Marine Anthraquinones: Pharmacological and Toxicological Issues




Abstract
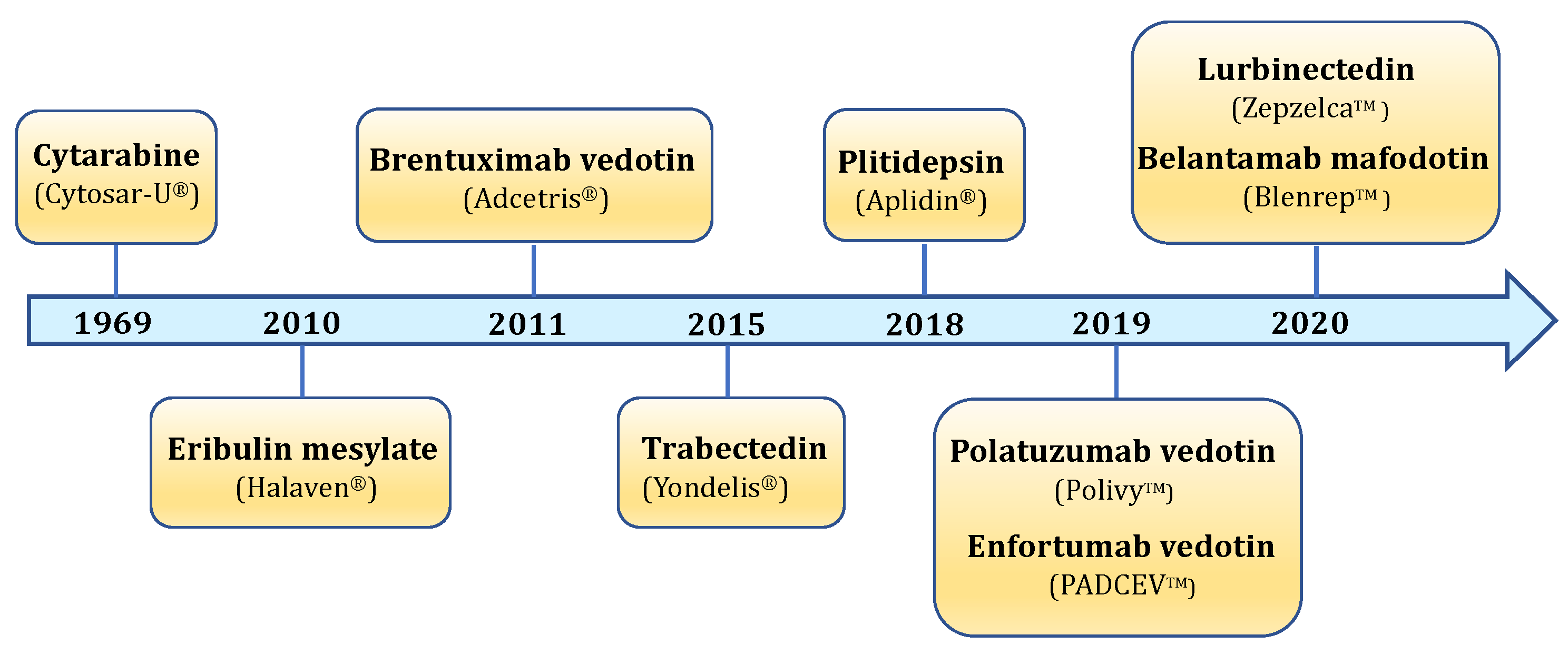
:1. Introduction
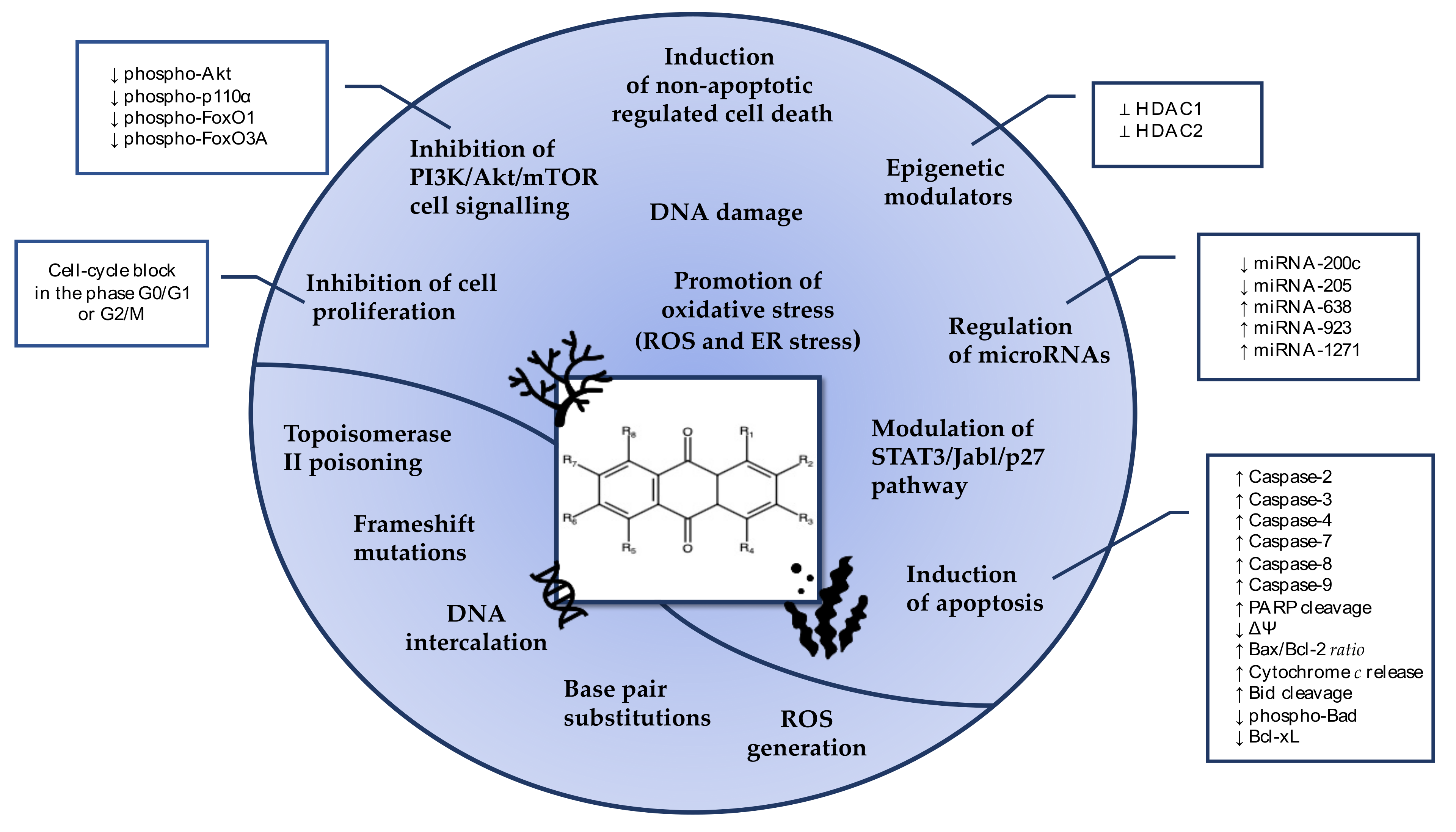
2. Anticancer Mechanisms of AQs Isolated from Marine Fungi
2.1. Emodin
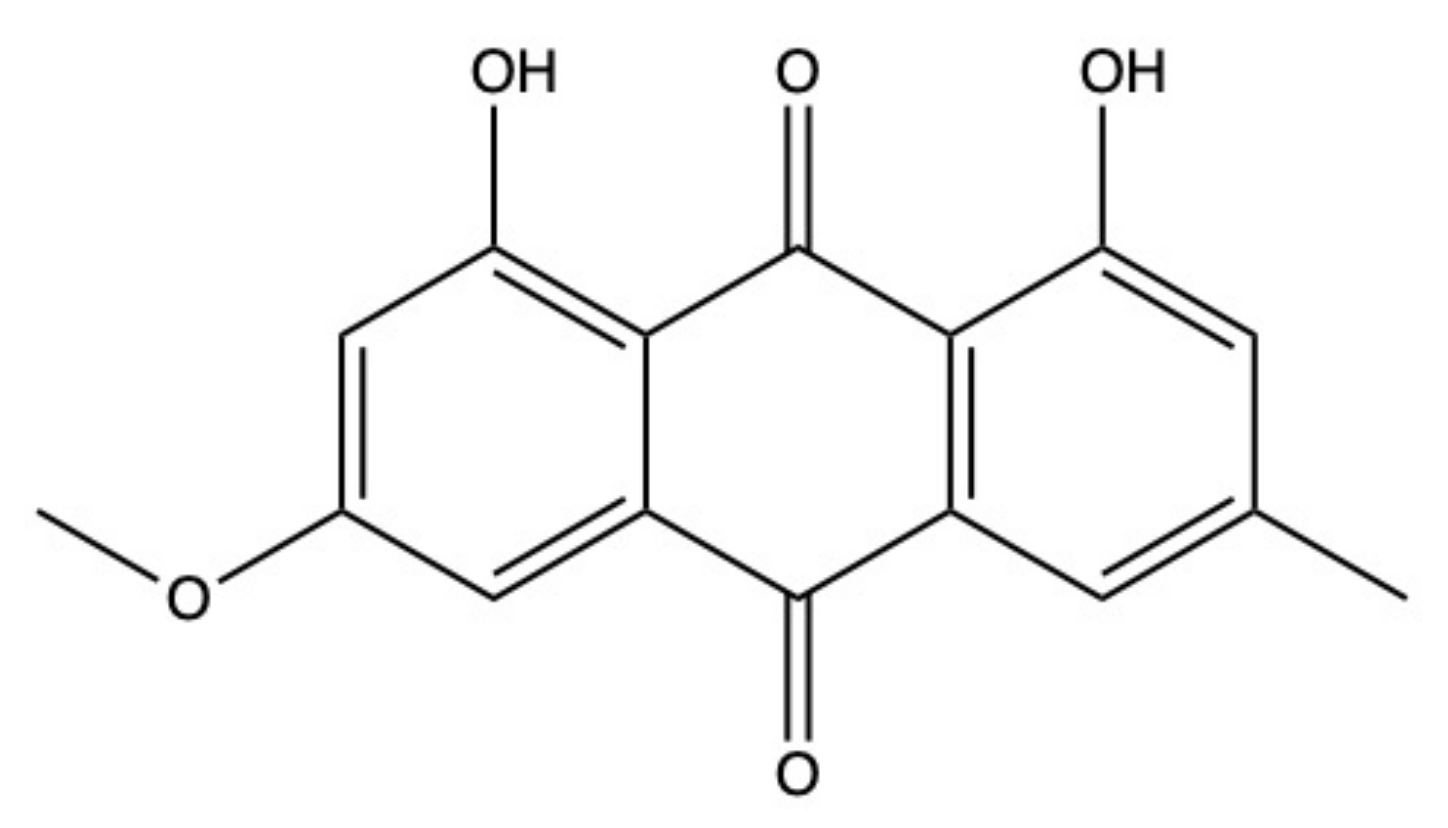
2.2. Physcion
2.3. Aspergiolide A
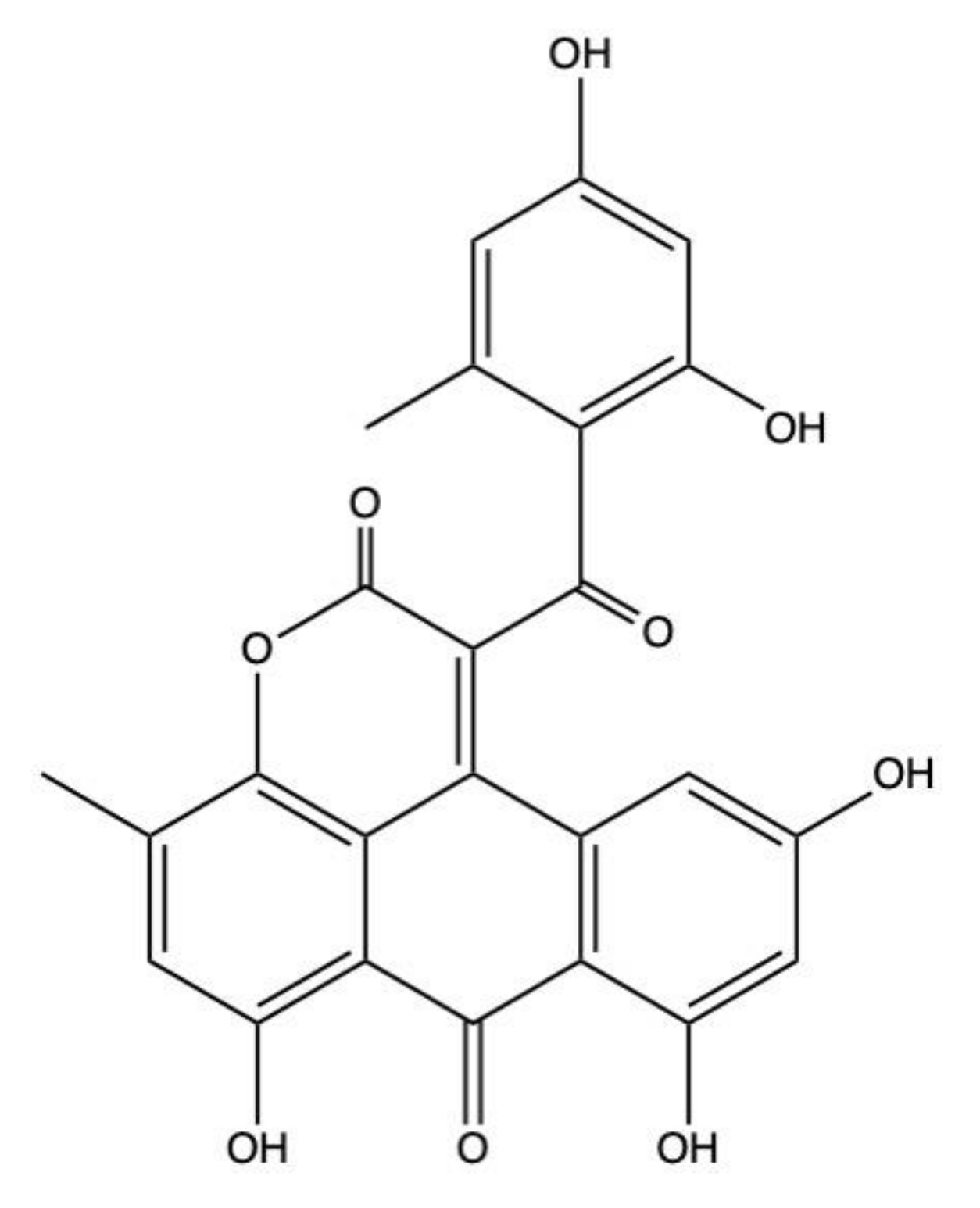
2.4. Alterporriols
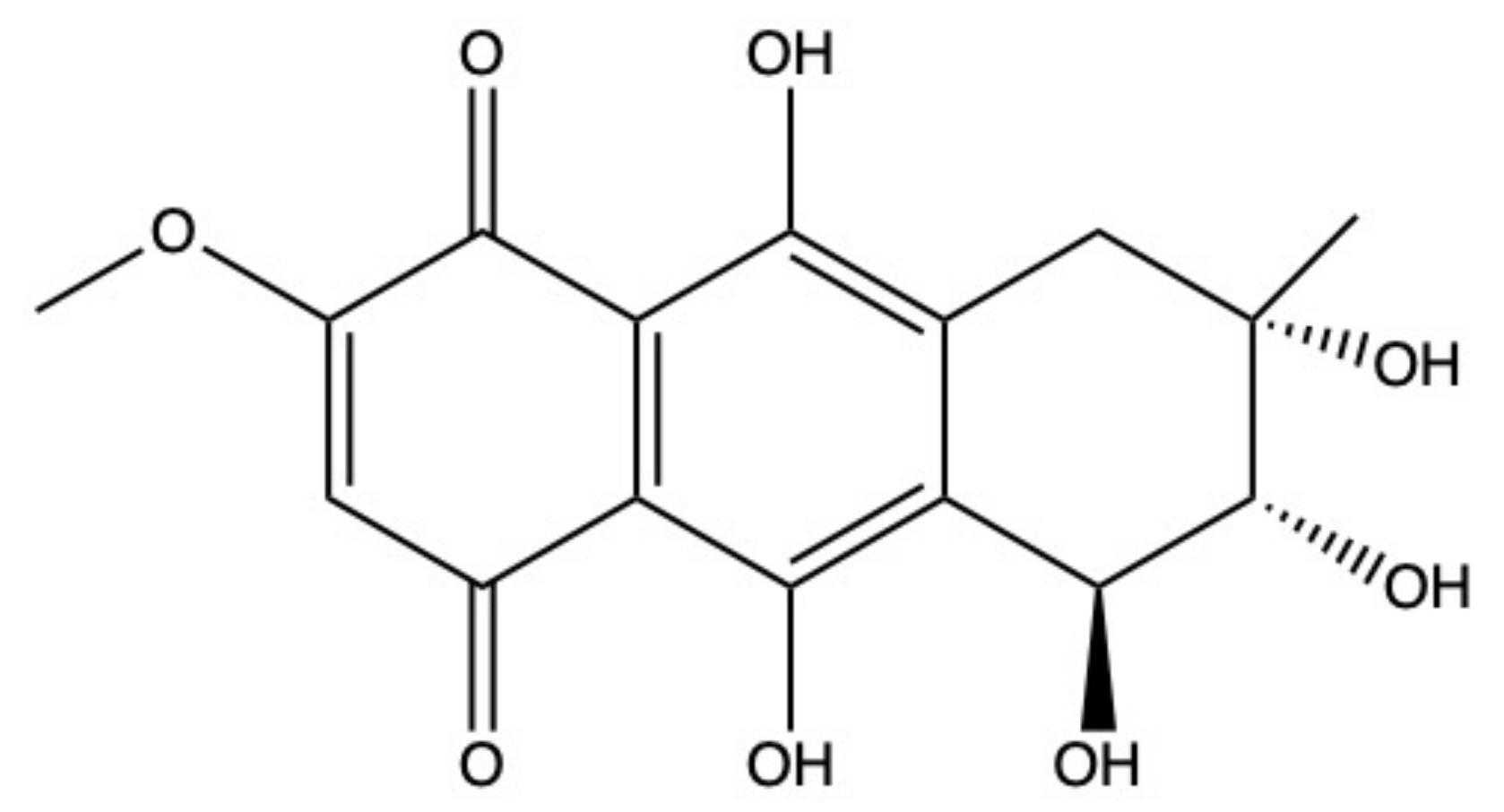
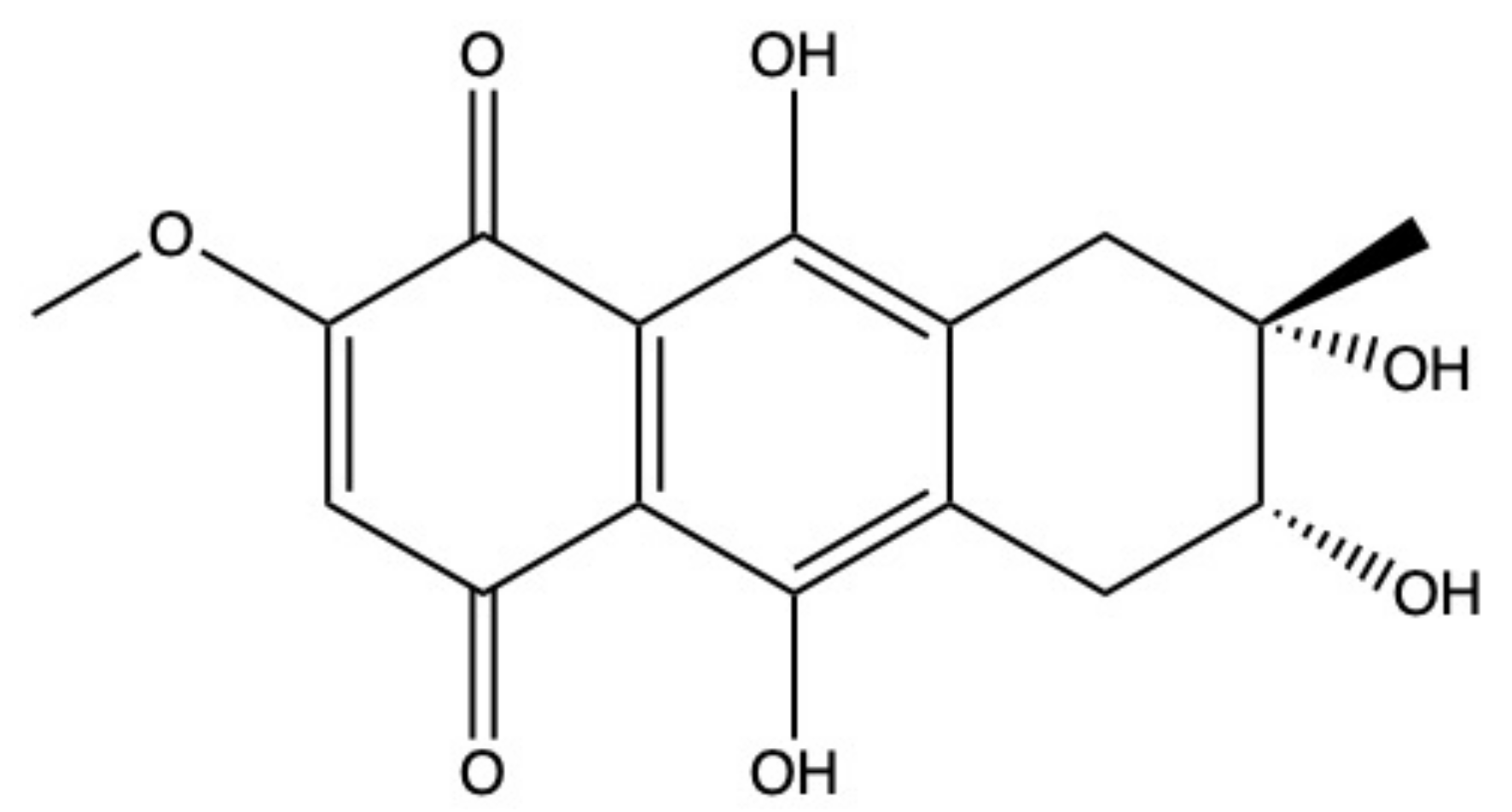
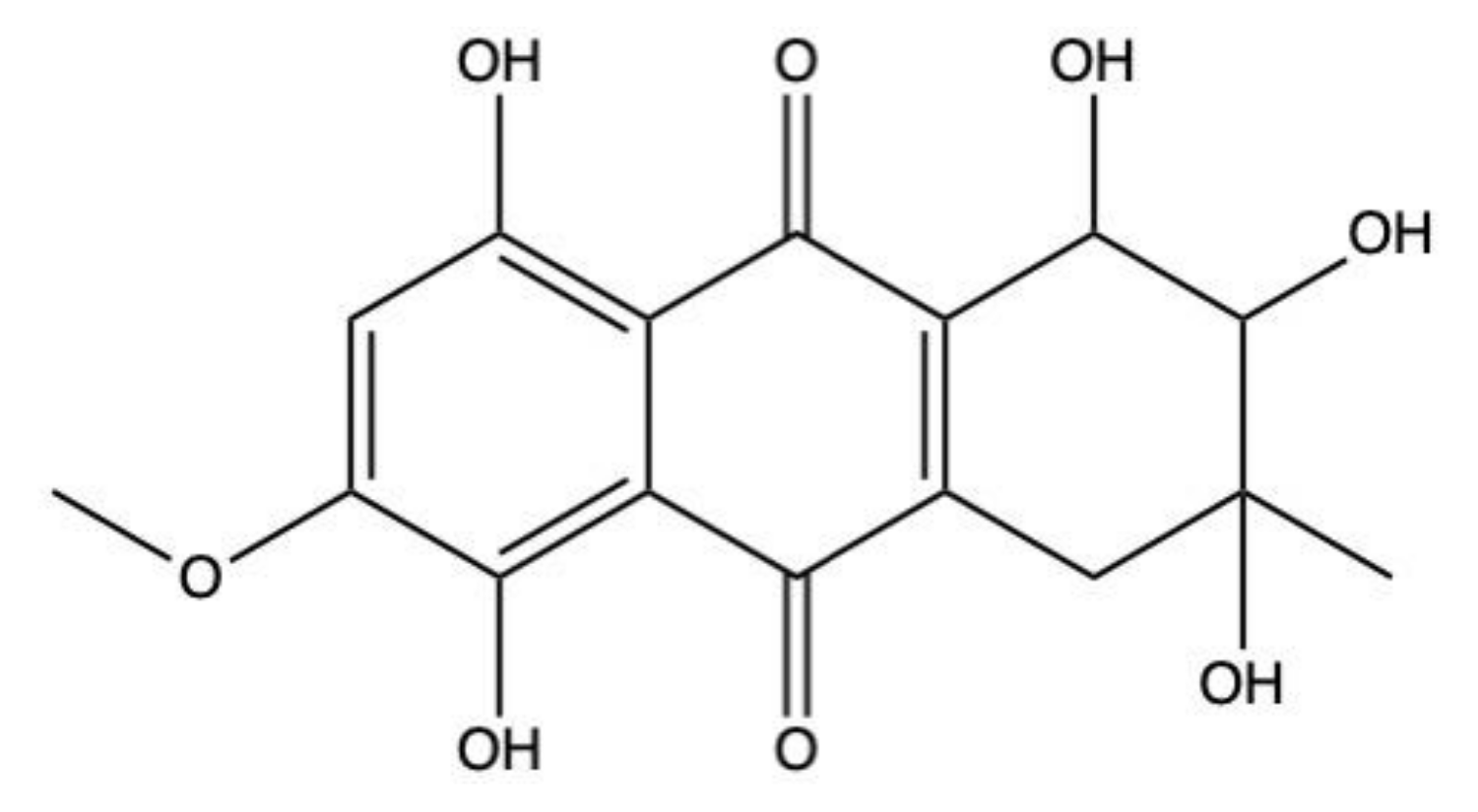
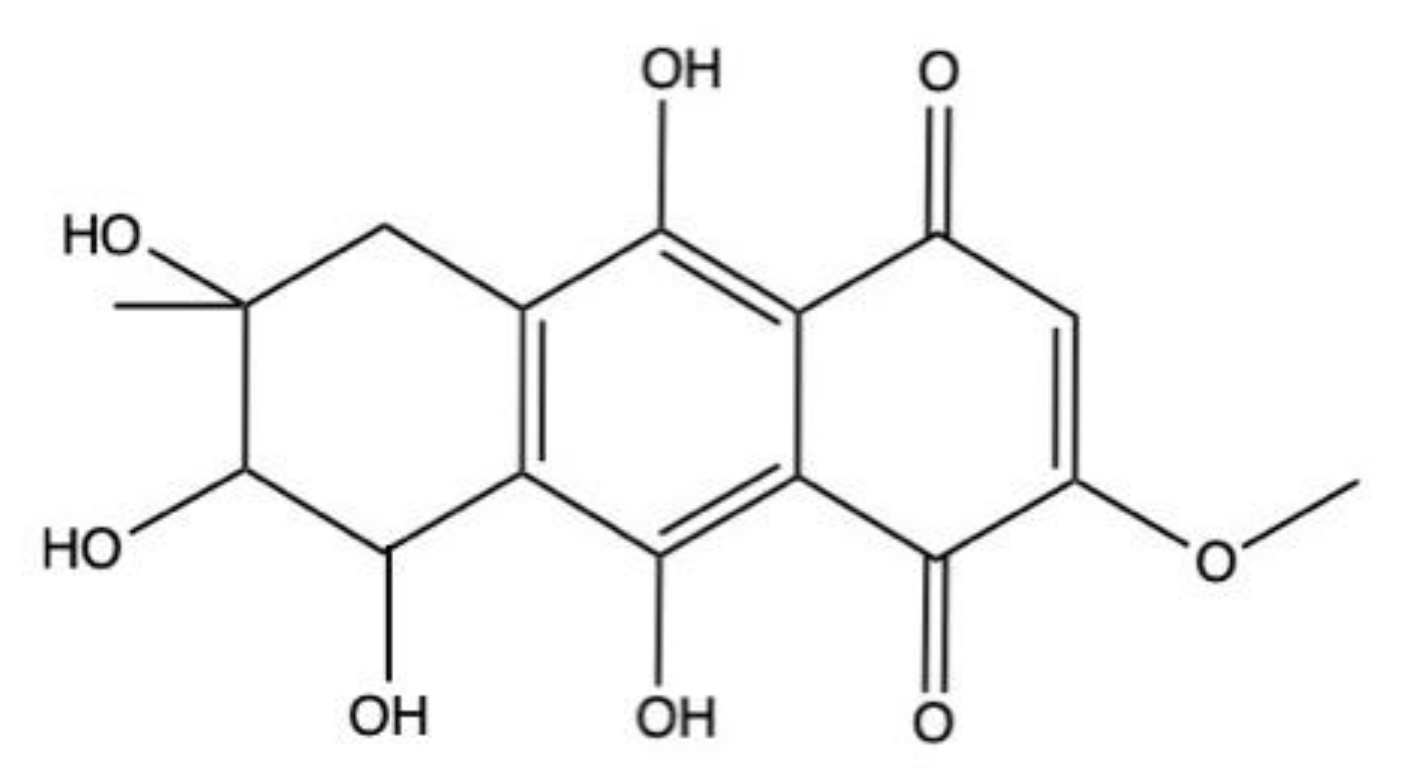
2.5. Bostrycin
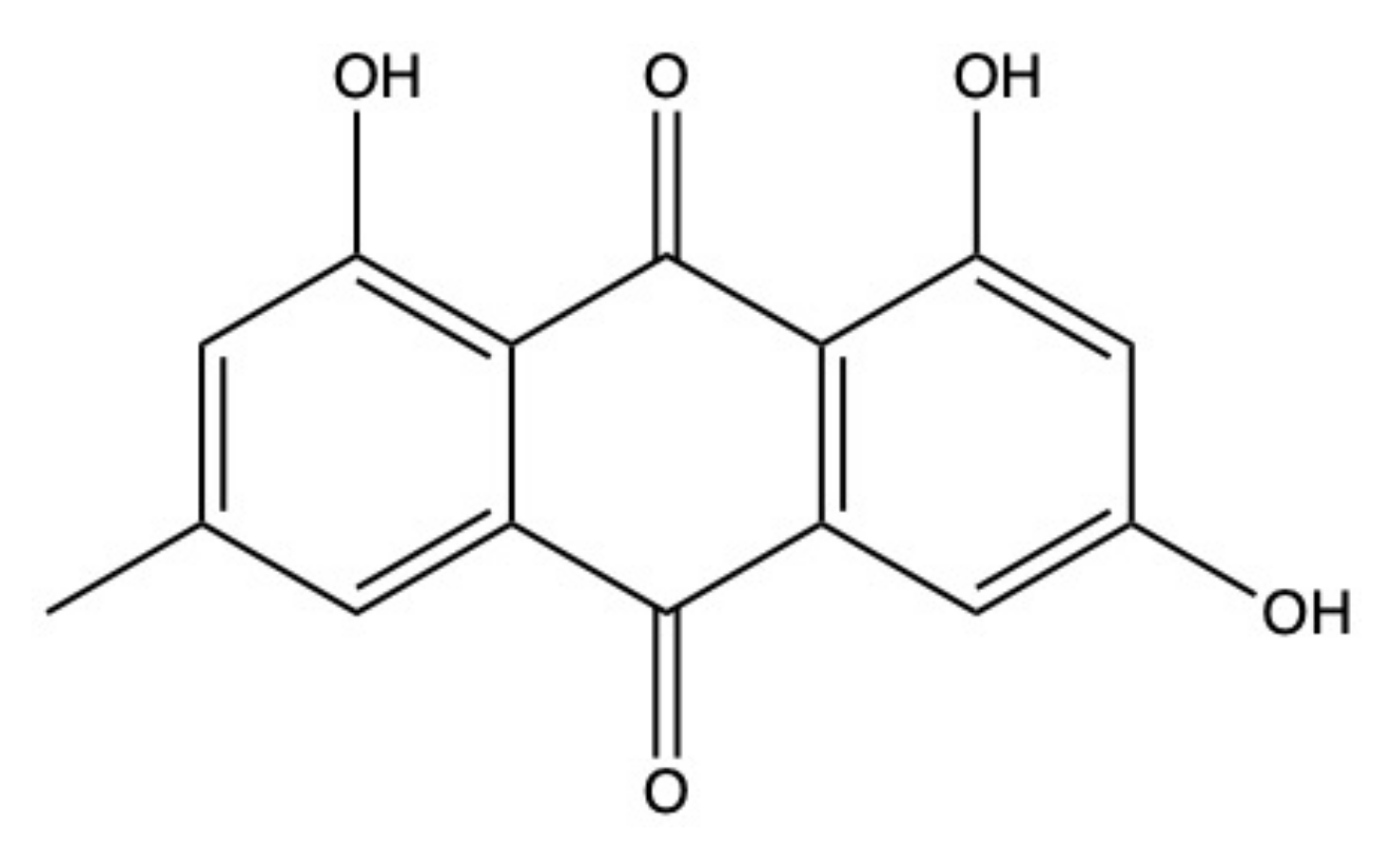
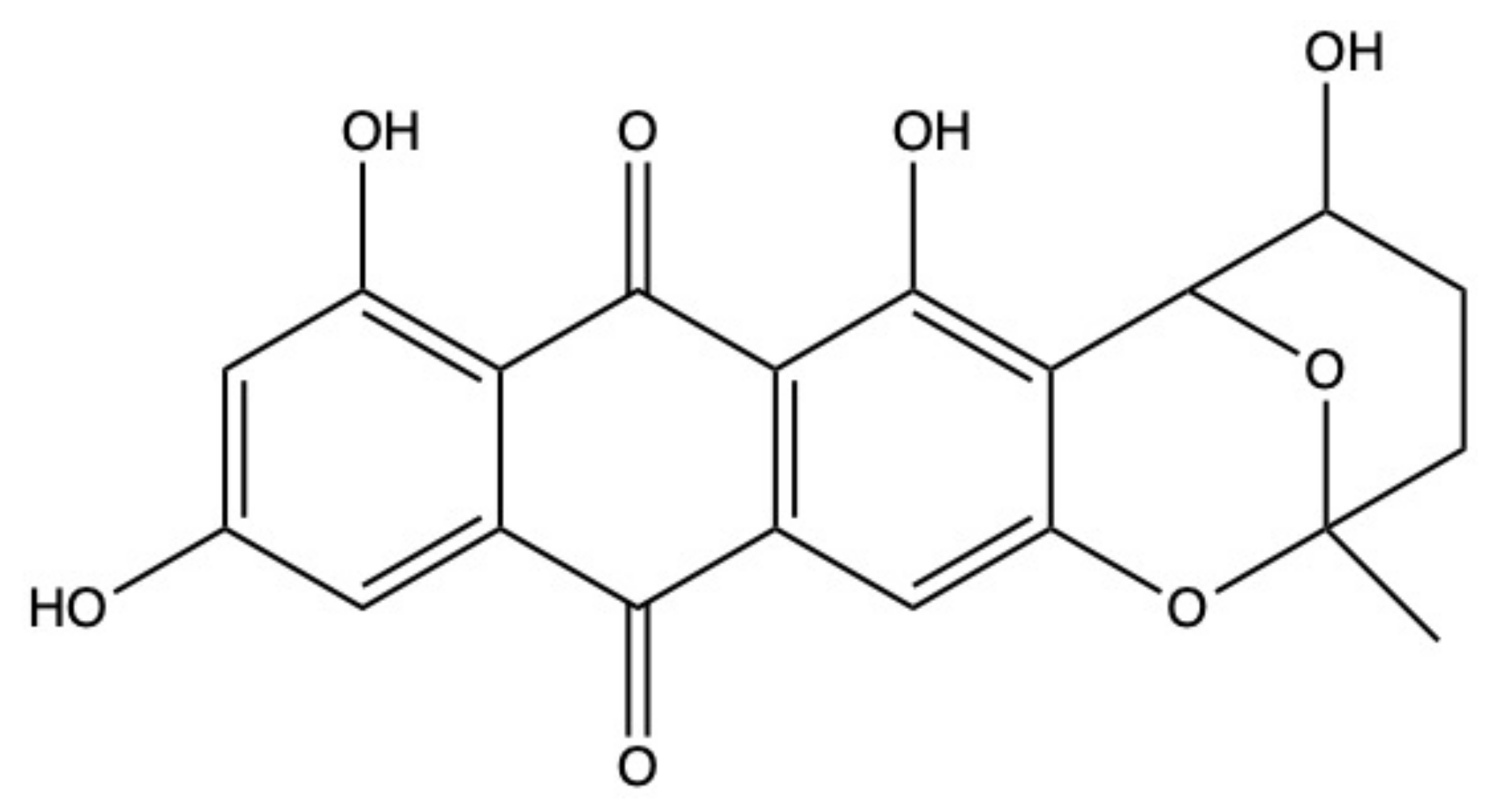
2.6. Nidurufin
2.7. G503
2.8. SZ-685C
2.9. 1403P-3
3. Anticancer Mechanisms of AQs from Other Marine Sources
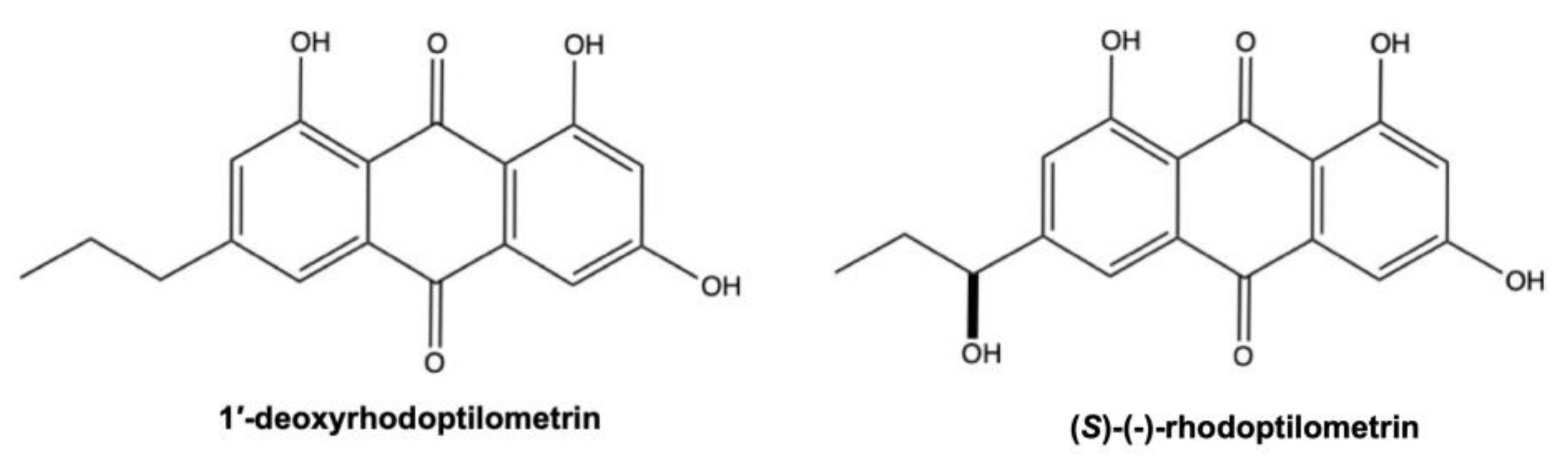
3.1. 1’-Deoxyrhodoptilometrin and (S)-(−)-Rhodoptilometrin
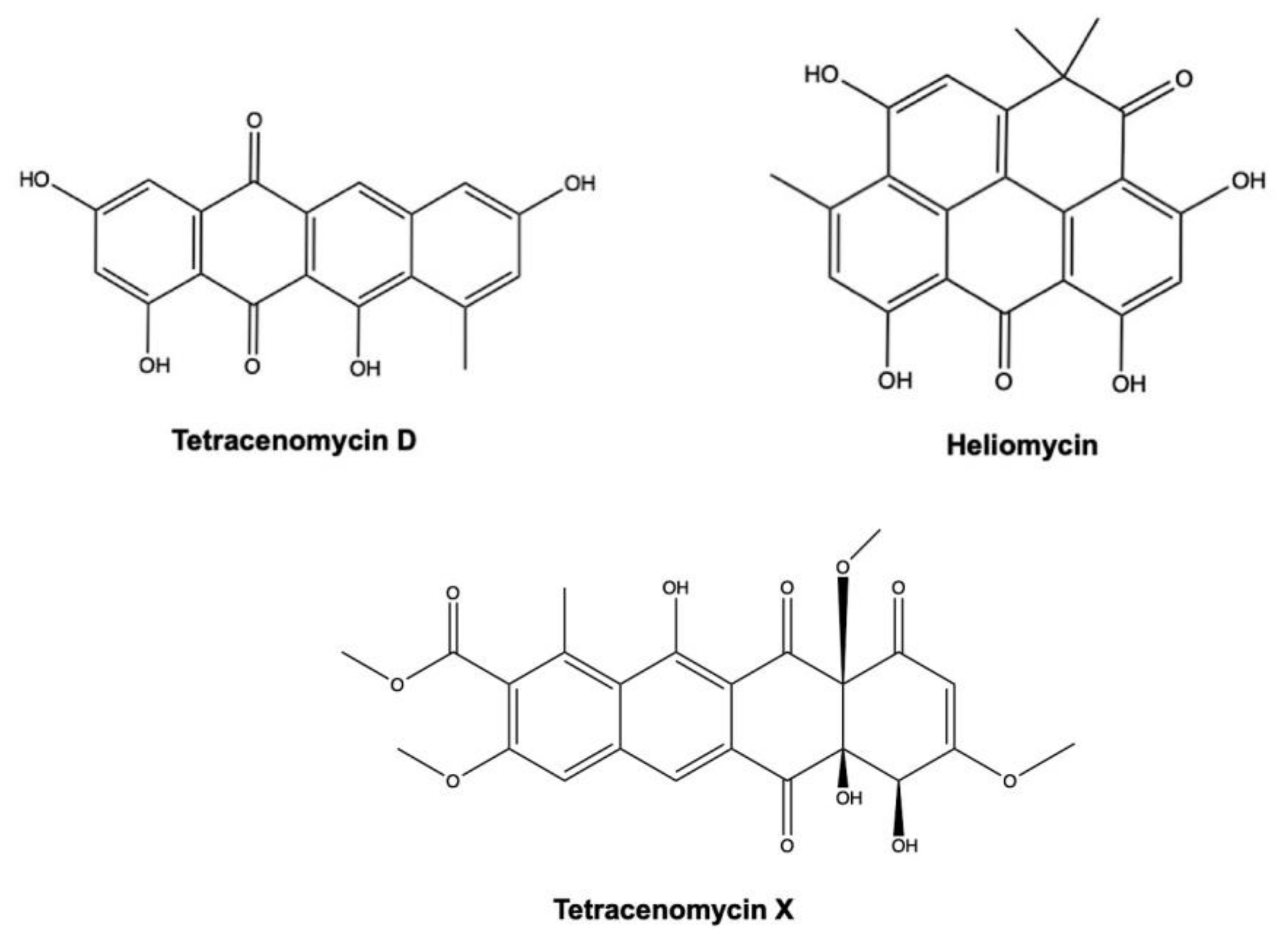
3.2. Tetracenomycin D and Heliomycin
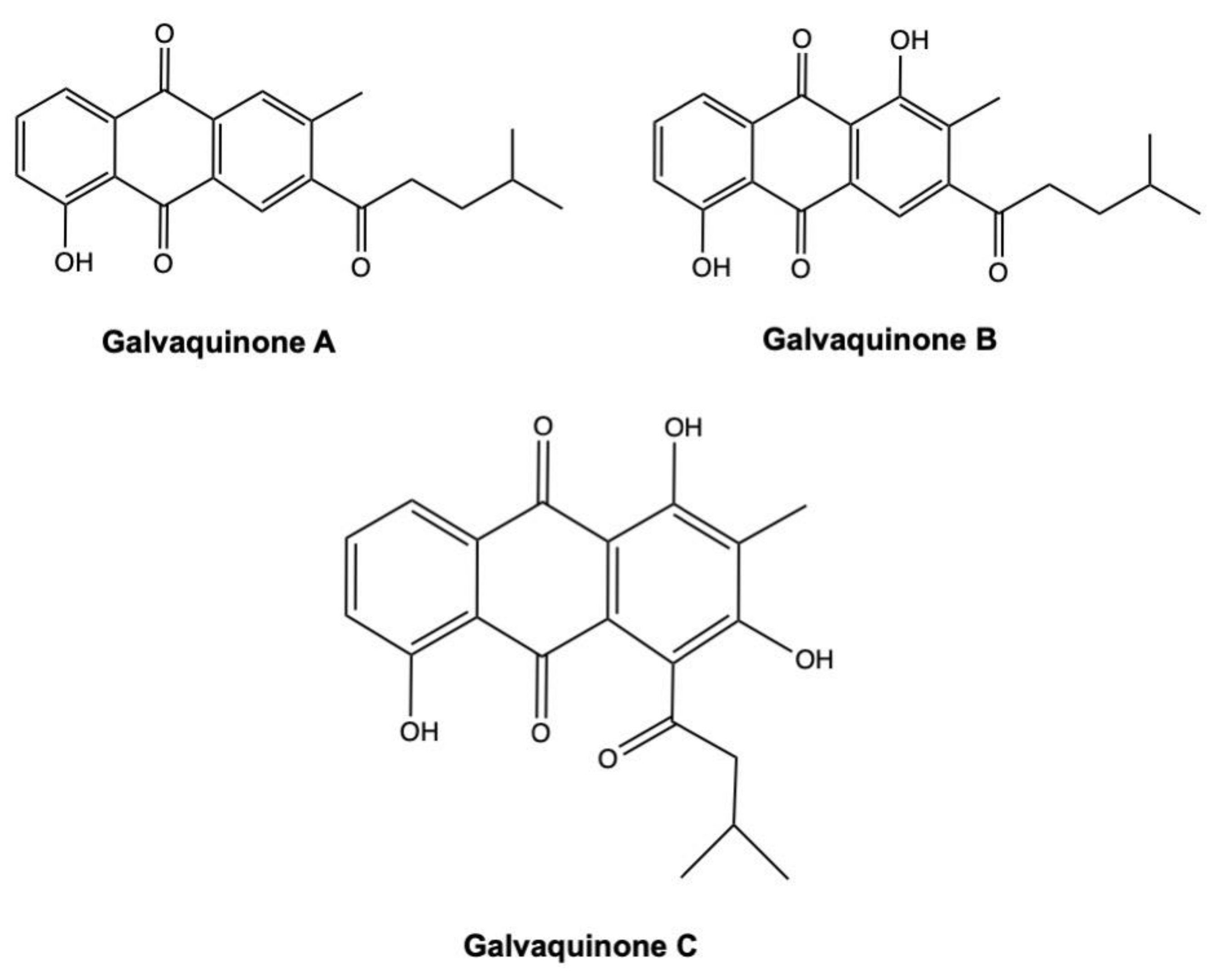
