Topsentinol L Trisulfate, a Marine Natural Product That Targets Basal-like and Claudin-Low Breast Cancers

 ,
, 
Abstract
:
1. Introduction
2. Results
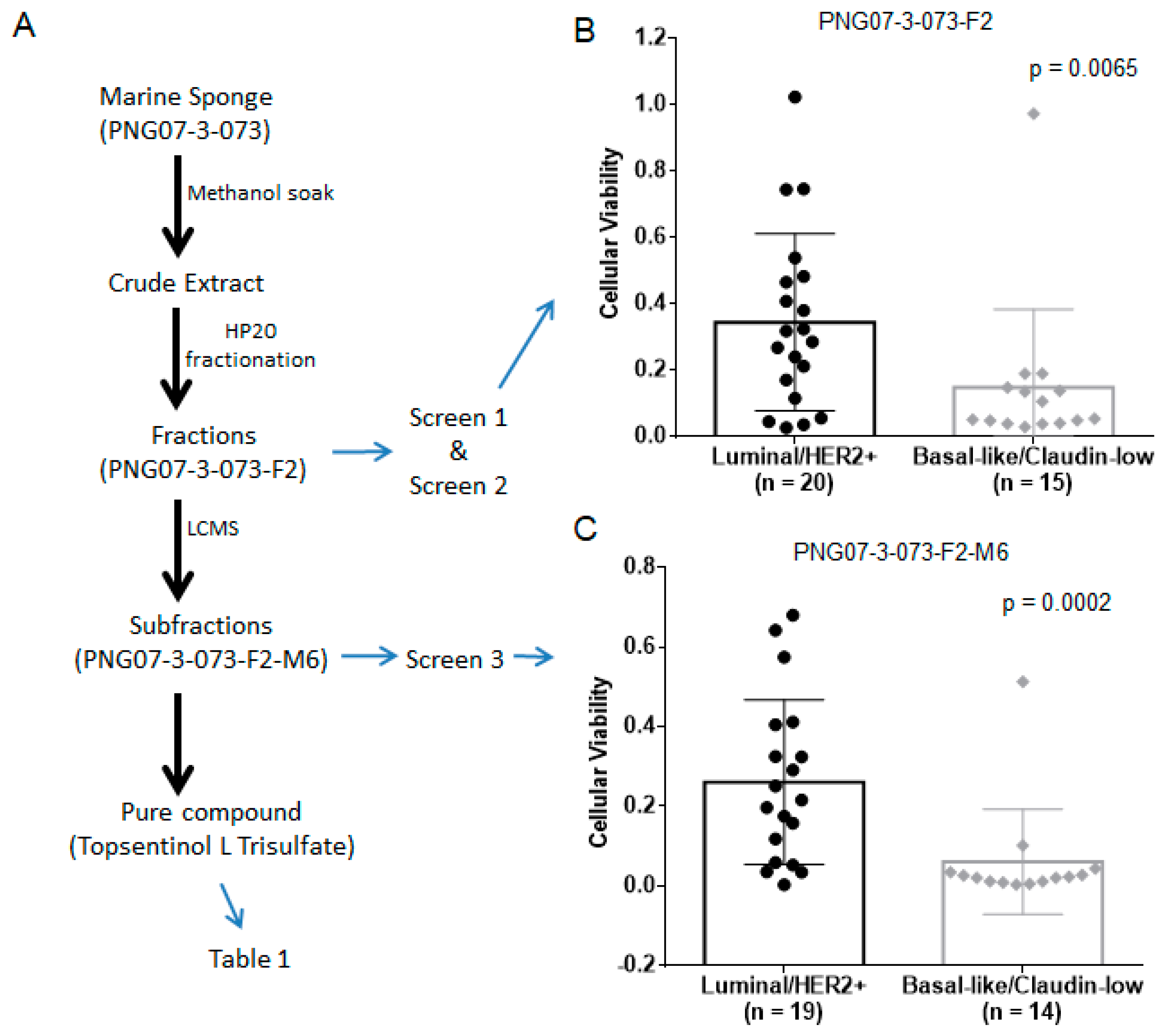
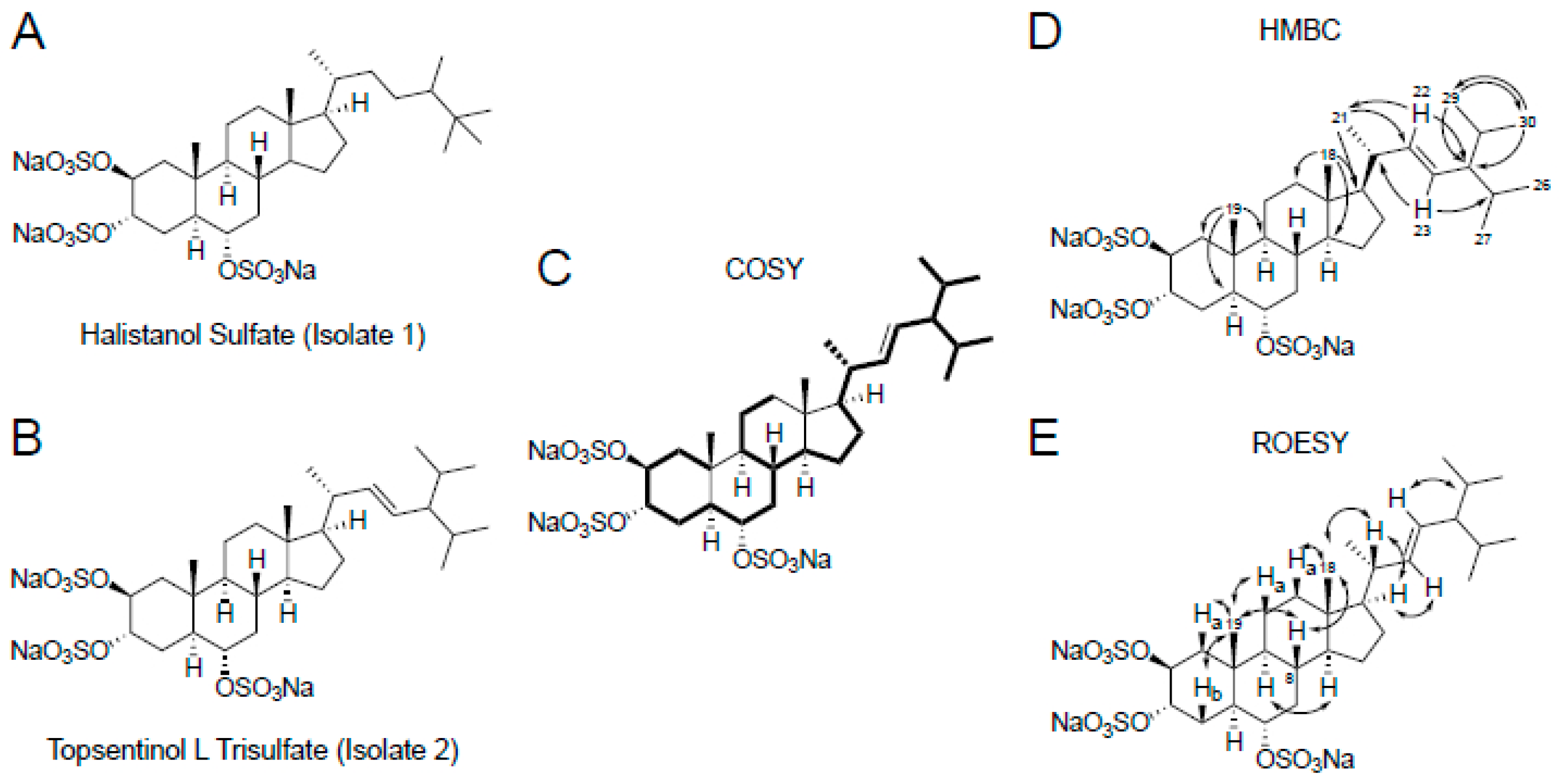
2.1. Identification of Topsentinol L Trisulfate as a Selective Inhibitor of Basal-Like and Claudin-Low BL-CL Breasst Cancers
2.2. Topsentinol L Trisulfate Treatment Inhibits AMPKα and CHK1 but Activates p38
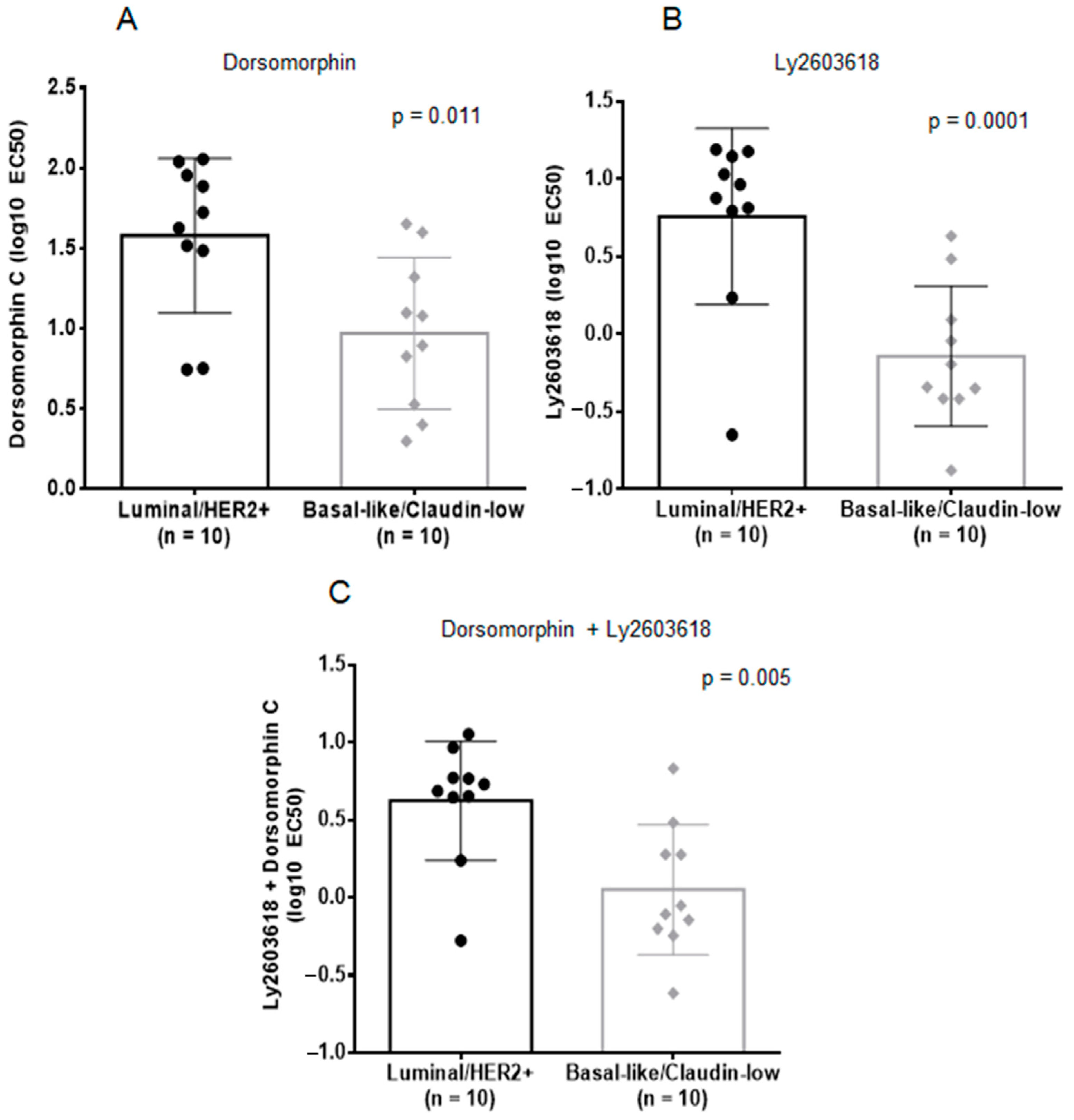
2.3. Inhibition of AMPK and CHK1, Alone or in Combination, is Effective against BL-CL Breast Cancers
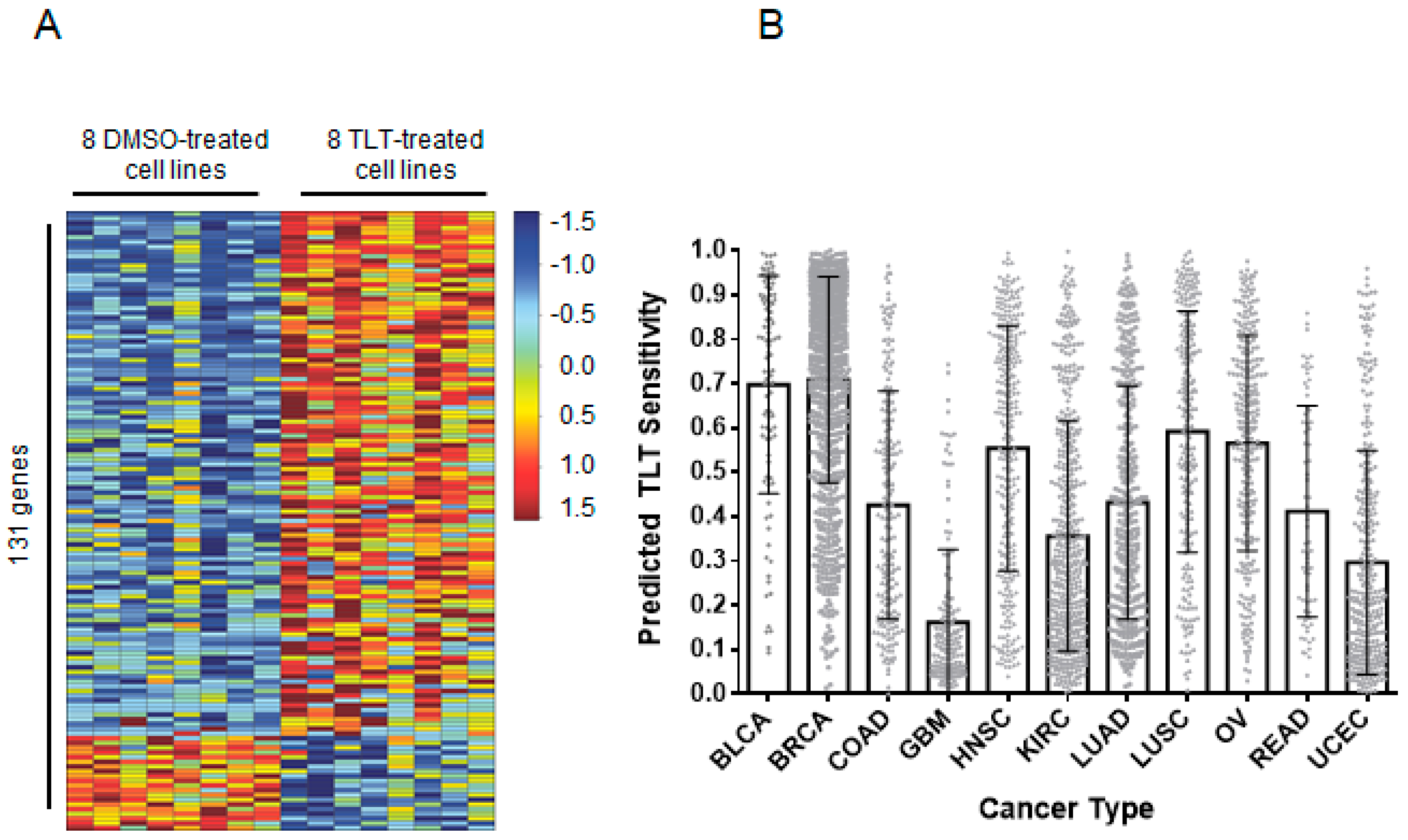
2.4. Topsentinol L Trisulfate (TLT) Sensitivity Signature Predicts Breast and Bladder Cancer Response in Human Tumors
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Viability Measurement
4.2. Marine Invertebrate Compound Library (MICL) Screens
4.3. Dose-Response Assays
4.4. Reverse Phase Protein Array
4.5. RNA Sequencing Data Acquisition
4.6. TLT Sensitivity Signature Generation and Analysis
4.7. Immunostaining
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef] [Green Version]
- Russnes, H.G.; Lingjaerde, O.C.; Borresen-Dale, A.L.; Caldas, C. Breast Cancer Molecular Stratification: From Intrinsic Subtypes to Integrative Clusters. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2010, 15 (Suppl. 5), 39–48. [Google Scholar] [CrossRef] [Green Version]
