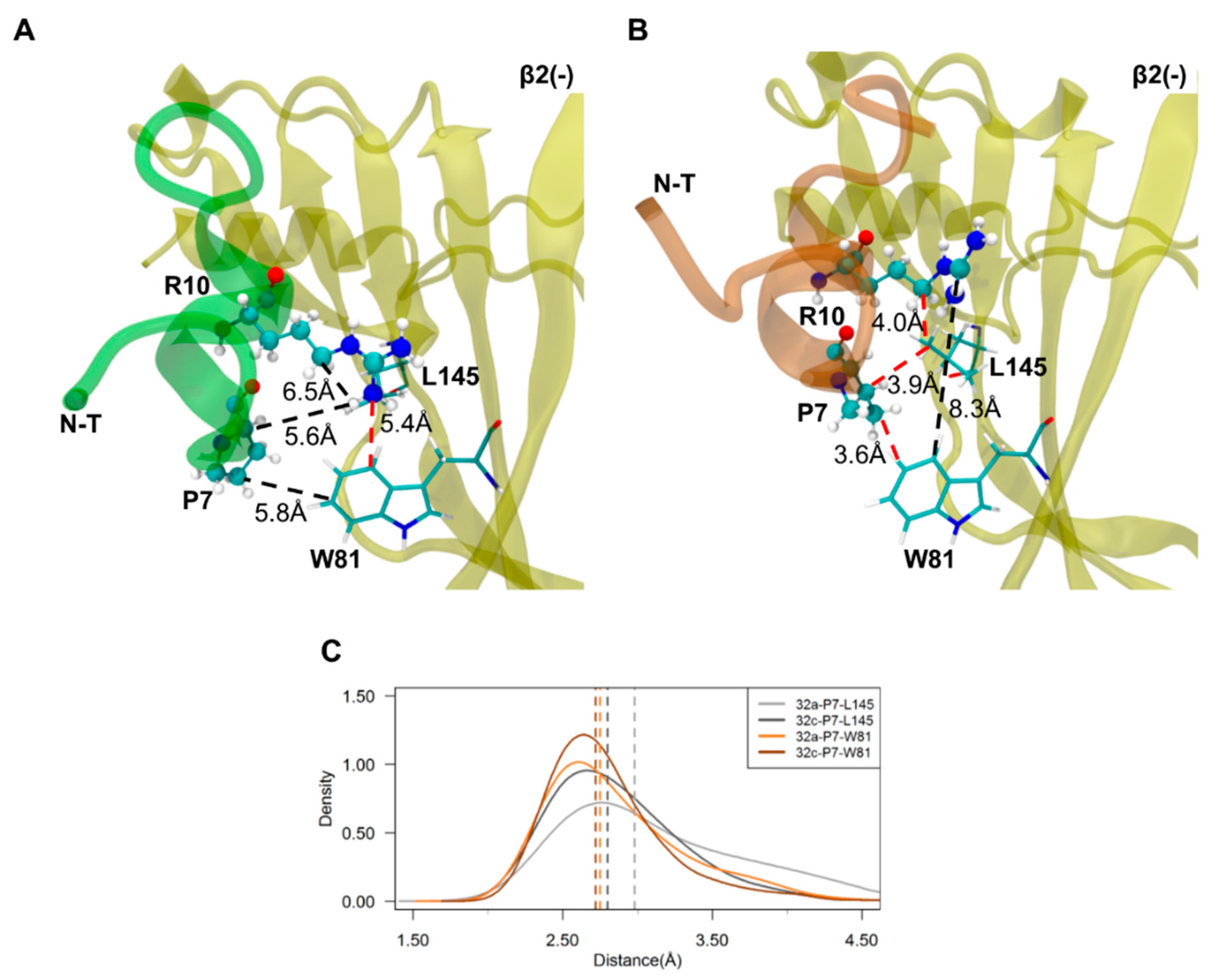
2.3. Important Pairwise Interactions of LsIA with Residues at α3(+)β2(−) Interfaces
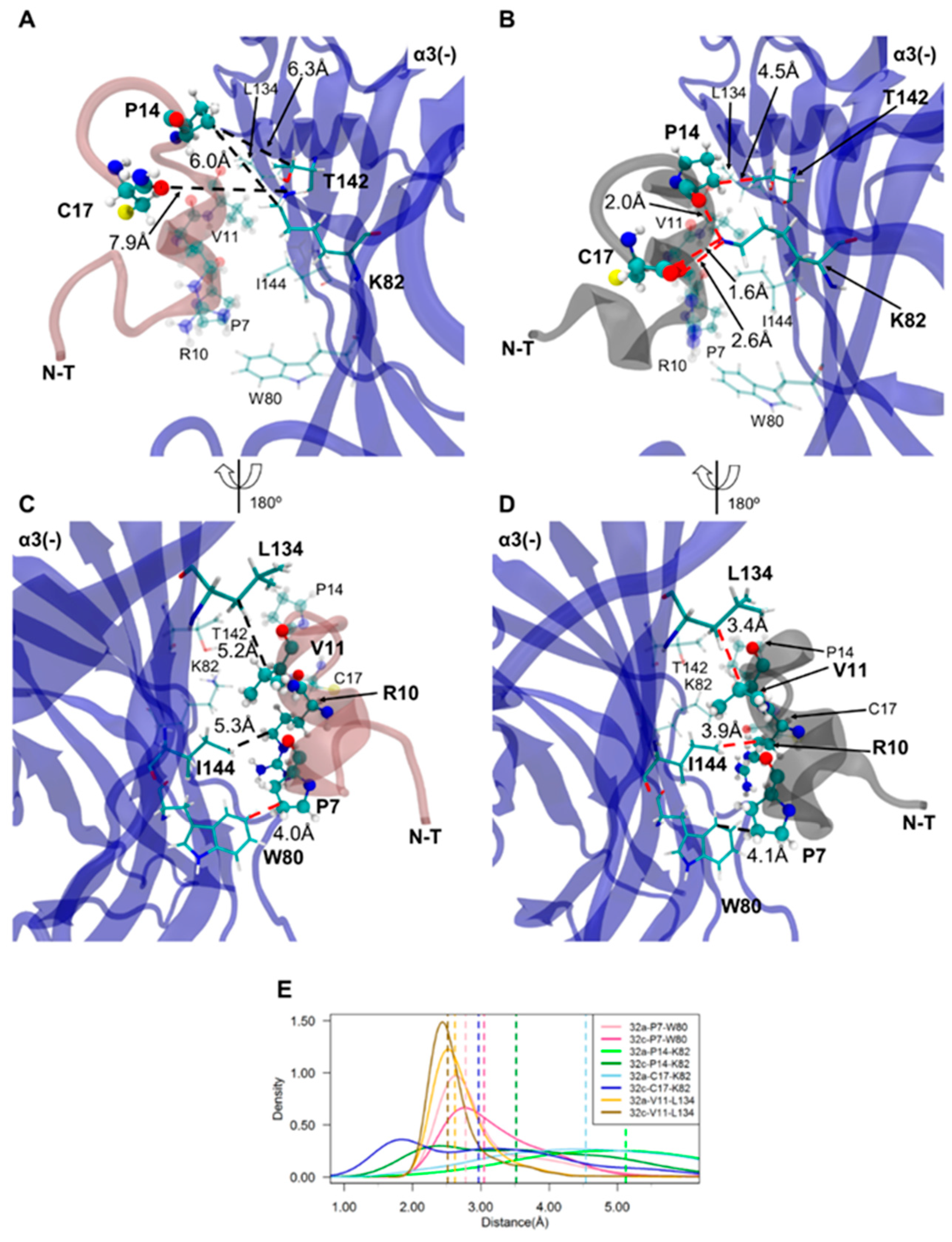
2.4. Important Pairwise Interactions of LsIA with Residues at β2(+)α3(−) Interfaces
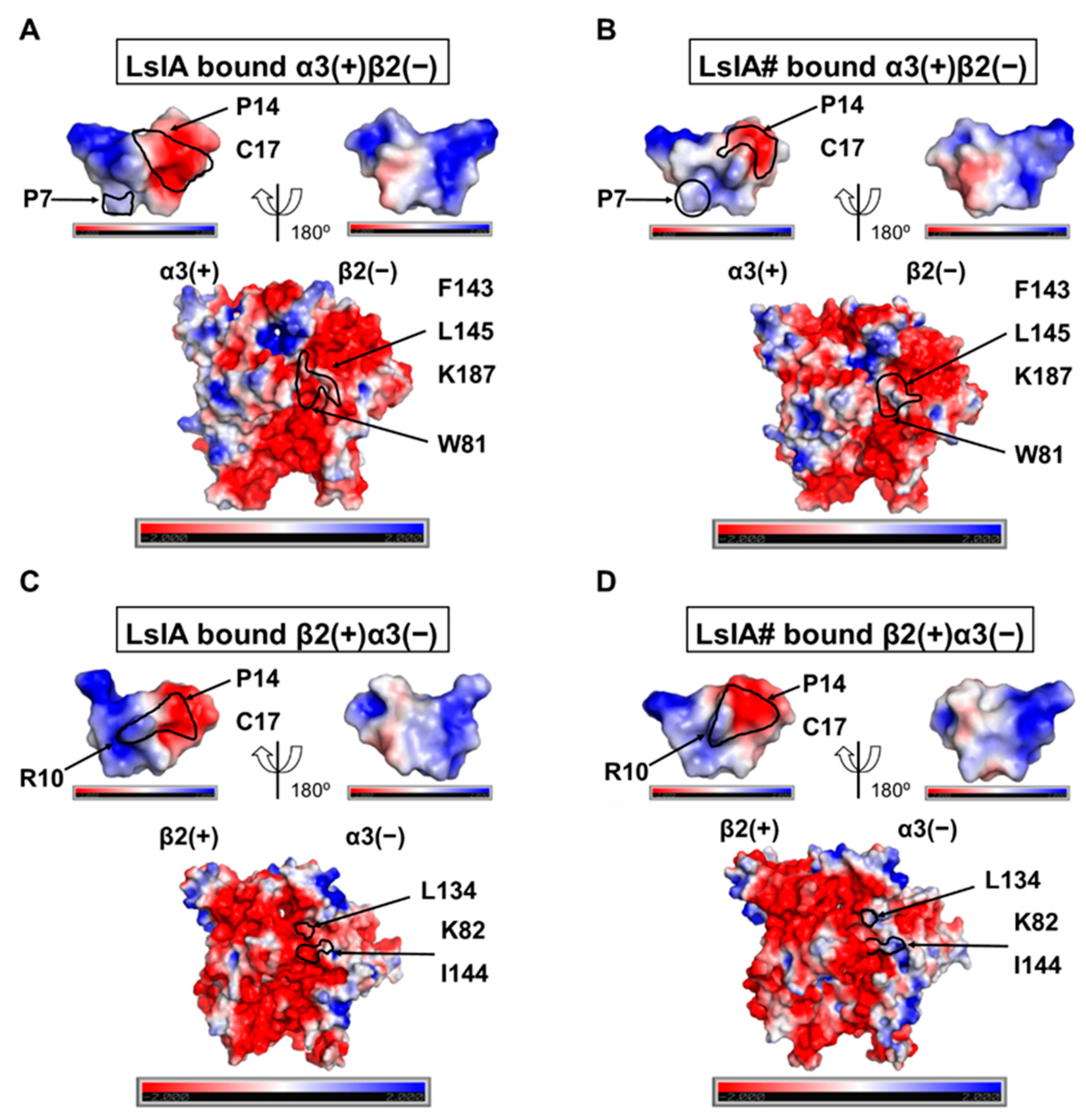
2.5. Understanding Changes in Electrostatic Potential by C-terminal Carboxylation of LsIA at α3(+)β2(−) and β2(−)α3(+) Interfaces
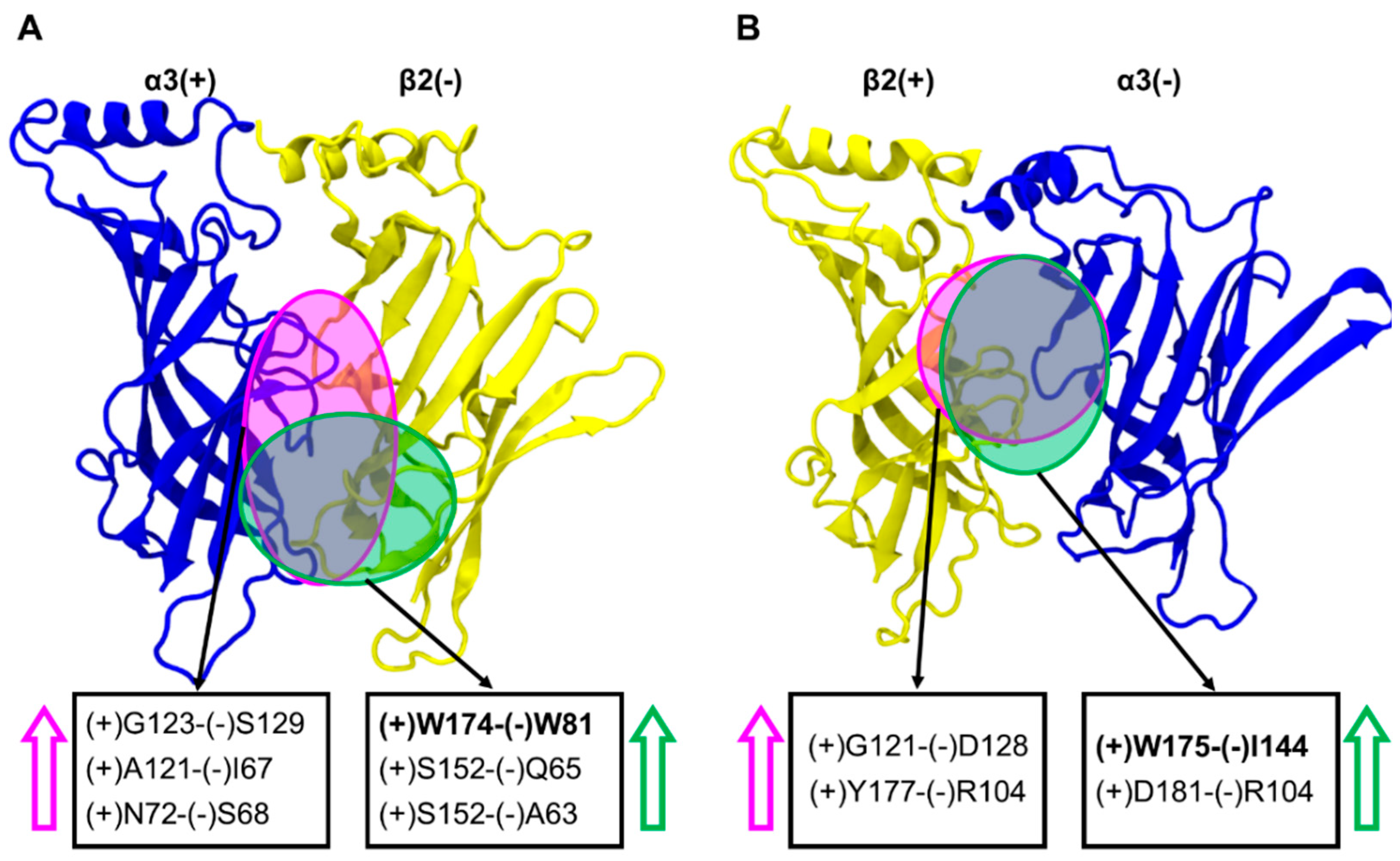
2.6. Inter-subunit Contacts at the α3(+)β2(−) and β2(+)α3(−) Interfaces
3. Discussion
3.1. Different Combinations of the Interfaces in Rat α3β2 may Affect the Distinct Stability and Flexibility of the Receptor and LsIA
3.2. C-terminal Carboxylation of LsIA Marginally Enhanced Overall α3β2 nAChR Contacts Versus LsIA
3.3. C-Terminal Carboxylation of LsIA Mainly Enhances the Contacts with Residues on the Complementary Subunit of both α3(+)β2(−) and β2(+)α3(−) Interfaces
4. Materials and Methods
4.1. Homology Modelling
4.2. Molecular Dynamics Simulation
4.2.1. MD Simulations on LsIA-α3β2 Complexes
4.2.2. MD Simulations on LsIA Only
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ellison, M.; Gao, F.; Wang, H.-L.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII target distinct regions of the human α7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 2004, 43, 16019–16026. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Moretti, M.; Bohr, I.; Ziabreva, I.; Vailati, S.; Longhi, R.; Riganti, L.; Gaimarri, A.; McKeith, I.G.; Perry, R.H.; et al. Selective nicotinic acetylcholine receptor subunit deficits identified in Alzheimer´s disease, Parkinson´s disease and dementia with Lewy bodies by immunoprecipitation. Neurobiol. Dis. 2006, 23, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Therapeut. 2013, 137, 22–54. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Paterson, D.; Nordberg, A. Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 2000, 61, 75–111. [Google Scholar] [CrossRef]
- Everhart, D.; Cartier, G.E.; Malhotra, A.; Gomes, A.V.; McIntosh, J.M.; Luetje, C.W. Determinants of potency on α-conotoxin MII, a peptide antagonist of neuronal nicotinic receptors. Biochemistry 2004, 43, 2732–2737. [Google Scholar] [CrossRef]
- McIntosh, J.M.; Azam, L.; Staheli, S.; Dowell, C.; Lindstrom, J.M.; Kuryatov, A.; Garrett, J.E.; Marks, M.J.; Whiteaker, P. Analogs of α-conotoxin MII are selective for α6-containing nicotinic acetylcholine receptors. Mol. Pharmacol. 2004, 65, 944–952. [Google Scholar] [CrossRef]