3.3. Galvaquinones
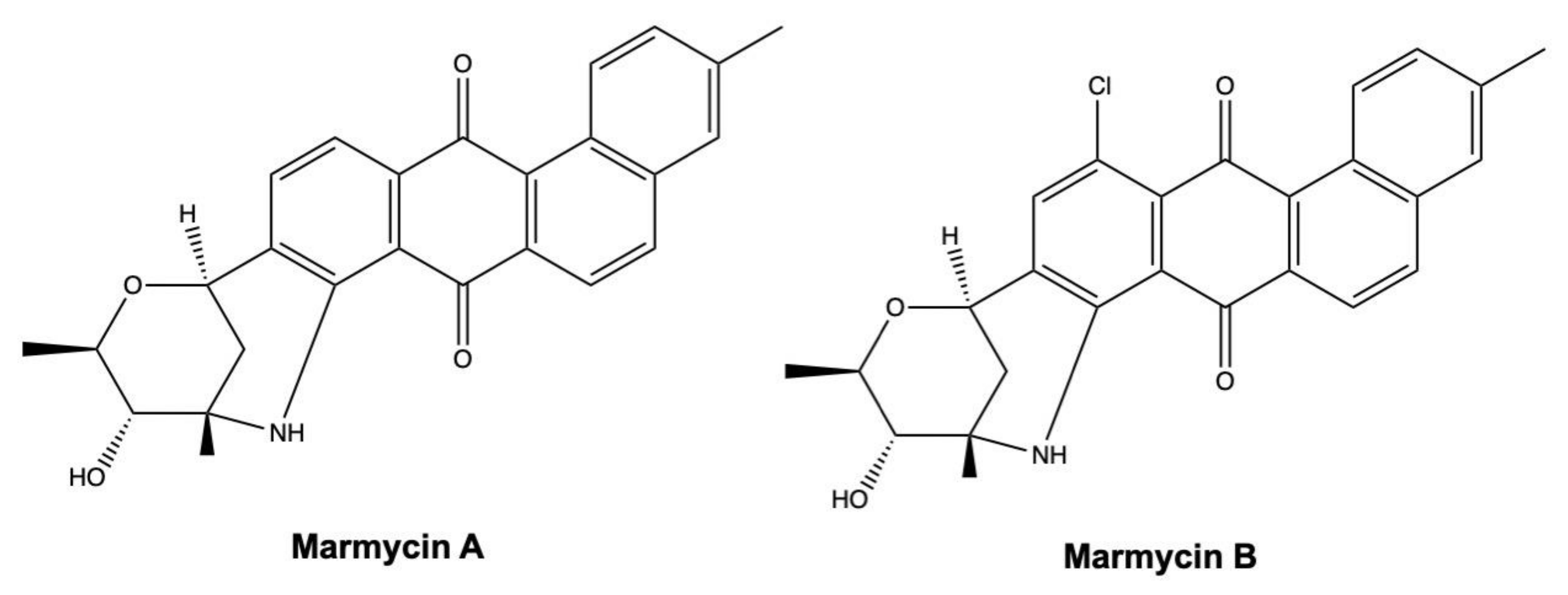
3.4. Angucyclines
4. Genotoxicity of AQs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- IARC–International Agency for Research on Cancer. Available online: https://www.iarc.who.int/ (accessed on 23 March 2021).
- Appeltans, W.; Ahyong, S.T.; Anderson, G.; Angel, M.V.; Artois, T.; Bailly, N.; Bamber, R.; Barber, A.; Bartsch, I.; Berta, A.; et al. The Magnitude of Global Marine Species Diversity. Curr. Biol. 2012, 22, 2189–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.-E.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine Natural Products: A Source of Novel Anticancer Drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [Green Version]
- Dyshlovoy, S.A.; Honecker, F. Marine Compounds and Cancer: Updates 2020. Mar. Drugs 2020, 18, 643. [Google Scholar] [CrossRef] [PubMed]
