1. Introduction
Marine-derived fungi are a known source of diketopiperazine-type alkaloids. Indole diketopiperazine alkaloids are characterized by certain products of condensation, including a complete tryptophan and a second amino acid such as L-tryptophan, L-proline, L-phenylalanine, L-histidine, or L-leucine, forming an indole diketopiperazine unit [
1]. Among the cyclo-(Try-Pro) prenylated alkaloids, there is a small group containing the rare 6/5/8/6/5 pentacyclic ring system, which includes (+)-deoxyisoaustamide [
2], deoxydihydroisoaustamide [
3], 16β-hydroxy-17β-methoxy-deoxydihydroisoaustamide [
3], carneamides B and C [
4], and versicamides A–F [
5].
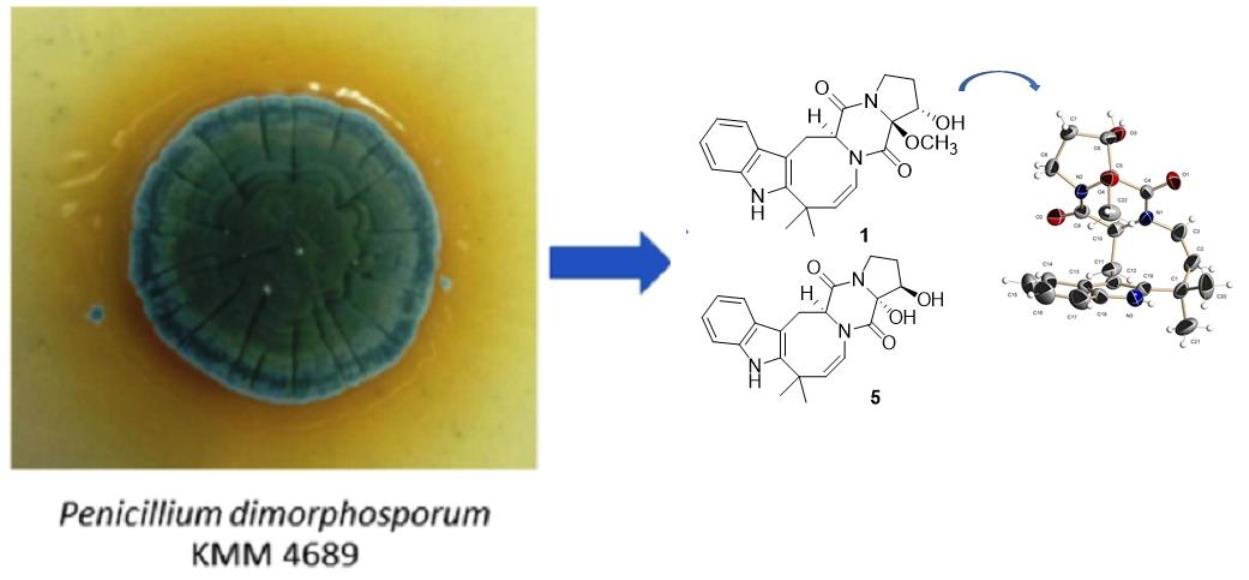
In our search for fungal secondary metabolites possessing novel chemical structures and/or biological activity, we have investigated the strain Penicillium dimorphosporum KMM 4689.
Fungi of the genus
Penicillium have a special position in nature and human life. The marine environment is no exception. These fungi were found in the extremely salty environment of the Dead Sea [
6]. The species
Penicillium dimorphosporum H.J. Swart was described upon the type strain, isolated from a mangrove swamp soil near Westernport Bay near Melbourne, Victoria, Australia, by H.J. Swart, in 1969. Until recently, the species
P. dimorphosporum assigned to the
P. restrictum series, the Monoverticillata section on account of the slow growth on most media, the short stipes arising from aerial hyphae, the presence of strongly echinulate globose conidia and monoverticillata conidiophores [
7]. Polyphasic taxonomy methods have shown that this species belongs to subgenus
Aspergilloides, section
Exilicaulis, series
Erubescentia [
8]. The strain
P. dimorphosporum KMM 4689 (Collection of Marine Microorganisms, RAS, WDCM #644) was isolated from an unidentified soft coral at South China Sea.
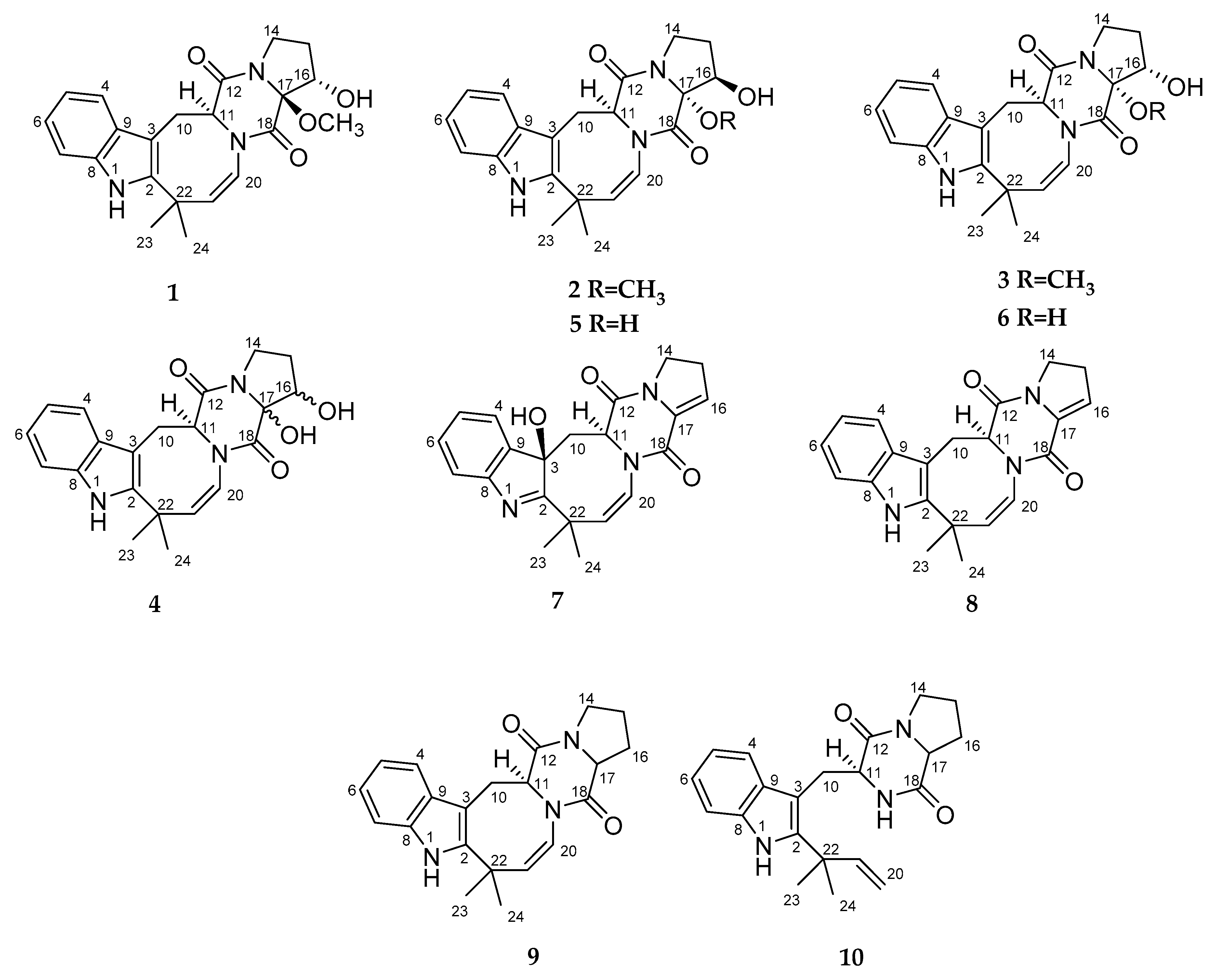
Herein, we report the isolation, structure determination, and biological assay results of the new prenylated indole alkaloids belonging to deoxyisoaustamide family (
1–
7), the known (+)-deoxyisoaustamide (
8) [
2,
9], deoxydihydroisoaustamide (
9) [
3] and desoxybrevianamide E (
10) [
2] produced by this fungus.
