
Structurally Diverse Polycyclic Salicylaldehyde Derivative Enantiomers from a Marine-Derived Fungus Eurotium sp. SCSIO F452

 , , , and
, , , and
Abstract
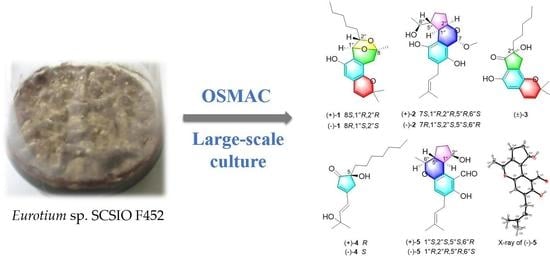
:
1. Introduction
2. Results
2.1. Structure Identification
2.2. Proposed Biosynthesis Pathway
2.3. Bioactivity Evaluation
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material, Fermentation, and Extraction
3.3. Purification
3.4. X-ray Crystallographic Analysis
3.5. ECD and 13C NMR Calculation Methods
3.6. Cytotoxicity, Antioxidative, and α-Glucosidase Inhibitory Activity, and Antimicrobial Activity Assays
3.6.1. Cytotoxicity Assay
3.6.2. Antioxidative Assay
3.6.3. α-Glucosidase Inhibitory Activity Assay
3.6.4. Antimicrobial Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carroll, A.R.A.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Aly, A.H.; Debbab, A.; Proksch, P. Fifty years of drug discovery from fungi. Fungal Divers. 2011, 50, 3–19. [Google Scholar] [CrossRef]
- Zhang, J.J.; Tang, X.; Moore, B.S. Genetic platforms for heterologous expression of microbial natural products. Nat. Prod. Rep. 2019, 36, 1313–1332. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ye, Y.; Zhang, Y. Large-scale culture as a complementary and practical method for discovering natural products with novel skeletons. Nat. Prod. Rep. 2021. [Google Scholar] [CrossRef]
- Zang, Y.; Gong, Y.; Gong, J.; Liu, J.; Chen, C.; Gu, L.; Zhou, Y.; Wang, J.; Zhu, H.; Zhang, Y. Fungal polyketides with three distinctive ring skeletons from the fungus Penicillium canescens uncovered by OSMAC and molecular networking strategies. J. Org. Chem. 2020, 85, 4973–4980. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Chen, C.; Xue, Y.; Tong, Q.; Li, X.N.; Chen, X.; Wang, J.; Yao, G.; Luo, Z.; Zhang, Y. Asperchalasine A, a cytochalasan dimer with an unprecedented decacyclic ring system, from Aspergillus flavipes. Angew. Chem. Int. Ed. 2015, 54, 13374–13378. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, F.; Luo, M.; Chen, Y.; Song, Y.; Zhang, W.; Zhang, S.; Ju, J. Halogenated anthraquinones from the marine-derived fungus Aspergillus sp. SCSIO F063. J. Nat. Prod. 2012, 75, 1346–1352. [Google Scholar] [CrossRef]
- Wang, F.Z.; Huang, Z.; Shi, X.F.; Chen, Y.C.; Zhang, W.M.; Tian, X.P.; Li, J.; Zhang, S. Cytotoxic indole diketopiperazines from the deep sea-derived fungus Acrostalagmus luteoalbus SCSIO F457. Bioorg. Med. Chem. Lett. 2012, 22, 7265–7267. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhong, W.M.; Chen, Y.C.; Xiang, Y.; Chen, X.Y.; Tian, X.P.; Zhang, W.M.; Zhang, S.; Wang, F.Z. A new butenolide derivative from the deep-sea fungus Aspergillus terreus SCSIO FZQ028. Nat. Prod. Res. 2020, 34, 1984–1991. [Google Scholar] [CrossRef]
- Xiang, Y.; Zeng, Q.; Mai, Z.M.; Chen, Y.C.; Shi, X.F.; Chen, X.Y.; Zhong, W.M.; Wei, X.Y.; Zhang, W.M.; Zhang, S.; et al. Asperorydines N-P, three new cyclopiazonic acid alkaloids from the marine-derived fungus Aspergillus flavus SCSIO F025. Fitoterapia 2021, 150, 104839. [Google Scholar] [CrossRef]