- Fouillaud, M.; Venkatachalam, M.; Girard-Valenciennes, E.; Caro, Y.; Dufossé, L. Anthraquinones and Derivatives from Marine-Derived Fungi: Structural Diversity and Selected Biological Activities. Mar. Drugs 2016, 14, 64. [Google Scholar] [CrossRef] [Green Version]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
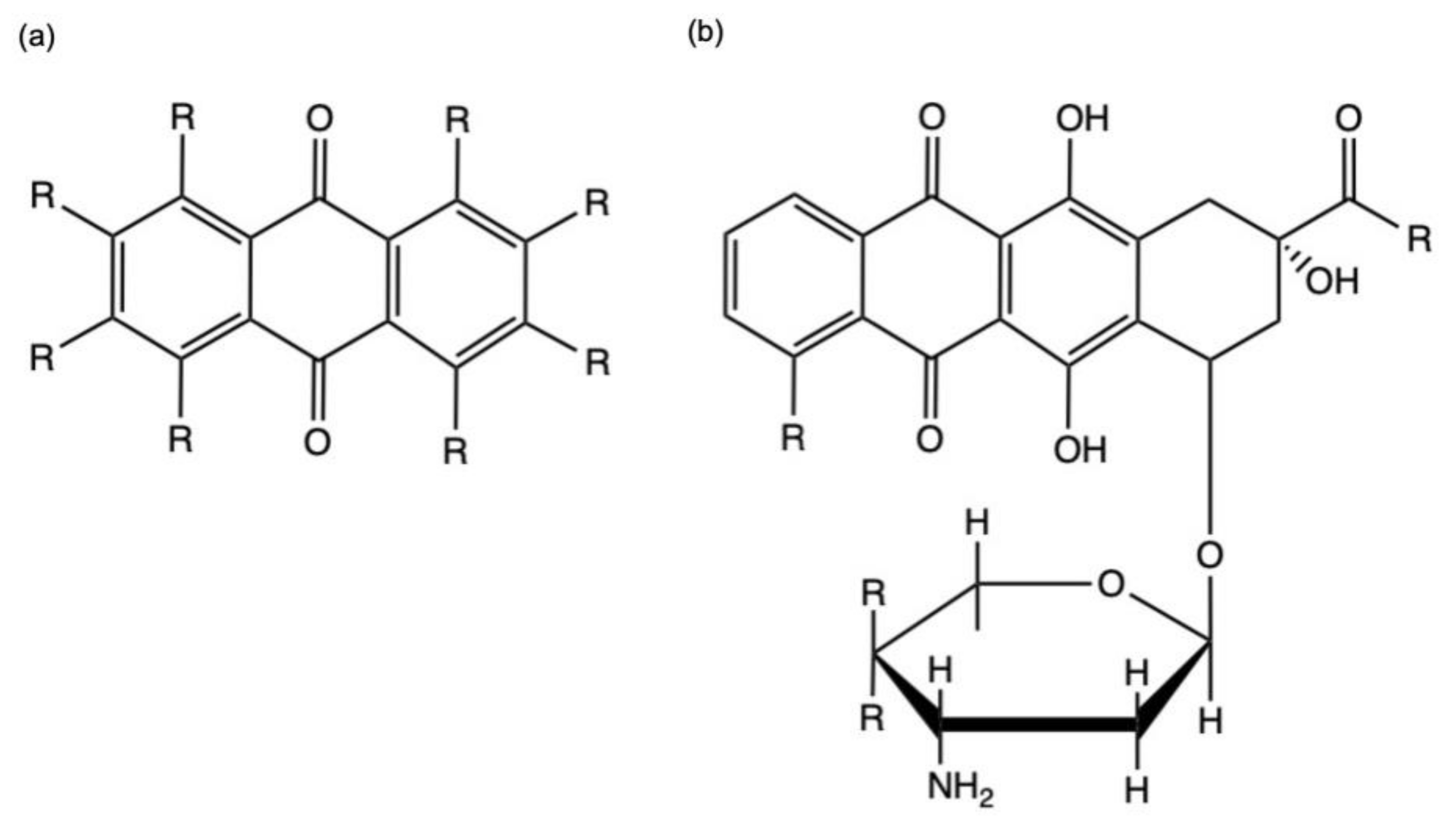
- Diaz-Muñoz, G.; Miranda, I.L.; Sartori, S.K.; de Rezende, D.C.; Diaz, M.A.N. Anthraquinones: An overview. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 58, pp. 313–338. ISBN 978-0-444-64056-7. [Google Scholar]
- Malik, E.M.; Müller, C.E. Anthraquinones as Pharmacological Tools and Drugs. Med. Res. Rev. 2016, 36, 705–748. [Google Scholar] [CrossRef]
- Wuthi-udomlert, M.; Kupittayanant, P.; Gritsanapan, W. In Vitro Evaluation of Antifungal Activity of Anthraquione Derivatives of Senna Alata. J. Health Res. 2010, 24, 117–122. [Google Scholar]