Diketopiperazine alkaloids have shown various biological activities, including cytotoxic [
10,
11,
12,
13], antiphytopathogenic [
14], anticancer [
15] and neuroprotective effects [
16]. Earlier it was reported that cryptoechinulin B and other echinuline-related compounds increased the viability of paraquat (PQ)-treated Neuro-2a cells, suppressing the upregulation of intracellular reactive oxygen species [
17]. PQ is a bipyridyl quaternary ammonium herbicide and PQ poisoning is one of the leading intoxications worldwide. PQ-induced toxicity has been associated with the induction of oxidative and endoplasmic reticulum stresses, apoptosis, mitochondrial damage, and inflammation, mediated by deregulation of various protein/signaling pathways [
18]. These pathological processes result in decreased cell viability and, finally, cell death. In this work, we evaluated the protective effects of the isolated compounds
1–
10 against the acute toxicity of PQ in murine neuroblastoma Neuro-2a cells which are commonly used for neuroprotective investigations [
19].
2. Results and Discussion
The fungus was cultured for 21 days on solid rice medium. The EtOAc extract of the mycelium was purified by a combination of a Si gel and an ODS-A column chromatography, and a reversed phase HPLC to yield compounds
1–
10 (
Figure 1).
The molecular formula of
1 was determined as C
22H
25N
3O
4 from the HRESIMS peak at m/z 394.1771 [M − H]
− and was in accordance with the
13C NMR data. The
1H and
13C NMR (
Table 1 and
Table 2;
Figures S21–S27), DEPT and HSQC spectra showed the presence of an NH proton (δ
H 10.69), a hydroxy proton (δ
H 5.39), three methyl groups (δ
H 1.34, 1.46, 1.53, δ
C 32.3, 47.6, 28.3), three sp
3 methylenes (δ
H 3.26, 3.60, 1.50, 1.83, 2.69, 3.94, δ
C 26.0, 28.8 and 42.6), two sp
3 methines (δ
H 3.90, 4.20, δ
C 74.2, 58.6), six olefinic methines (δ
H 5.81 (2), 6.90, 6.95, 7.19, 7.32, δ
C 140.6, 121.9, 118.6, 120.5, 110.4, 117.4), four sp
2(δ
C 102.8, 128.3, 134.9 and 141.1) and a sp
3(δ
C 37.3) quaternary carbons, an oxygenated quaternary carbon (δ
C 93.9) and two amide carbonyls (δ
C 162.1 and 166.3).
The
1H-
13C HMBC correlations (
Figure S26) from H-1 to C-2 (
δC 141.1), C-3 (
δC 102.8), C-8 (
δC 134.9) and C-9 (
δC 128.3), from H-4 to C-3, C-6 (
δC 120.5), C-8, С-9, and from H-7 to C-5 (
δC 118.6), and C-9 together with the
1H–
1H COSY correlations of H-4/H-5/H-6/H-7 indicated the presence of a disubstituted indole core in
1. The characteristic NMR data and HMBC correlations from H-11 (δ
H 4.20)to C-12 (
δC 166.3) and C-18 (
δC 162.1) suggested the presence of diketopiperazine ring in
1. The correlations observed in the COSY and HSQC spectra and long-range correlations from H-14α (δ
H 3.94) to C-12 and C-15 (δ
C 28.8), from H-16 (δ
H 3.90) to C-14 (
δC 42.6), C-15 and C-17 (δ
C 93.9), and from 17-OCH
3(δ
H 1.46, δ