- Gao, F.; Mer, G.; Tonelli, M.; Hansen, S.B.; Burghardt, T.P.; Taylor, P.; Sine, S.M. Solution NMR of acetylcholine binding protein reveals agonist-mediated conformational change of the C-loop. Mol. Pharmacol. 2006, 70, 1230–1235. [Google Scholar] [CrossRef] [Green Version]
- Inserra, M.C.; Kompella, S.N.; Vetter, I.; Brust, A.; Daly, N.L.; Cuny, H.; Craik, D.J.; Alewood, P.F.; Adams, D.J.; Lewis, R.J. Isolation and characterization of α-conotoxin LsIA with potent activity at nicotinic acetylcholine receptors. Biochem. Pharmacol. 2013, 86, 791–799. [Google Scholar] [CrossRef]
- Cox, B.C.; Marritt, A.M.; Perry, D.C.; Kellar, K.J. Transport of multiple nicotinic acetylcholine receptors in the rat optic nerve: High densities of receptors containing α6 and β3 subunits. J. Neurochem. 2008, 105, 1924–1938. [Google Scholar] [CrossRef] [Green Version]
- Millar, N.S.; Gotti, C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, T.F.; Dougherty, D.A.; Lester, H.A. The 5-HT3AB receptor shows an A3B2 stoichiometry at the plasma membrane. Biophys. J. 2013, 105, 887–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamotte d´Incamps, B.; Zorbaz, T.; Dingova, D.; Krejci, E.; Ascher, P. Stoichiometry of the heteromeric nicotinic receptors of the Renshaw cell. J. Neurosci. Res. 2018, 38, 4943–4956. [Google Scholar] [CrossRef] [PubMed]
- Moroni, M.; Bermudez, I. Stoichiometry and pharmacology of two human α4β2 nicotinic receptor types. J. Mol. Neurosci. 2006, 30, 95–96. [Google Scholar] [CrossRef]
- Wu, X.; Tae, H.-S.; Huang, Y.-H.; Adams, D.J.; Craik, D.J.; Kaas, Q. Stoichiometry dependent inhibition of rat α3β4 nicotinic acetylcholine receptor by the ribbon isomer of α-conotoxin AuIB. Biochem. Pharmacol. 2018, 155, 288–297. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, S.; Benallegue, N.; Carbone, A.; Gasparri, F.; Vijayan, R.; Biggin, P.C.; Moroni, M.; Bermudez, I. Additional acetylcholine (ACh) binding site at α4/α4 interface of (α4β2) 2α4 nicotinic receptor influences agonist sensitivity. J. Biol. Chem. 2011, 286, 31043–31054. [Google Scholar] [CrossRef] [Green Version]
- Harpsøe, K.; Ahring, P.K.; Christensen, J.K.; Jensen, M.L.; Peters, D.; Balle, T. Unraveling the high- and low-sensitivity agonist responses of nicotinic acetylcholine receptors. J. Neurosci. Res. 2011, 31, 10759–10766. [Google Scholar] [CrossRef] [Green Version]