- Eroles, P.; Bosch, A.; Perez-Fidalgo, J.A.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sorlie, T. Re-definition of claudin-low as a breast cancer phenotype. Nat. Commun. 2020, 11, 1787. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Borresen-Dale, A.L. Systems biology and genomics of breast cancer. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression featuRes. between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugni, T.S.; Richards, B.; Bhoite, L.; Cimbora, D.; Harper, M.K.; Ireland, C.M. Marine natural product libraries for high-throughput screening and rapid drug discovery. J. Nat. Prod. 2008, 71, 1095–1098. [Google Scholar] [CrossRef] [Green Version]
- Bugni, T.S.; Harper, M.K.; McCulloch, M.W.B.; Reppart, J.; Ireland, C.M. Fractionated Marine Invertebrate Extract Libraries for Drug Discovery. Molecules 2008, 13, 1372–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortholand, J.Y.; Ganesan, A. Natural products and combinatorial chemistry: Back to the future. Curr. Opin. Chem. Biol. 2004, 8, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Bindseil, K.U.; Jakupovic, J.; Wolf, D.; Lavayre, J.; Leboul, J.; van der Pyl, D. Pure compound libraries; a new perspective for natural product based drug discovery. Drug Discov. Today 2001, 6, 840–847. [Google Scholar] [CrossRef]
- Grabowski, K.; Baringhaus, K.H.; Schneider, G. Scaffold diversity of natural products: Inspiration for combinatorial library design. Nat. Prod. Rep. 2008, 25, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Fusetani, N.; Matsunaga, S.; Konosu, S. Bioactive marine metabolites II. Halistanol sulfate, as antimicrobial novel steroid sulfate from the marine sponge Halichondria cf. moorei Bergquist. Tetrahedron Lett. 1981, 22, 1985–1988. [Google Scholar] [CrossRef]