C 47.6) to C-17 indicated that the oxidized proline moiety forms a diketopiperazine ring. The HMBC correlations from H
2-10 (δ
H 3.26, 3.60) to C-2, C-3, C-9, C-11 (δ
C 58.6) and C-12, from H-21 (δ
H 5.81) to C-2, C-20 (
δC 121.9), C-22 (
δC 37.3) and C-24 (
δC 32.3), and COSY correlations H-11/H
2-10 and H-20/H21 indicated the formation of a closed cycle between the indole and diketopiperazine parts in
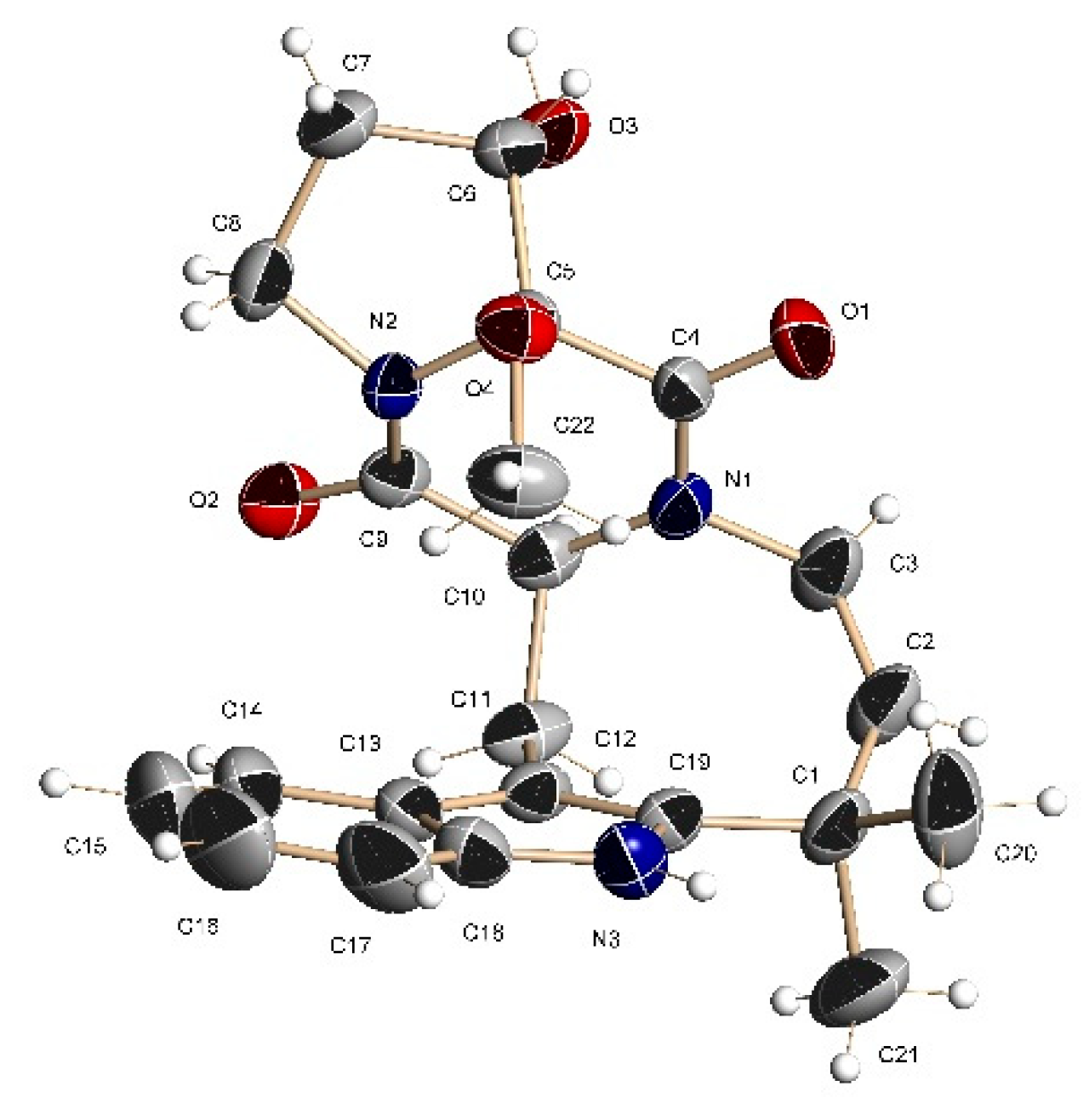
1. The structure and relative configuration of
1 were further confirmed by X-ray crystallographic analysis carried out for a single crystal obtained by recrystallization from acetonitrile-water (
Figure 2 and
Tables S1–S3).
The upfield shift of the 17-OCH3 signal (δH 1.46) in 1H NMR spectrum of 1 is thought to be due to the magnetic anisotropy of the coplanar C-18 carbonyl group.
The configuration of the chiral center C-11 in
1 as
S was established based on obvious biogenetic relationships with (+)-deoxyisoaustamide (
8), the optical rotation value of which was in full agreement with the literature data [
2,
9]. Thus, the absolute configuration of
1 was established as 11
S,16
S,17
S. Compound
1 was named 16α-hydroxy-17β-methoxy-deoxydihydroisoaustamide.
The HRESIMS of
2 and
3 showed the quasimolecular ions at m/z 394.1773 [M - H]
− and m/z 394.1771 [M - H]
−, respectively. These data, coupled with
13C NMR spectral data (DEPT), established the molecular formula of
2 and
3 as C
22H
25N
3O
4 for both. A close inspection of the
1H and
13C NMR data (
Table 1 and
Table 2 and
Figures S28–S41) of
2 and
3 by DEPT and HSQC revealed the presence of disubstituted indole and oxidized proline moieties, diketopiperazine and azocane rings. The main COSY and HMBC correlations showed in
Figure 3 indicate that compounds
2 and
3 have the same coplanar structure as
1.
The absolute configurations of the chiral centers in
2 were defined based on NOESY (
Figure S34) correlations 17-OCH
3(δ
H 3.12)/H-11 (δ
H 4.56), H-20 (δ
H 6.02), H-15α (δ
H 1.90), H-16 (δ
H 4.01); 16-OH (δ
H 4.72)/H-15β (δ
H 1.39), H
3-23(δ
H 1.45) and H-11 /H-20, and biogenetic considerations as 11
S,16
R,17
R. Compound
2 was named 16β-hydroxy-17α-methoxy-deoxydihydroisoaustamide.
NOE cross-peaks (
Figure S41) 17-OCH
3(δ
H 3.12)/H-11 (δ
H 4.30), H-20 (δ
H 5.78) and the upfield shift and magnitude of the coupling constant of the H-16 signal (
δH 2.01, t, 8.8) suggested the α-orientation of methoxy group at C-17 and α-orientation of hydroxy group at C-16 in
3. The absolute configurations of the chiral centers in
3 were defined as 11
S,16
S,17
R based on biogenetic consideration. Compound
3 was named 16α-hydroxy-17α-methoxy-deoxydihydroisoaustamide.
The HRESIMS of 4, 5 and 6 showed the quasimolecular ions at m/z 404.1575 [M + Na]+, 404.1583 [M + Na]+ and 404.1581 [M + Na]+, respectively. These data, coupled with 13C NMR spectral data (DEPT), established the molecular formula of all compounds as C21H23N3O4.