- Boffi, J.C.; Marcovich, I.; Gill-Thind, J.K.; Corradi, J.; Collins, T.; Lipovsek, M.M.; Moglie, M.; Plazas, P.V.; Craig, P.O.; Millar, N.S.; et al. Differential contribution of subunit interfaces to α9α10 nicotinic acetylcholine receptor function. Mol. Pharmacol. 2017, 91, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Indurthi, D.C.; Pera, E.; Kim, H.-L.; Chu, C.; McLeod, M.D.; McIntosh, J.M.; Absalom, N.L.; Chebib, M. Presence of multiple binding sites on α9α10 nAChR receptors alludes to stoichiometric-dependent action of the α-conotoxin, Vc1.1. Biochem. Pharmacol. 2014, 89, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Sambasivarao, S.V.; Roberts, J.; Bharadwaj, V.S.; Slingsby, J.G.; Rohleder, C.; Mallory, C.; Groome, J.R.; McDougal, O.M.; Maupin, C.M. Acetylcholine promotes binding of α-Conotoxin MII at α3β2 nicotinic acetylcholine receptors. ChemBioChem 2014, 15, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Dutertre, S.; Nicke, A.; Lewis, R.J. β2 subunit contribution to 4/7 α-conotoxin binding to the nicotinic acetylcholine receptor. J. Biol. Chem. 2005, 280, 30460–30468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiembob, D.L.; Roberts, R.L.; Luetje, C.W.; McIntosh, J.M. Determinants of α-conotoxin BuIA selectivity on the nicotinic acetylcholine receptor β subunit. Biochemistry 2006, 45, 11200–11207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhangsun, D.; Zhu, X.; Wu, Y.; Hu, Y.; Kaas, Q.; Craik, D.J.; McIntosh, J.M.; Luo, S. Key residues in the nicotinic acetylcholine receptor β2 subunit contribute to α-conotoxin LvIA binding. J. Biol. Chem. 2015, 290, 9855–9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, R.M., Jr.; Roh, S.-H.; Gharpure, A.; Morales-Perez, C.L.; Teng, J.; Hibbs, R.E. Structural principles of distinct assemblies of the human α4β2 nicotinic receptor. Nature 2018, 557, 261–265. [Google Scholar] [CrossRef]
- Di Cesare Mannelli, L.; Cinci, L.; Micheli, L.; Zanardelli, M.; Pacini, A.; McIntosh, J.M.; Ghelardini, C. α-conotoxin RgIA protects against the development of nerve injury-induced chronic pain and prevents both neuronal and glial derangement. Pain 2014, 155, 1986–1995. [Google Scholar] [CrossRef] [Green Version]
- Nicke, A.; Loughnan, M.L.; Millard, E.L.; Alewood, P.F.; Adams, D.J.; Daly, N.L.; Craik, D.J.; Lewis, R.J. Isolation, structure, and activity of GID, a novel α4/7-conotoxin with an extended N-terminal sequence. J. Biol. Chem. 2003, 278, 3137–3144. [Google Scholar] [CrossRef] [Green Version]