- Wang, F.Z.; Huang, Z.; Shi, X.F.; Chen, Y.C.; Zhang, W.M.; Tian, X.P.; Li, J.; Zhang, S. Analysis of secondary metabolites produced by Eurotium sp. SCSIO F452 isolated from the South China Sea sediment. Zhongguo Haiyang Yaowu 2013, 32, 7–12. [Google Scholar]
- Zhong, W.; Wang, J.; Wei, X.; Chen, Y.; Fu, T.; Xiang, Y.; Huang, X.; Tian, X.; Xiao, Z.; Zhang, W.; et al. Variecolortins A-C, three pairs of spirocyclic diketopiperazine enantiomers from the marine-derived fungus Eurotium sp. SCSIO F452. Org. Lett. 2018, 20, 4593–4596. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, J.; Wei, X.; Fu, T.; Chen, Y.; Zeng, Q.; Huang, Z.; Huang, X.; Zhang, W.; Zhang, S.; et al. Three pairs of new spirocyclic alkaloid enantiomers from the marine-derived fungus Eurotium sp. SCSIO F452. Front. Chem. 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.-M.; Wang, J.-F.; Wei, X.-Y.; Zeng, Q.; Chen, X.-Y.; Xiang, Y.; Tian, X.-P.; Zhang, S.; Long, L.-J.; Wang, F.-Z. (+)- and (−)-Eurotone A: A pair of enantiomeric polyketide dimers from a marine-derived fungus Eurotium sp. SCSIO F452. Tetrahedron Lett. 2019, 60, 1600–1603. [Google Scholar] [CrossRef]
- Zhong, W.; Chen, Y.; Wei, X.; Wang, J.; Zhang, W.; Wang, F.; Zhang, S. Salicylaldehyde derivatives from a marine-derived fungus Eurotium sp. SCSIO F452. J. Antibiot. 2020, 74, 273–279. [Google Scholar] [CrossRef]
- Zhong, W.; Chen, Y.; Mai, Z.; Wei, X.; Wang, J.; Zeng, Q.; Chen, X.; Tian, X.; Zhang, W.; Wang, F.; et al. Euroticins A and B, two pairs of highly constructed salicylaldehyde derivative enantiomers from a marine-derived fungus Eurotium sp. SCSIO F452. J. Org. Chem. 2020, 85, 12754–12759. [Google Scholar] [CrossRef]
- Zhong, W.; Chen, Y.; Wei, X.; Wang, J.; Zeng, Q.; Tian, X.; Zhang, W.; Wang, F.; Zhang, S. Euroticins C–E, three pairs of polycyclic salicylaldehyde derivative enantiomers from a marine-derived fungus Eurotium sp. SCSIO F452. Org. Chem. Front. 2021, 8, 1466–1473. [Google Scholar] [CrossRef]
- Li, D.L.; Li, X.M.; Li, T.G.; Dang, H.Y.; Proksch, P.; Wang, B.G. Benzaldehyde derivatives from Eurotium rubrum, an endophytic fungus derived from the mangrove plant Hibiscus tiliaceus. Chem. Pharm. Bull. 2008, 56, 1282–1285. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Shao, C.-L.; Wang, K.-L.; Xu, Y.; She, Z.-G.; Wang, C.-Y. Dihydroisocoumarin derivatives with antifouling activities from a gorgonian-derived Eurotium sp. fungus. Tetrahedron 2014, 70, 9132–9138. [Google Scholar] [CrossRef]
- Karplus, M. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 1959, 30, 11–15. [Google Scholar] [CrossRef]
- Karplus, M. Vicinal proton coupling in nuclear magnetic resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Liang, X.; Huang, Z.H.; Ma, X.; Zheng, Z.H.; Zhang, X.X.; Lu, X.H.; Qi, S.H. Mycotoxins as inhibitors of protein tyrosine phosphatases from the deep-sea-derived fungus Aspergillus puniceus SCSIO z021. Bioorg. Chem. 2021, 107, 104571. [Google Scholar] [CrossRef] [PubMed]