The general features of
1H and
13C NMR spectra (
Table 1 and
Table 2;
Figures S42–S49) of
4 showed a close similarity of the proton and carbon chemical shifts to the ones for
1, with the exception of the C-17 and C-18 carbon signals, and the absence of signals from the methoxy group. The molecular mass difference of 14 mass units between
1 and
4 and
1H-
13C HMBC correlations from 17-OH (δ
H 4.21) to C-16 (δ
C 74.3) and C-17 (δ
C 88.5) and correlation 17-OH/N-13 (δ
N 142.3) in
1H-
15N GHMBC spectrum (
Figure S48) indicate the presence of a hydroxy group at C-17 in
4.
NOE cross-peak (
Figure S49) 17-OH (δ
H 4.21)/H-16 (δ
H 3.94) indicated that the hydroxy group at C-17 and the proton at C-16 are on the same side of the proline ring in
4. The data of NOESY and CD (
Figure S4) spectra did not allow to unambiguously establish the configurations of the C-16 and C-17 asymmetric centers in compound
4. The configuration of the chiral center C-11 in
4 as
S was established on the basis of biogenetic relationships with (+)-deoxyisoaustamide (
8). Compound
4 was named 16,17-dihydroxy-deoxydihydroisoaustamide.
The planar structures of the compounds
5 and
6 were found by extensive NMR spectroscopy (
1H,
13C, DEPT, HSQC and HMBC)(
Table 1 and
Table 2 and
Figures S50–S63) to be the same as those of
4. The general features of
1H and
13C NMR spectra of
5 and
6 closely resembled those of
2 and
3, respectively.
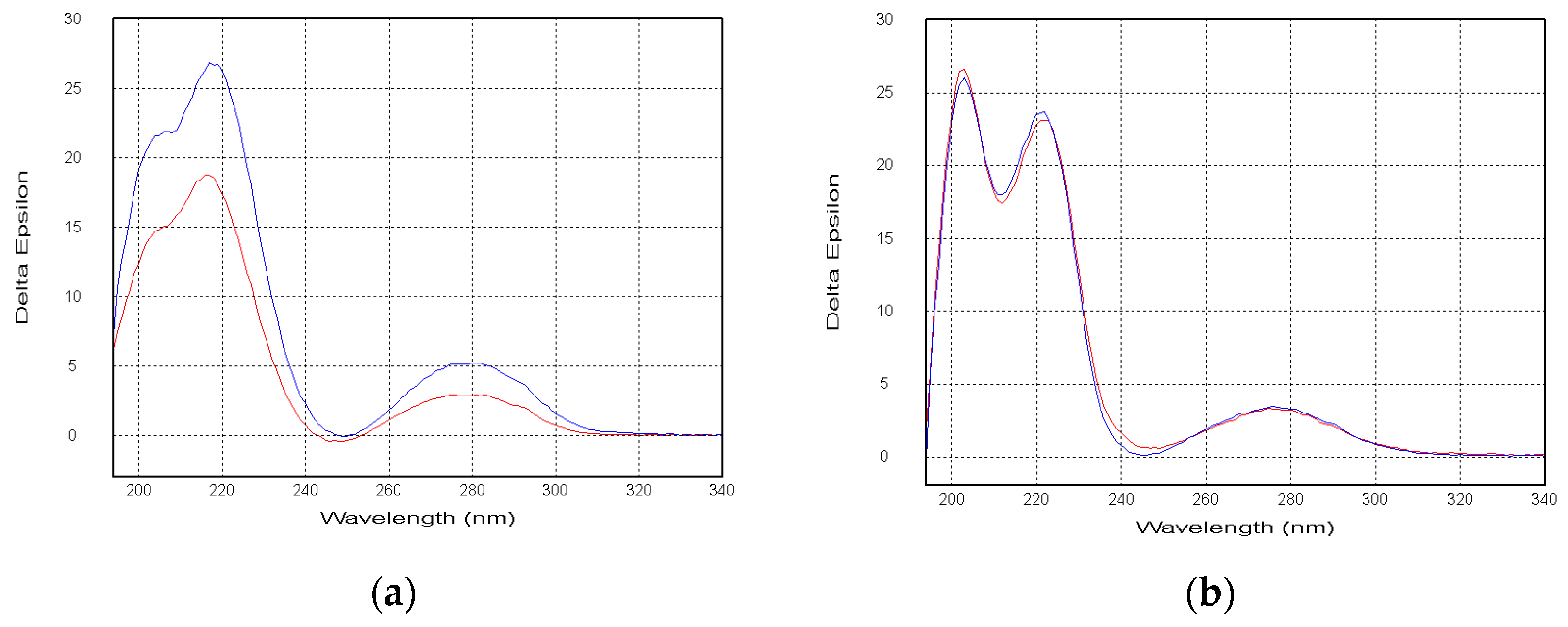
Compounds
5 and
6 exhibited the nearly identical CD spectra to those of
2 and
3, respectively (
Figure 4). Thus, the absolute configurations of the
5 and
6 were established as 11
S,16
R,17
R and 11
S16
S17
R, respectively. Compounds
5 and
6 were named 16β,17α-dihydroxy-deoxydihydroisoaustamide and 16α,17α-dihydroxy-deoxydihydroisoaustamide, respectively.
The HRESIMS of
7 showed the quasimolecular ion at m/z 362.1511 [M - H]
−. These data, coupled with
13C NMR spectral data (DEPT), established the molecular formula of
7 as C
21H
21N
3O
3. The
1H and
13C NMR (
Table 1 and
Table 2 and
Figures S64–S70), DEPT and HSQC spectra showed the presence of a hydroxy proton (δ
H 5.78), two methyl groups (δ
H 1.36, 1.47, δ
C 28.3, 26.2), three sp
3 methylenes (δ
C 27.2, 38.7 and 45.2), a methine (δ
H 4.08, δ
C 58.5), seven olefinic methines (δ
H 5.61, 5.66, 5.84, 7.14, 7.20, 7.25, 7.27, δ
C 121.3, 117.7, 141.8, 125.6, 124.3, 120.5, 129.2), six sp
2(δ
C 132.6, 140.2, 153.2, 154.5, 160.5 and 187.5) and one sp
3(δ
C 41.8) quaternary carbon and one oxygenated sp
3 quaternary carbon (δ
C 84.7).