- Abraham, N.; Healy, M.; Ragnarsson, L.; Brust, A.; Alewood, P.F.; Lewis, R.J. Structural mechanisms for α-conotoxin activity at the human α3β4 nicotinic acetylcholine receptor. Sci. Rep. 2017, 7, 45466. [Google Scholar] [CrossRef] [Green Version]
- Loughnan, M.L.; Nicke, A.; Jones, A.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Chemical and functional identification and characterization of novel sulfated α-conotoxins from the cone snail Conus anemone. J. Med. Chem. 2004, 47, 1234–1241. [Google Scholar] [CrossRef]
- Armishaw, C.J. Synthetic α-conotoxin mutants as probes for studying nicotinic acetylcholine receptors and in the development of novel drug leads. Toxins 2010, 2, 1471. [Google Scholar] [CrossRef] [Green Version]
- Cartier, G.E.; Yoshikami, D.; Gray, W.R.; Luo, S.; Olivera, B.M.; McIntosh, J.M. A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J. Biol. Chem. 1996, 271, 7522–7528. [Google Scholar] [CrossRef] [Green Version]
- Dutertre, S.; Nicke, A.; Tyndall, J.D.A.; Lewis, R.J. Determination of α-conotoxin binding modes on neuronal nicotinic acetylcholine receptors. J. Mol. Recognit. 2004, 17, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Akondi, K.B.; Zhangsun, D.; Wu, Y.; Zhu, X.; Hu, Y.; Christensen, S.; Dowell, C.; Daly, N.L.; Craik, D.J.; et al. Atypical α-conotoxin LtIA from Conus Litteratus targets a novel microsite of the α3β2 nicotinic receptor. J. Biol. Chem. 2010, 285, 12355–12366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellisanti, C.D.; Yao, Y.; Stroud, J.C.; Wang, Z.-Z.; Chen, L. Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 Å resolution. Nat. Neurosci. 2007, 10, 953. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Ulens, C.; Büttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; et al. AChBP-targeted α-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar] [CrossRef]
- Celie, P.H.N.; Kasheverov, I.E.; Mordventisev, D.Y.; Hogg, R.C.; van Nierop, P.; van Elk, R.; van Rossum-Fikkert, S.E.; Zhmak, M.N.; Bertrand, D.; Tsetlin, V.; et al. Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an α-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 2005, 12, 582–588. [Google Scholar] [CrossRef]