- Malmir, M.; Serrano, R.; Silva, O. Anthraquinones as potential antimicrobial agents-a review. In Antimicrobial Research: Novel Bioknowledge and Educational Programs; Mendez-Vilas, A., Ed.; Formatex Research Center S.L.: Badajoz, Spain, 2017; pp. 55–61. [Google Scholar]
- Osman, C.P.; Ismail, N.H. Antiplasmodial Anthraquinones from Medicinal Plants: The Chemistry and Possible Mode of Actions. Nat. Prod. Commun. 2018, 13, 1934578X1801301. [Google Scholar] [CrossRef] [Green Version]
- Chien, S.-C.; Wu, Y.-C.; Chen, Z.-W.; Yang, W.-C. Naturally Occurring Anthraquinones: Chemistry and Therapeutic Potential in Autoimmune Diabetes. Evid-Based Compl. Alt. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, A.D.; Panchal, P.V.; Harle, U.N.; Nanda, R.K.; Shaikh, H.M. Anti-Inflammatory and Antiarthritic Activity of Anthraquinone Derivatives in Rodents. Int. J. Inflam. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-M.; Wu, S.-C.; Chung, W.-J.; Lin, H.-C.; Chen, K.-T.; Chen, Y.-C.; Hsu, M.-F.; Yang, J.-M.; Wang, J.-P.; Lin, C.