The
1H-
13C HMBC (
Figure S69) correlations from H-16 (δ
H 5.66) to C-14 (
δC 45.2), C-15 (
δC 27.2), C-17 (
δC 132.6) and C-18 (
δC 154.5), from H-11 (δ
H 4.08) to C-12 (
δC 160.5) and C-18 and
1H–
1H COSY correlations H
2-14/H
2-15/H-16 revealed the presence of an unsaturated proline moiety and a diketopiperazine ring in
7. The long-range correlations from 3-OH to C-2 (
δC 187.5), C-3 (
δC 84.7), C-9 (
δC 140.2) and C-10 (
δC 38.7), from H-4 (δ
H 7.20) to C-3, C-6 (
δC 129.2), C-8 (
δC 153.2), from H-7 (δ
H 7.25) to C-5 (
δC 125.6), C-9 and from H-21 to C-2, C-20 (
δC 121.3), C-22 (
δC 41.8), C-23 (
δC 26.2), С-24 (
δC 28.3) and C-18 together with
1H–
1H COSY correlations of H-4/H-5/H-6/H-7, H
2-10/H-11 and H-20/H-21 indicated the location of the hydroxy group at C-3 and established the structures of disubstituted indole core and azocane ring in
7.
The absolute configurations of the chiral centers in
7 were defined based on NOESY correlations 3-OH (δ
H 5.78)/H-21 (δ
H 5.84), H
3-23 (δ
H 1.47); H
3-23 (δ
H 1.47)/H-10β (δ
H 2.82), H-21; H-11 (δ
H 4.08)/H-20 (δ
H 5.61)(
Figure S70) and biogenetic considerations as 3
R,11
S. Compound
7 was named 3β-hydroxy-deoxyisoaustamide.
Of note, we have tried to determine the absolute configuration of the asymmetric centers at C-16 for compounds 1, 2, 4 and 5 using the modified Mosher’s method. Esterification of compounds with (S)-MTPA chloride at the C-16 hydroxy groups produced (R)-MTPA esters. However, the attempts to obtain (S)-MTPA esters from (R)-MTPA-Cl were unsuccessful.
The structures of known compounds (+)-deoxyisoaustamide (
8) [
2], deoxydihydroisoaustamide (
9) [
3] and desoxybrevianamide E (
10) [
2] were determined based on HRESIMS and NMR data as well as comparison with literature data.
We further examined potential protective effects of the isolated compounds
1–
10 against the acute toxicity of paraquat (PQ) in murine neuroblastoma Neuro-2a cells, which is a well-established model for neuroprotective activity studies [
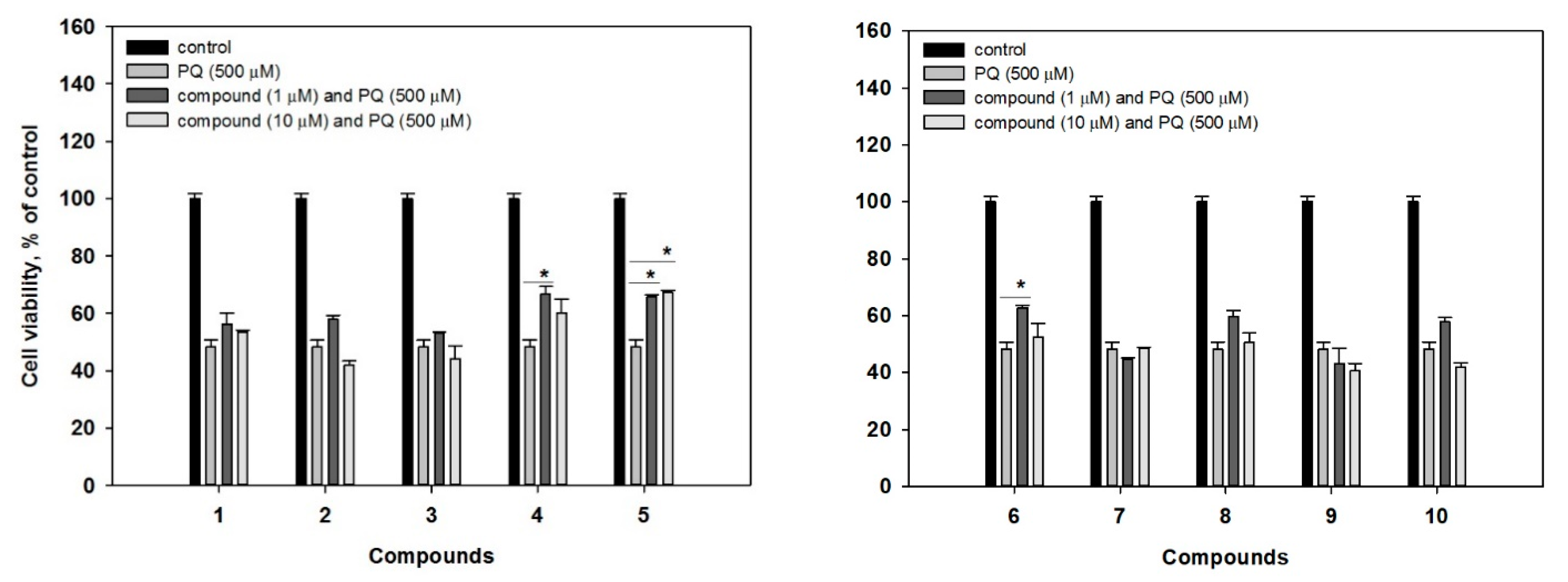
16]. In our experiments, treatment of Neuro-2a cells with 500 μM of PQ induced a decrease of cell viability by 51.8% (
Figure 5). Whereas co-treatment with 1 μM of
4 and
6 could increase a viability of PQ-treated cells by 38.6% and 30.3%, respectively. Compound
5 increased a viability of the cells by 36.5% and 39.4% at concentrations at 1 μM and 10 μM, respectively. At the same time, the investigated compounds were non-cytotoxic to Neuro-2a cells, as well as to human prostate epithelial PNT-2 cells (compounds
1,
2,
4,
8–
10,
Figure S71), used as non-cancer human cell lines to demonstrate the lack of cytotoxic activity to human cells (IC
50s > 100 μM, data not shown).
This is the first report on neuroprotective activity of deoxyisoaustamides. Earlier, it was reported that (+)-deoxyisoaustamide (
8) and deoxydihydroisoaustamide (
9) were studied in glutamate and t-BHP-induced cytotoxicity assays [
3]. In addition, inhibitory effects of the metabolites on nitrite production of LPS-stimulated RAW264.7 and BV2 cells were evaluated. These compounds showed no significant effects on cytoprotection or nitrite inhibition [
3]. In our experiments, new deoxyisoaustamides
4–
6 revealed a statistical increase of PQ-treated Neuro-2a cell viability.