- Grishin, A.A.; Cuny, H.; Hung, A.; Clark, R.J.; Brust, A.; Akondi, K.; Alewood, P.F.; Craik, D.J.; Adams, D.J. Identifying key amino acid residues that affect α-conotoxin AuIB inhibition of α3β4 nicotinic acetylcholine receptors. J. Biol. Chem. 2013, 288, 34428–34442. [Google Scholar] [CrossRef] [Green Version]
- Suresh, A.; Hung, A. Molecular simulation study of the unbinding of α-conotoxin [ϒ4E]GID at the α7 and α4β2 neuronal nicotinic acetylcholine receptors. J. Mol. Graph. Model. 2016, 70, 109–121. [Google Scholar] [CrossRef]
- Xu, M.; Zhu, X.; Yu, J.; Yu, J.; Luo, S.; Wang, X. The crystal structure of Ac-AChBP in complex with α-conotoxin LvIA reveals the mechanism of its selectivity towards different nAChR subtypes. Protein Cell 2017. [Google Scholar] [CrossRef]
- Ulens, C.; Hogg, R.C.; Celie, P.H.; Bertrand, D.; Tsetlin, V.; Smit, A.B.; Sixma, T.K. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc. Natl. Acad. Sci. USA 2006, 103, 3615–3620. [Google Scholar] [CrossRef] [Green Version]
- Zouridakis, M.; Papakyriakou, A.; Ivanov, I.A.; Kasheverov, I.E.; Tsetlin, V.; Tzartos, S.; Giastas, P. Crystal structure of the monomeric extracellular domain of α9 nicotinic receptor subunit in complex with α-conotoxin RgIA: Molecular dynamics insights into RgIA binding to α9α10 nicotinic receptors. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Bourne, Y.; Talley, T.T.; Hansen, S.B.; Palmer, T.; Marchot, P. Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J. 2005, 24, 1512–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.-Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Tjong, H.; Zhou, H.-X. Spontaneous conformational change and toxin binding in α7 acetylcholine receptor: Insight into channel activation and inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 8280–8285. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal α7-nAChR by α-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef] [Green Version]
- Sahu, B.S.; Obbineni, J.M.; Sahu, G.; Singh, P.K.; Sonawane, P.J.; Sasi, B.K.; Allu, P.K.R.; Maji, S.K.; Bera, A.K.; Senapati, S.; et al. Molecular interactions of the physiological anti-hypertensive peptide catestatin with the neuronal nicotinic acetylcholine receptor. J. Cell Sci. 2012, 125, 2323–2337. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.-W.; Liao, M.-L.; Meng, X.-L.; Somero, G.N. Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc. Natl. Acad. Sci. USA 2018, 115, 1274–1279. [Google Scholar] [CrossRef] [Green Version]