- Dai, J.; Sorribas, A.; Yoshida, W.Y.; Kelly, M.; Williams, P.G. Topsentinols, 24-isopropyl steroids from the marine sponge Topsentia sp. J. Nat. Prod. 2010, 73, 1597–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibes, R.; Qiu, Y.; Lu, Y.; Hennessy, B.; Andreeff, M.; Mills, G.B.; Kornblau, S.M. Reverse phase protein array: Validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 2006, 5, 2512–2521. [Google Scholar] [CrossRef] [Green Version]
- Lizcano, J.M.; Goransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Ha, J.; Daniel, S.; Broyles, S.S.; Kim, K.H. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J. Biol. Chem. 1994, 269, 22162–22168. [Google Scholar]
- Jiang, K.; Pereira, E.; Maxfield, M.; Russell, B.; Goudelock, D.M.; Sanchez, Y. Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J. Biol. Chem. 2003, 278, 25207–25217. [Google Scholar] [CrossRef] [Green Version]
- Albiges, L.; Goubar, A.; Scott, V.; Vicier, C.; Lefebvre, C.; Alsafadi, S.; Commo, F.; Saghatchian, M.; Lazar, V.; Dessen, P.; et al. Chk1 as a new therapeutic target in triple-negative breast cancer. Breast 2014, 23, 250–258. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.A.; Ouchi, T. Cellular commitment to reentry into the cell cycle after stalled DNA is determined by site-specific phosphorylation of Chk1 and PTEN. Mol. Cancer Ther. 2008, 7, 2509–2516. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. USA 2014, 111, E435–E444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- McNeely, S.; Beckmann, R.; Bence Lin, A.K. CHEK again: Revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol. Ther. 2014, 142, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bild, A.H.; Yao, G.; Chang, J.T.; Wang, Q.; Potti, A.; Chasse, D.; Joshi, M.B.; Harpole, D.; Lancaster, J.M.; Berchuck, A.; et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006, 439, 353–357. [Google Scholar] [CrossRef]
- Brady, S.W.; McQuerry, J.A.; Qiao, Y.; Piccolo, S.R.; Shrestha, G.; Jenkins, D.F.; Layer, R.M.; Pedersen, B.S.; Miller, R.H.; Esch, A.; et al. Combating subclonal evolution of resistant cancer phenotypes. Nat. Commun. 2017, 8, 1231. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.L.; Soldi, R.; Zhang, H.; Gustafson, A.M.; Wilcox, R.; Welm, B.E.; Chang, J.T.; Johnson, E.; Spira, A.; Jeffrey, S.S.; et al. A pharmacogenomic method for individualized prediction of drug sensitivity. Mol. Syst. Biol. 2011, 7, 513. [Google Scholar] [CrossRef]
- Omberg, L.; Ellrott, K.; Yuan, Y.; Kandoth, C.; Wong, C.; Kellen, M.R.; Friend, S.H.; Stuart, J.; Liang, H.; Margolin, A.A. Enabling transparent and collaborative computational analysis of 12 tumor types within The Cancer Genome Atlas. Nat. Genet 2013, 45, 1121–1126. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Damrauer, J.S.; Hoadley, K.A.; Chism, D.D.; Fan, C.; Tiganelli, C.J.; Wobker, S.E.; Yeh, J.J.; Milowsky, M.I.; Iyer, G.; Parker, J.S.; et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc. Natl. Acad. Sci. USA 2014, 111, 3110–3115. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Chai, S.; Mose, L.E.; Selitsky, S.R.; Krishnan, B.; Saito, R.; Iglesia, M.D.; Milowsky, M.I.; Parker, J.S.; Kim, W.Y.; et al. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 2016, 1, e85902. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Carling, D.; Gamblin, S.J. AMP-activated protein kinase: Also regulated by ADP? Trends Biochem. Sci. 2011, 36, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2014. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Pierotti, M.A.; Berrino, F.; Gariboldi, M.; Melani, C.; Mogavero, A.; Negri, T.; Pasanisi, P.; Pilotti, S. Targeting metabolism for cancer treatment and prevention: Metformin, an old drug with multi-faceted effects. Oncogene 2013, 32, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Rios, M.; Foretz, M.; Viollet, B.; Prieto, A.; Fraga, M.; Costoya, J.A.; Senaris, R. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013, 73, 2628–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.U.; Suy, S.; Danner, M.; Dailey, V.; Zhang, Y.; Li, H.; Hyduke, D.R.; Collins, B.T.; Gagnon, G.; Kallakury, B.; et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol. Cancer Ther. 2009, 8, 733–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M.; et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010, 38, e178. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Vogt, J.; Traynor, R.; Sapkota, G.P. The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signal 2011, 23, 1831–1842. [Google Scholar] [CrossRef] [Green Version]