-N. Antiplatelet Effect and Selective Binding to Cyclooxygenase (COX) by Molecular Docking Analysis of Flavonoids and Lignans. Int. J. Mol. Sci. 2007, 8, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Seo, E.J.; Ngoc, T.M.; Lee, S.-M.; Kim, Y.S.; Jung, Y.-S. Chrysophanol-8-O-Glucoside, an Anthraquinone Derivative in Rhubarb, Has Antiplatelet and Anticoagulant Activities. J. Pharmacol. Sci. 2012, 118, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Jackson, T.C.; Verrier, J.D.; Kochanek, P.M. Anthraquinone-2-Sulfonic Acid (AQ2S) Is a Novel Neurotherapeutic Agent. Cell Death Dis. 2013, 4, e451. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Wang, C.; Li, D.; Hou, H. Novel Anthraquinone Compounds as Anticancer Agents and Their Potential Mechanism. Future Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Babykutty, S.; Sathiadevan, P.P.; Srinivas, P. Molecular Mechanism of Emodin Action: Transition from Laxative Ingredient to an Antitumor Agent. Med. Res. Rev. 2007, 27, 591–608. [Google Scholar] [CrossRef]
- Huang, Q.; Lu, G.; Shen, H.-M.; Chung, M.C.M.; Ong, C.N. Anti-Cancer Properties of Anthraquinones from Rhubarb. Med. Res. Rev. 2007, 27, 609–630. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeyer, J.; Kohlmeyer, E. Marine Mycology; Elsevier: London, UK, 1979; p. 704. [Google Scholar]
- Pang, K.-L.; Overy, D.P.; Jones, E.B.G.; da Luz Calado, M.; Burgaud, G.; Walker, A.K.; Johnson, J.A.; Kerr, R.G.; Cha, H.-J.; Bills, G.F. ‘Marine Fungi’ and ‘Marine-Derived Fungi’ in Natural Product Chemistry Research: Toward a New Consensual Definition. Fungal Biol. Rev. 2016, 30, 163–175. [Google Scholar] [CrossRef]
- Rateb, M.E.; Ebel, R. Secondary Metabolites of Fungi from Marine Habitats. Nat. Prod. Rep. 2011, 28, 290. [Google Scholar] [CrossRef]
- Richards, T.A.; Jones, M.D.M.; Leonard, G.; Bass, D. Marine Fungi: Their Ecology and Molecular Diversity. Ann. Rev. Mar. Sci. 2012, 4, 495–522. [Google Scholar] [CrossRef]