- Meher, B.R.; Wang, Y. Exploring the drug resistance of V32I and M46L mutant HIV-1 protease to inhibitor TMC114: Flap dynamics and binding mechanism. J. Mol. Graph. Model. 2015, 56, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Lerner, M.; Carlson, H. APBS Plugin for PyMOL. University of Michigan. Ann. Arbor. 2006, 7, 14. [Google Scholar]
- Dilip, A.; Lešnik, S.; Štular, T.; Janežič, D.; Konc, J. Ligand-based virtual screening interface between PyMOL and LiSiCA. J. Cheminform. 2016, 8, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [PubMed]
- Talley, T.T.; Olivera, B.M.; Han, K.-H.; Christensen, S.B.; Dowell, C.; Tsigelny, I.; Ho, K.-Y.; Taylor, P.; McIntosh, J.M. α-Conotoxin OmIA is a potent ligand for the acetylcholine-binding protein as well as α3β2 and α7 nicotinic acetylcholine receptors. J. Biol. Chem. 2006, 281, 24678–24686. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System. Available online: http://www.pymol.org (accessed on 20 June 2020).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kouvatsos, N.; Giastas, P.; Chroni-Tzartou, D.; Poulopoulou, C.; Tzartos, S.J. Crystal structure of a human neuronal nAChR extracellular domain in pentameric assembly: Ligand-bound α2 homopentamer. Proc. Natl. Acad. Sci. USA 2016, 113, 9635–9640. [Google Scholar] [CrossRef] [Green Version]
- Henchman, R.H.; Wang, H.-L.; Sine, S.M.; Taylor, P.; McCammon, A.J. Ligand-induced conformational change in the α7 nicotinic receptor ligand binding domain. Biophys. J. 2005, 88, 2564–2576. [Google Scholar] [CrossRef] [Green Version]
- Gharpure, A.; Teng, J.; Zhuang, Y.; Noviello, C.M.; Walsh, R.M., Jr.; Cabuco, R.; Howard, R.J.; Zaveri, N.T.; Lindahl, E.; Hibbs, R.E. Agonist selectivity and ion permeation in the α3β4 ganglionic nicotinic receptor. Neuron 2019, 104, 501–511. [Google Scholar] [CrossRef]
- Wen, J.; Hung, A. Effects of C-terminal carboxylation on α-conotoxin LsIA interactions with human α7 nicotinic acetylcholine receptor: Molecular simulation studies. Mar. Drugs 2019, 17, 206. [Google Scholar] [CrossRef] [Green Version]
- Kompella, S.N.; Cuny, H.; Hung, A.; Adams, D.J. Molecular basis for differential sensitivity of α-conotoxin RegIIA at rat and human neuronal nicotinic acetylcholine receptors. Mol. Pharmacol. 2015, 88, 993–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, R.C.; Miranda, L.P.; Craik, D.J.; Lewis, R.J.; Alewood, P.F.; Adams, D.J. Single amino acid substitutions in α-conotoxin pnia shift selectivity for subtypes of the mammalian neuronal nicotinic acetylcholine receptor. J. Biol. Chem. 1999, 274, 36559–36564. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Nguyen, T.A.; Cartier, G.E.; Olivera, B.M.; Yoshikami, D.; McIntosh, J.M. Single-residue alteration in α-conotoxin pnia switches its nachr subtype selectivity. Biochemistry 1999, 38, 14542–14548. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Plazas, P.V.; Watkins, M.; Gomez-Casati, M.E.; Olivera, B.M.; Elgoyhen, A.B. A novel α-conotoxin, PeIA, cloned from conus pergrandis, discriminates between rat α9α10 and α7 nicotinic cholinergic receptors. J. Biol. Chem. 2005, 280, 30107–30112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, M.S.; Zwart, R.; Ursu, D.; Jensen, M.M.; Pinborg, L.H.; Gilmour, G.; Wu, J.; Sher, E.; Mikkelsen, J.D. α7 and β2 nicotinic acetylcholine receptor subunits form heteromeric receptor complexes that are expressed in the human cortex and display distinct pharmacological properties. PLoS ONE 2015, 10, e0130572. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Cuny, H.; Yu, R.; Tae, H.-S.; Kompella, S.N.; Adams, D.J. α-Conotoxins active at α3-containing nicotinic acetylcholine receptors and their molecular determinants for selective inhibition. Br. J. Pharmacol. 2017, 175, 1855–1868. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.-G.; Jiang, N.; Huang, Y.-B.; Ma, X.-K.; Brek Eaton, J.; Gao, M.; Chang, Y.-C.; Lukas, R.J.; Whiteaker, P.; Neisewander, J.; et al. Cocaine potently blocks neuronal α3β4 nicotinic acetylcholine receptors in SH-SY5Y cells. Acta Pharmacol. Sin. 2020, 41, 163–172. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K.S. Bringing molecular dynamics simulation data into view. Trends Biochem. Sci. 2019, 44, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Leffler, A.E.; Kuryatov, A.; Zebroski, H.A.; Powell, S.R.; Filipenko, P.; Hussein, A.K.; Gorson, J.; Heizmann, A.; Lyskov, S.; Tsien, R.W.; et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. Proc. Natl. Acad. Sci. USA 2017, 114, E8100–E8109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, A.L.; Moroni, M.; Groot-Kormelink, P.J.; Bermudez, I. Pentameric concatenated (α4)2(β2)3 and (α4)3(β2)2 nicotinic acetylcholine receptors: Subunit arrangement determines functional expression. Br. J. Pharmacol. 