- Iadevaia, S.; Lu, Y.; Morales, F.C.; Mills, G.B.; Ram, P.T. Identification of optimal drug combinations targeting cellular networks: Integrating phospho-proteomics and computational network analysis. Cancer Res. 2010, 70, 6704–6714. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- West, M.; Blanchette, C.; Dressman, H.; Huang, E.; Ishida, S.; Spang, R.; Zuzan, H.; Olson, J.A., Jr.; Marks, J.R.; Nevins, J.R. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc. Natl. Acad. Sci. USA 2001, 98, 11462–11467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benito, M.; Parker, J.; Du, Q.; Wu, J.; Xiang, D.; Perou, C.M.; Marron, J.S. Adjustment of systematic microarray data biases. Bioinformatics 2004, 20, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Gene Expression Subtype | Topsentinol L Trisulfate EC50 (µM) |
|---|---|---|
| BT20 | Basal-like | >100 |
| HCC1143 | Basal-like | 74 |
| HCC1395 | Basal-like | 77 |
| HCC1806 | Basal-like | >100 |
| HCC1937 | Basal-like | >100 |
| HCC70 | basal-like | 23 |
| MD468 | Basal-like | 69 |
| HCC38 | Claudin-low | >100 |
| Hs578T | Claudin-low | 99 |
| MD157 | Claudin-low | 50 |
| MD231 | Claudin-low | 34 |
| MD436 | Claudin-low | 87 |
| HCC1569 | Basal/HER2 positive | >100 |
| HCC1954 | Basal/HER2 positive | 63 |
| JMT-1 | Basal/HER2 positive | 41 |
| MD453 | Luminal | >100 |
| HCC2218 | Luminal | >100 |
| Cama-1 | Luminal | >100 |
| MD134VI | Luminal | >100 |
| MD175VII | Luminal | >100 |
| MD415 | Luminal | >100 |
| ZR75-1 | Luminal | >100 |
| MCF7 | Luminal | >100 |
| T47D | Luminal | 26 |
| BT483 | Luminal | >100 |
| AU565 | Luminal/HER2 positive | >100 |