The analyses of structure-activity relationships suggest a key role of both hydroxy groups at C-16 and C-17 for the neuroprotective activity of investigated deoxyisoaustamide alkaloids. Indeed, the presence of the mentioned feature in structures 4–6 correlates with a higher activity of the compounds.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 343 polarimeter (Perkin Elmer, Waltham, MA, USA). UV spectra were recorded on a Shimadzu UV-1601PC spectrometer (Shimadzu Corporation, Kyoto, Japan) in methanol. CD spectra were measured with a Chirascan-Plus CD spectrometer (Leatherhead, UK) in methanol. NMR spectra were recorded in CD3OD and DMSO-d6, on a Bruker DPX-500 (Bruker BioSpin GmbH, Rheinstetten, Germany) and a Bruker DRX-700 (Bruker BioSpin GmbH, Rheinstetten, Germany) spectrometer, using TMS as an internal standard. HRESIMS spectra were measured on a Maxis impact mass spectrometer (Bruker Daltonics GmbH, Rheinstetten, Germany). Microscopic examination and photography of fungal cultures were performed with Olympus CX41 microscope fitted with (equipped with) an Olympus SC30 digital camera. Detailed examination of ornamentation of the fungal conidia was performed by scanning electron microscopy (SEM) EVO 40.
Low-pressure liquid column chromatography was performed using Si gel (60/100 μm, Imid Ltd., Russia) and Gel ODS-A (12 nm, S—75 um, YMC Co., Ishikawa, Japan). Plates precoated with Si gel (5–17 μm, 4.5 × 6.0 cm, Imid Ltd., Russia) and Si gel 60 RP-18 F254S (20 × 20 cm, Merck KGaA, Germany) were used for thin-layer chromatography. Preparative HPLC was carried out on an Agilent 1100 chromatograph (Agilent Technologies, USA) using a YMC ODS-AM (YMC Co., Ishikawa, Japan)(5 µm, 10 × 250 mm), YMC ODS-A (YMC Co., Ishikawa, Japan)(5 µm, 4.6 × 250 mm) and Supelco Discovery C-18 (5 μm, 250 × 4.6 mm) columns with an Agilent 1100 refractometer (Agilent Technologies, Santa Clara, CA, USA).
3.2. Fungal Strain
Soft coral samples were collected using a Van Veen bottom grab at various points in the South China Sea. The samples were collected in individual sterile plastic bags and stored frozen (−18 °C) before use. Isolation of fungal colonies was made by the plating methods using Tubaki agar. Isolation of pure cultures was made by transferring of inoculums to the slant wort agar. Macroscopical characters were studied on the agar media Czapek yeast extract agar (CYA), yeast extract agar (YES) and malt extract agar (MEA). Preparation of these media is detailed by Frisvad and Samson (2004). The strains were inoculated at three points on 9-cm Petri dishes and incubated for 7 d at 25 °C in darkness. In addition, inoculated CYA plates were incubated for 7 d at 37 °C according to the recommendations of Pitt. Color names are from Ridgway. To determine the degree of halotolerance, the strains were grown on MEA supplemented with 5, 10, 15 and 20% NaCl at 25 °C for 7 days.
The cultures used for the molecular studies were grown on malt extract agar under 25 °C for 7 d. DNA extraction was performed by DNA kit (DNA-TechnologyLtd., Moscow, Russia) according to the manufacturer’s instructions. Fragments containing the ITS regions were amplified using primers ITS1 and ITS4 [
20].Newly generated sequences were compared to the available sequences of the National Center for Biotechnology Informatic (NCBI) by using BLAST. BLAST search results indicated that the sequence was 99% identical with the sequenceof
Penicillium dimorphosporum strain CBS 456.70(GenBank accession numberMH859796.1). The sequence was deposited in GenBank nucleotide sequence database under MW325972 code.
3.3. Cultivation of Fungus
The fungus was cultured at 22 °C for three weeks in 60 × 500 mL Erlenmeyer flasks, each containing rice (20.0 g), yeast extract (20.0 mg), KH2PO4(10 mg) and natural sea water from the Marine Experimental Station of PIBOC, Troitsa (Trinity) Bay, Sea of Japan (40 mL).
3.4. Extraction and Isolation
At the end of the incubation period, the mycelia and medium were homogenized and extracted with EtOAc (1 L). The obtained extract was concentrated to dryness. The residue (2.8 g) was dissolved in H2O−EtOH (4:1)(100 mL) and was extracted with n-hexane (0.2 L × 3) and EtOAc (0.2 L × 3). After evaporation of the EtOAc layer, the residual material (1.5 g) was passed over a silica column (3 × 14 cm), which was eluted first with n-hexane (200 mL) followed by a step gradient from 5% to 50% EtOAc in n-hexane (total volume 20 L). Fractions of 250 mL were collected and combined on the basis of TLC (Si gel, toluene–isopropanol 6:1 and 3:1, v/v).
The n-hexane-EtOAc fraction (70:30, 250 mg) was separated on a Gel ODS-A column (1.5 × 8 cm), which was eluted by a step gradient from 40% to 80% CH3CN in H2O (total volume 1 L) to yield subfractions I and II. Subfraction I (60% CH3CN, 150 mg) was purified by RP HPLC on a YMC ODS-AM column eluting with CH3CN-H2O (50:50) to yield subfraction I.I (50 mg) and compounds 8(17 mg) and 10(40 mg). Subfraction I.I was purified and separated by RP HPLC on a YMC ODS-A column eluting at first with CH3CN-H2O (45:55) to yield 1(19 mg) and 2(5 mg) and then with CH3CN-H2O (40:60) to yield 3(7.5 mg) and 9(5.5 mg). Subfraction II (40% CH3CN, 150 mg) was purified by RP HPLC on a YMC ODS-AM column eluting with CH3CN-H2O (35:65) and then on a Supelco Discovery C-18 column eluting at first with CH3CN-H2O (35:65) to yield 4 (8 mg), 5(5.2 mg), 6(3.5 mg) and then with CH3CN-H2O (30:70) to yield 7(6 mg).