- Kamada, T.; Kang, M.C.; Phan, C.S.; Zanil, I.I.; Jeon, Y.J.; Vairappan, C.S. Bioactive cembranoids from the soft foral genus Sinularia sp. in Borneo. Mar. Drugs 2018, 16, 99. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, A.; Liu, J.; Bao, S.; Peng, R.; Hu, Y.; Yuan, T.; Hou, S.; Xie, T.; Zhang, Q.; et al. Chartspiroton, a tetracyclic spiro-naphthoquinone derivative from a medicinal plant endophytic Streptomyces. Org. Lett. 2020, 22, 3739–3743. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Zhang, L.; Zhang, Q.; Zhang, W.; Chen, Y.; Zhang, W.; Zhang, H.; Zhang, C. Dassonmycins A and B, polycyclic thioalkaloids from a marine sponge-derived Nocardiopsis dassonvillei SCSIO 40065. Org. Lett. 2021, 23, 2858–2862. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Neese, F. The ORCA program system. WIREs Comput. Molecul. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Molecul. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Sun, Z.H.; Gu, J.; Ye, W.; Wen, L.X.; Lin, Q.B.; Li, S.N.; Chen, Y.C.; Li, H.H.; Zhang, W.M. Geospallins A-C: New thiodiketopiperazines with inhibitory activity against angiotensin-converting enzyme from a deep-sea-derived fungus Geosmithia pallida FS140. Mar. Drugs 2018, 16, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| No. | δCa | δH (J, Hz) b | δCa | δH (J, Hz) b |
| 1 | 120.6 | 121.1 | ||
| 2 | 143.0 | 144.6 | ||
| 3 | 120.1 | 127.7 | ||
| 4 | 110.3 | 6.36, s | 117.0 | 6.51, s |
| 5 | 145.7 | 148.2 | ||
| 6 | 125.6 | 122.4 | ||
| 7 | 35.8 | 2.66, d (17.3) | 95.9 | 5.58, s |
| 2.49, overlap | ||||
| 8 | 106.2 | |||
| 9 | 25.8 | 1.53, s | ||
| 1′ | 122.6 | 6.27, d (9.7) | 29.2 | 3.27, m |
| 2′ | 131.6 | 5.67, d (9.7) | 123.8 | 5.28, br t (7.4) |
| 3′ | 75.7 | 132.6 | ||
| 4′ | 28.0 | 1.31, s | 25.9 | 1.72, s |
| 5′ | 27.8 | 1.29, s | 17.8 | 1.69, s |
| 1″ | 74.1 | 5.09, s | 41.3 | 2.30, dd (12.0, 8.1) |
| 2″ | 84.8 | 3.75, t (6.7) | 67.9 | 4.61, ddd (8.1, 7.7, 5.0) |
| 3″ | 35.1 | 1.45, m | 35.0 | 2.49, m |
| 1.71, overlap | ||||
| 4″ | 25.2 | 1.32, overlap | 29.5 | 1.92 m |
| 1.39, m | ||||
| 5″ | 31.5 | 1.27, overlap | 49.8 | 1.33, m |
| 6″ | 22.5 | 1.28, overlap | 80.8 | 4.31, dq (10.4, 6.2) |
| 7″ | 14.4 | 0.86, t (6.7) | 21.8 | 1.30, d (6.2) |
| 5-OH | 8.99, s | |||
| 7-OMe | 55.4 | 3.45, s | ||
| 3 | 4 | |||
|---|---|---|---|---|
| No. | δCa | δH (J, Hz) b | δCc | δH (J, Hz) d |
| 1 | 128.3 | 41.1 | 2.80, d (17.8) | |
| 2.51, overlap | ||||
| 2 | 140.1 | 169.4 | ||
| 3 | 130.8 | 126.7 | 6.01, s | |
| 4 | 112.9 | 6.60, s | 209.5 | |
| 5 | 158.1 | 76.4 | ||
| 6 | 112.9 | |||
| 7 | 57.3 | 4.99, d (16.3) | ||
| 4.81, d (16.3) | ||||
| 1′ | 122.4 | 6.46, d (9.9) | 121.7 | 6.68, d (15.8) |
| 2′ | 138.0 | 6.07, d (9.9) | 149.5 | 6.49, d (15.8) |
| 3′ | 77.1 | 69.5 | ||
| 4′ | 28.0 | 1.43, s | 29.5 | 1.24, s |
| 5′ | 27.7 | 1.42, s | 29.5 | 1.24, s |