2009, 156, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemecz, A.; Taylor, P. Creating an α7 nicotinic acetylcholine recognition domain from the acetylcholine-binding protein: Crystallographic and ligand selectivity analyses. J. Biol. Chem. 2011, 286, 42555–42565. [Google Scholar] [CrossRef] [Green Version]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human α4β2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.G.; Santos, R.N.D.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Newcombe, J.; Chatzidaki, A.; Sheppard, T.D.; Topf, M.; Millar, N.S. Diversity of nicotinic acetylcholine receptor positive allosteric modulators revealed by mutagenesis and a revised structural model. Mol. Pharmacol. 2018, 93, 128–140. [Google Scholar] [CrossRef]
- Gonzalez-Gutierrez, J.P.; Hodar, M.; Viscarra, F.; Paillali, P.; Guerra-Díaz, N.; Pessoa-Mahana, H.; Hernández-Morantes, J.J.; Pérez-Sánchez, H.; Bermúdez, I.; Reyes-Parada, M.; et al. Minimal structural changes determine full and partial nicotinic receptor agonist activity for nicotine analogues. Molecules 2019, 24, 2684. [Google Scholar] [CrossRef] [Green Version]
- Norleans, J.; Wang, J.; Kuryatov, A.; Leffler, A.; Doebelin, C.; Kamenecka, T.M.; Lindstrom, J. Discovery of an intrasubunit nicotinic acetylcholine receptor binding site for the positive allosteric modulator Br-PBTC. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.; Adams, D.J.; Hung, A. Interactions of the α3β2 Nicotinic Acetylcholine Receptor Interfaces with α-Conotoxin LsIA and its Carboxylated C-terminus Analogue: Molecular Dynamics Simulations. Mar. Drugs 2020, 18, 349. https://doi.org/10.3390/md18070349
Wen J, Adams DJ, Hung A. Interactions of the α3β2 Nicotinic Acetylcholine Receptor Interfaces with α-Conotoxin LsIA and its Carboxylated C-terminus Analogue: Molecular Dynamics Simulations. Marine Drugs. 2020; 18(7):349. https://doi.org/10.3390/md18070349
Chicago/Turabian StyleWen, Jierong, David J. Adams, and Andrew Hung. 2020. "Interactions of the α3β2 Nicotinic Acetylcholine Receptor Interfaces with α-Conotoxin LsIA and its Carboxylated C-terminus Analogue: Molecular Dynamics Simulations" Marine Drugs 18, no. 7: 349. https://doi.org/10.3390/md18070349
APA StyleWen, J., Adams, D. J., & Hung, A. (2020). Interactions of the α3β2 Nicotinic Acetylcholine Receptor Interfaces with α-Conotoxin LsIA and its Carboxylated C-terminus Analogue: Molecular Dynamics Simulations. Marine Drugs, 18(7), 349. https://doi.org/10.3390/md18070349