3.5. Spectral Data
16α-hydroxy-17β-methoxy-deoxydihydroisoaustamide (
1): colorless crystal; mp 309–310 °C; [α]
D20 + 171.4 (
c 0.07 CH
3OH); CD (
c 3.0 × 10
−4, CH
3OH), λ
max(∆ε) 206 (+20.77), 213 (+19.90), 276 (+5.32) nm, see
Supplementary Figure S1; UV (CH
3OH)
λmax(log
ε) 283 (3.83), 223 (4.48) and 198 (4.46) nm, see
Supplementary Figure S11;
1H and
13C NMR data, see
Table 1 and
Table 2,
Supplementary Figures S21–S27; HRESIMS m/z 394.1771 [M − H]
− (calcd. for C
22H
24N
3O
4, 394.1772, Δ +0.3 ppm), 418.1733 [M + Na]
+ (calcd. for C
22H
25N
3O
4Na, 418.1737, Δ +1.0 ppm).
16β-hydroxy-17α-methoxy-deoxydihydroisoaustamide (
2): amorphous solids; [α]
D20 +211.1 (
c 0.09 CH
3OH); CD (
c 3.8 × 10
−4, CH
3OH), λ
max(∆ε) 207 (+21.88), 218 (+26.66), 280 (+5.17) nm, see
Supplementary Figure S2; UV (CH
3OH)
λmax(log
ε) 283 (3.79), 224 (4.43) and 198 (4.40) nm, see
Supplementary Figure S12;
1H and
13C NMR data, see
Table 1 and
Table 2,
Supplementary Figures S28–S34; HRESIMS m/z 394.1773 [M − H]
− (calcd. for C
22H
24N
3O
4, 394.1772, Δ −0.1 ppm), 418.1733 [M + Na]
+(calcd. for C
22H
25N
3O
4Na, 418.1737, Δ +1.0 ppm).
16α-hydroxy-17α-methoxy-deoxydihydroisoaustamide (
3): amorphous solids; [α]
D20 +198.0 (
c 0.1 CH
3OH); CD (
c 4.2 × 10
−4, CH
3OH), λ
max(∆ε) 203 (+26.54), 222 (+23.02), 275 (+3.29) nm, see
Supplementary Figure S3; UV (CH
3OH)
λmax(log
ε) 283 (3.65), 224 (4.33) and 198 (4.23) nm, see
Supplementary Figure S13;
1H and
13C NMR data, see
Table 1 and
Table 2,
Supplementary Figures S35–S41; HRESIMS m/z 394.1771 [M − H]− (calcd. for C
22H
24N
3O
4, 394.1772, Δ +0.4 ppm), 418.1733 [M + Na]
+(calcd. for C
22H
25N
3O
4Na, 418.1737, Δ +0.5 ppm).
16,17-dihydroxy-deoxydihydroisoaustamide (
4): amorphous solids; [α]
D20 +153.7 (
c 0.08 CH
3OH); CD (
c 4.4 × 10
−4, CH
3OH), λ
max(∆ε) 200 (+27.59), 223 (+14.08), 274 (+3.26) nm, see
Supplementary Figure S4; UV (CH
3OH)
λmax(log
ε) 282 (3.79), 223 (4.41) and 198 (4.35) nm, see
Supplementary Figure S14;
1H and
13C NMR data, see
Table 1 and
Table 2,
Supplementary Figures S42–S49; HRESIMS m/z 380.1617 [M − H]
− (calcd. for C
21H
22N
3O
4, 380.1616, Δ −0.2 ppm), 404.1575 [M + Na]
+(calcd. for C
21H
23N
3O
4Na, 404.1581, Δ +1.5 ppm).
16β,17α-dihydroxy-deoxydihydroisoaustamide (
5)
: amorphous solids; [α]
D20 +141.7 (
c 0.12 CH
3OH); CD (
c 5.2 × 10
−4, CH
3OH), λ
max(∆ε) 206 (+15.03), 216 (+18.72), 281 (+2.92) nm, see
Supplementary Figure S5; UV (CH
3OH)
λmax(log
ε) 282 (3.68), 276 (3.65) and 223 (4.23) nm, see
Supplementary Figure S15;
1H and
13C NMR data, see
Table 1 and
Table 2,
Supplementary Figures S50–S56; HRESIMS m/z 380.1617 [M − H]
− (calcd. for C
21H
22N
3O
4, 380.1616, Δ −0.2 ppm), 404.1575 [M + Na]
+(calcd. for C
21H
23N
3O
4Na, 404.1581, Δ +1.5 ppm).
16α,17α-dihydroxy-deoxydihydroisoaustamide (
6): amorphous solids; [α]
D20 +195.4 (
c 0.13 CH
3OH); CD (
c 5.2 × 10
−4, CH
3OH), λ
max(∆ε) 203 (+26.02), 222 (+19.85), 276 (+3.43) nm, see
Supplementary Figure S6; UV (CH
3OH)
λmax(log
ε) 283 (3.84), 275 (3.80) and 223 (4.41) nm, see
Supplementary Figure S16;
1H and
13C NMR data, see
Table 1 and
Table 2,
Supplementary Figures S57–S63; HRESIMS m/z 382.1764 [M + H]
+ (calcd. for C
21H
24N
3O
4, 382.1761, Δ −0.6 ppm), 404.1583 [M + Na]
+(calcd. for C
21H
23N
3O
4Na, 404.1581, Δ −0.4 ppm).
3β-hydroxy-deoxyisoaustamide (
7): amorphous solids; [α]
D20 +182.0 (
c 0.08 CH
3OH); CD (
c 3.7 × 10
−4, CH
3OH), λ
max(∆ε) 219 (–53.92), 237 (+25.27), 272 (+17.00), 295 (+15.50) nm, see
Supplementary Figure S7; UV (CH
3OH)
λmax(log
ε) 274 (3.85), 223 (4.34) and 195 (4.18) nm, see
Supplementary Figure S17;
1H and
13C NMR data, see
Supplementary Figures S64–S70; HRESIMS m/z 404.1581 [M + Na]
+(calcd. for C
21H
23N
3O
4Na, 404.1581, Δ +0.0 ppm).