| 1″ | 197.5 | 37.4 | 1.50, dt (12.6, 3.9) | |
| 1.39, dt (12.6, 4.8) | ||||
| 2″ | 92.3 | 23.2 | 1.25 overlap | |
| 1.10 m | ||||
| 3″ | 37.0 | 2.11, m | 29.5 | 1.21, overlap |
| 1.83, m | ||||
| 4″ | 23.6 | 1.53, m | 28.7 | 1.22, overlap |
| 1.35, overlap | ||||
| 5″ | 32.8 | 1.30, overlap | 31.2 | 1.20, overlap |
| 6″ | 23.2 | 1.29, overlap | 22.1 | 1.24, overlap |
| 7″ | 14.3 | 0.88, t (7.0) | 14.0 | 0.84, t (7.2) |
| 5-OH | 11.2, s | |||
| 2″-OH | 5.90, br s | |||
| Compounds | IC50 (μM) | |||
|---|---|---|---|---|
| SF-268 | MCF-7 | HepG2 | A549 | |
| (+)-1 | 21.88 ± 0.96 | 27.17 ± 2.03 | 28.00 ± 1.68 | 33.78 ± 0.34 |
| (−)-1 | 37.31 ± 2.46 | 28.00 ± 1.57 | 30.72 ± 3.55 | 33.43 ± 1.50 |
| (+)-2 | >100 | >100 | >100 | >100 |
| (−)-2 | >100 | >100 | >100 | >100 |
| (±)-3 | >100 | >100 | >100 | >100 |
| (+)-4 | 12.74 ± 0.46 | 20.51 ± 1.54 | 19.88 ± 5.09 | 16.90 ± 0.92 |
| (−)-4 | 23.73 ± 3.88 | 23.56 ± 2.99 | 19.53 ± 2.70 | 22.15 ± 1.54 |
| (+)-5 | 21.98 ± 0.88 | 55.59 ± 4.85 | 41.18 ± 2.63 | 47.34 ± 0.71 |
| (−)-5 | 35.65 ± 1.79 | 50.67 ± 2.48 | 40.69 ± 2.76 | 42.61 ± 1.43 |
| Adriamycin | 1.19 ± 0.03 | 2.02 ± 0.04 | 1.99 ± 0.07 | 1.73 ± 0.04 |
| Compounds | Antioxidative Activity EC50 (μM) | α-Glucosidase Inhibitory IC50 (μM) |
|---|---|---|
| (+)-1 | 42.34 ± 3.85 | >100 |
| (−)-1 | 41.40 ± 2.68 | >100 |
| (+)-2 | 76.90 ± 0.99 | 38.04 ± 2.73 |
| (−)-2 | 77.07 ± 1.88 | 79.71 ± 1.74 |
| (±)-3 | >100 | 16.31 ± 1.68 |
| (+)-4 | >100 | >100 |
| (−)-4 | >100 | >100 |
| (+)-5 | >100 | 89.41 ± 7.86 |
| (−)-5 | >100 | >100 |
| Ascorbic acid a | 11.35 ± 0.56 | |
| Acarbose b | 32.92 ± 1.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, W.-M.; Wei, X.-Y.; Chen, Y.-C.; Zeng, Q.; Wang, J.-F.; Shi, X.-F.; Tian, X.-P.; Zhang, W.-M.; Wang, F.-Z.; Zhang, S. Structurally Diverse Polycyclic Salicylaldehyde Derivative Enantiomers from a Marine-Derived Fungus Eurotium sp. SCSIO F452. Mar. Drugs 2021, 19, 543. https://doi.org/10.3390/md19100543
Zhong W-M, Wei X-Y, Chen Y-C, Zeng Q, Wang J-F, Shi X-F, Tian X-P, Zhang W-M, Wang F-Z, Zhang S. Structurally Diverse Polycyclic Salicylaldehyde Derivative Enantiomers from a Marine-Derived Fungus Eurotium sp. SCSIO F452. Marine Drugs. 2021; 19(10):543. https://doi.org/10.3390/md19100543
Chicago/Turabian StyleZhong, Wei-Mao, Xiao-Yi Wei, Yu-Chan Chen, Qi Zeng, Jun-Feng Wang, Xue-Feng Shi, Xin-Peng Tian, Wei-Min Zhang, Fa-Zuo Wang, and Si Zhang. 2021. "Structurally Diverse Polycyclic Salicylaldehyde Derivative Enantiomers from a Marine-Derived Fungus Eurotium sp. SCSIO F452" Marine Drugs 19, no. 10: 543. https://doi.org/10.3390/md19100543
APA StyleZhong, W. -M., Wei, X. -Y., Chen, Y. -C., Zeng, Q., Wang, J. -F., Shi, X. -F., Tian, X. -P., Zhang, W. -M., Wang, F. -Z., & Zhang, S. (2021). Structurally Diverse Polycyclic Salicylaldehyde Derivative Enantiomers from a Marine-Derived Fungus Eurotium sp. SCSIO F452. Marine Drugs, 19(10), 543. https://doi.org/10.3390/md19100543