| HCC1419 | Luminal/HER2 positive | >100 |
| SKBR3 | Luminal/HER2 positive | >100 |
| BT474 | Luminal/HER2 positive | 84 |
| UACC812 | Luminal/HER2 positive | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Chaar, N.N.; Smith, T.E.; Shrestha, G.; Piccolo, S.R.; Harper, M.K.; Van Wagoner, R.M.; Lu, Z.; Venancio, A.R.; Ireland, C.M.; Bild, A.H.; et al. Topsentinol L Trisulfate, a Marine Natural Product That Targets Basal-like and Claudin-Low Breast Cancers. Mar. Drugs 2021, 19, 41. https://doi.org/10.3390/md19010041
El-Chaar NN, Smith TE, Shrestha G, Piccolo SR, Harper MK, Van Wagoner RM, Lu Z, Venancio AR, Ireland CM, Bild AH, et al. Topsentinol L Trisulfate, a Marine Natural Product That Targets Basal-like and Claudin-Low Breast Cancers. Marine Drugs. 2021; 19(1):41. https://doi.org/10.3390/md19010041
Chicago/Turabian StyleEl-Chaar, Nader N., Thomas E. Smith, Gajendra Shrestha, Stephen R. Piccolo, Mary Kay Harper, Ryan M. Van Wagoner, Zhenyu Lu, Ashlee R. Venancio, Chris M. Ireland, Andrea H. Bild, and et al. 2021. "Topsentinol L Trisulfate, a Marine Natural Product That Targets Basal-like and Claudin-Low Breast Cancers" Marine Drugs 19, no. 1: 41. https://doi.org/10.3390/md19010041
APA StyleEl-Chaar, N. N., Smith, T. E., Shrestha, G., Piccolo, S. R., Harper, M. K., Van Wagoner, R. M., Lu, Z., Venancio, A. R., Ireland, C. M., Bild, A. H., & Moos, P. J. (2021). Topsentinol L Trisulfate, a Marine Natural Product That Targets Basal-like and Claudin-Low Breast Cancers. Marine Drugs, 19(1), 41. https://doi.org/10.3390/md19010041