(+)-deoxyisoaustamide (
8): amorphous solids; [α]
D20 +192.0 (
c 0.13 CH
3OH); CD (
c 4.8 × 10
−4, CH
3OH), λ
max(∆ε) 203 (+4.82), 226 (+11.44), 371 (+7.31) nm, see
Supplementary Figure S8; UV (CH
3OH)
λmax(log
ε) 273 (3.76), 222 (4.37) and 199 (4.35) nm, see
Supplementary Figure S18.
deoxydihydroisoaustamide (
9): amorphous solids; CD (
c 4.8 × 10
−4, CH
3OH), λ
max(∆ε) 203 (+9.24), 223 (+20.09), 274 (+5.59) nm, see
Supplementary Figure S9; UV (CH
3OH)
λmax(log
ε) 283 (3.68), 224 (4.34) and 201 (4.26) nm, see
Supplementary Figure S19.
desoxybrevianamide E (
10): amorphous solids; CD (
c 4.7 × 10
−4, CH
3OH), λ
max(∆ε) 215 (+7.55), 234 (+2.87), 283 (+2.77) nm, see
Supplementary Figure S10; UV (CH
3OH)
λmax(log
ε) 283 (3.96), 224 (4.53) and 200 (4.49) nm, see
Supplementary Figure S20.
3.6. X-ray Crystal Data for 1
Experimental intensity data for
1 were collected at T = 298(2) K on a BRUKER Kappa APEX2 diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Intensity data were corrected for absorption using the multi-scan method. The structure was solved using direct methods and refined by least-squares calculation in anisotropic approximation for non-hydrogen atoms. Hydrogen atoms were placed in geometrically idealized positions and refined in the riding-model approximation. Data collection, reduction, and refinement of the lattice parameters were performed using the Apex2 software package [
21]. All calculations were performed with SHELXL/PC program [
22,
23]. Main crystallographic data and details of refinement of the crystal structure of 1 are shown in
Tables S1–S3. Supplementary crystallographic data (accession numbers CCDC 2048738) can be obtained free of charge from the Cambridge Crystallographic Data Center via
http://www.ccdc.cam.ac. uk/data_request/cif (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, UK; Fax: +44-1223-336-033 or Email:
[email protected]).
3.7. Cell Lines and Culture Conditions
The cells of mouse neuroblastoma cell line Neuro-2a and human prostate non-cancer cells PNT-2 were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). Neuro-2a were cultured in DMEM medium containing 10% fetal bovine serum (Biolot, St. Petersburg, Russia) and 1% penicillin/streptomycin (Biolot, St. Petersburg, Russia) at 37 °C in a humidified atmosphere with 5% (
v/v) CO
2. PNT-2 were cultured and handled as previously described [
24]. The cells were incubated in cultural flasks until sub-confluent (~80%).
3.8. MTT Cell Viability Assay
The Neuro-2a cells (1 × 104 cells/well) or PNT-2 cells (0.6 × 104 cells/well)were seeded in 96-well plate, incubated overnight. Then, the culture medium was exchanged to the fresh corresponding medium containing the different concentrations of the investigated compounds and the cells were further incubated for an additional 24 h. After that, cell viability was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method according to the manufacturer’s instructions (Sigma-Aldrich, St. Louis, MO, USA). Absorbance of the converted formazan was measured using a Multiskan FC microplate photometer (Thermo Scientific, Waltham, MA, USA) at λ = 570 nm with background subtraction at λ = 630–690 nm. The results were presented as percentages of control data.
3.9. Paraquat-Induced Neurotoxicity
The Neuro-2a cells (1 × 104 cells/well of 96-well plate) were pretreated with the studied compounds at concentrations of 1 and 10 µM for 1 h, and then 500 µM of PQ (Sigma-Aldrich, St. Louis, MO, USA) was added (500 µM, final concentration) to the neuroblastoma cells. Cells incubated without PQ and compounds, and with PQ alone, were used as positive and negative controls, respectively. The viability of cells was measured after 24 h using MTT method. The results were presented as percentages of positive control data.
All data were obtained in three independent replicates and calculated values were expressed as mean ± SEM. Student’s t-test was performed using SigmaPlot 14.0 (Systat Software Inc., San Jose, CA, USA) to determine statistical significance.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
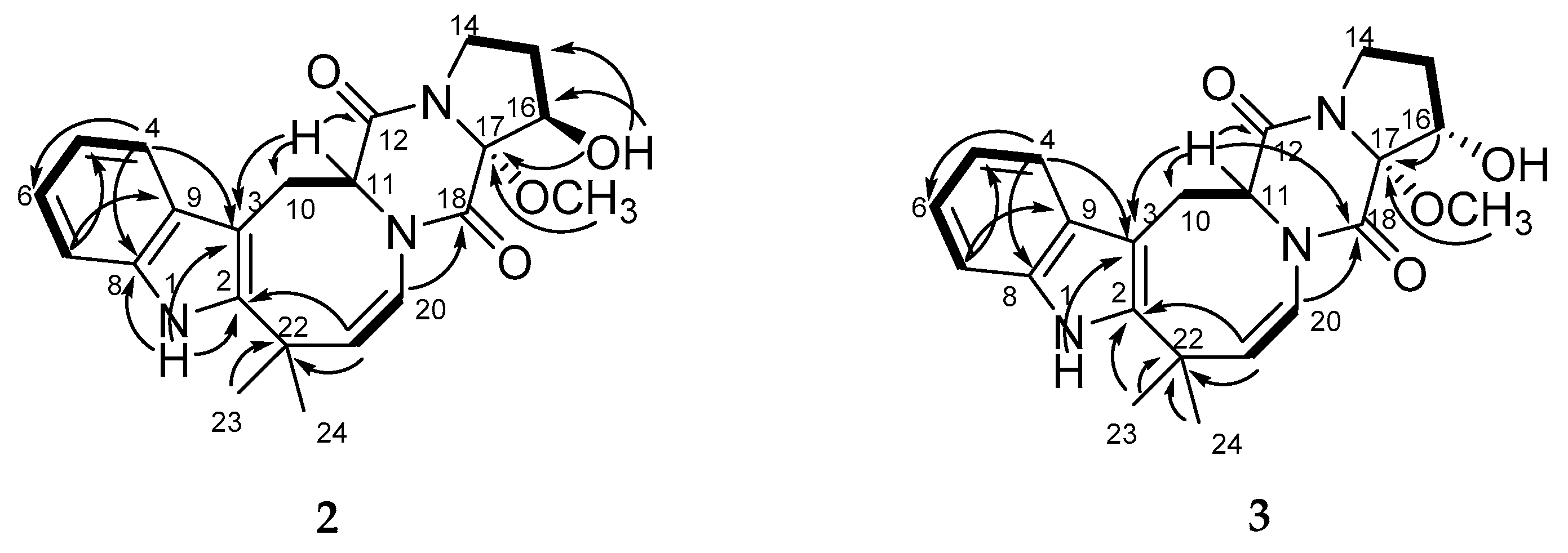{kind=link}
{kind=link}
{kind=link}