- Liu, L.; Zheng, Y.-Y.; Shao, C.-L.; Wang, C.-Y. Metabolites from Marine Invertebrates and Their Symbiotic Microorganisms: Molecular Diversity Discovery, Mining, and Application. Mar. Life Sci. Technol. 2019, 1, 60–94. [Google Scholar] [CrossRef] [Green Version]
- Gessler, N.N.; Egorova, A.S.; Belozerskaya, T.A. Fungal Anthraquinones. Appl. Biochem. Microbiol. 2013, 49, 85–99. [Google Scholar] [CrossRef]
- Huang, H.; Wang, F.; Luo, M.; Chen, Y.; Song, Y.; Zhang, W.; Zhang, S.; Ju, J. Halogenated Anthraquinones from the Marine-Derived Fungus Aspergillus Sp. SCSIO F063. J. Nat. Prod. 2012, 75, 1346–1352. [Google Scholar] [CrossRef]
- Du, L.; Zhu, T.; Liu, H.; Fang, Y.; Zhu, W.; Gu, Q. Cytotoxic Polyketides from a Marine-Derived Fungus Aspergillus Glaucus. J. Nat. Prod. 2008, 71, 1837–1842. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liao, Y.; Tang, C.; Huang, X.; Luo, Z.; Chen, J.; Cai, P. Cytotoxic and Antibacterial Compounds from the Coral-Derived Fungus Aspergillus Tritici SP2-8-1. Mar. Drugs 2017, 15, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.-K.; Huang, H.-R.; She, Z.-G.; Shao, C.-L.; Liu, F.; Cai, X.-L.; Vrijmoed, L.L.P.; Lin, Y.-C. 1H And13C NMR Assignments for Five Anthraquinones from the Mangrove Endophytic Fungus Halorosellinia Sp. (No. 1403). Magn. Reson. Chem. 2007, 45, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.-Z.; Gao, S.; Gao, Y.-X.; Wang, A.-R.; Xu, Y.-B.; Sun, R.; Hu, P.-G.; Yang, G.-F.; Li, A.-J.; Zhong, D.; et al. Novel Dibenzo[b,e]Oxepinones from the Freshwater-Derived Fungus Chaetomium Sp. YMF 1.02105. Planta Med. 2012, 78, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, H.; Xia, X.; Liang, Y.; Yan, Y.; She, Z.; Lin, Y.; Fu, L. Anthracenedione Derivative 1403P-3 Induces Apoptosis in KB and KBv200 Cells via Reactive Oxygen Species-Independent Mitochondrial Pathway and Death Receptor Pathway. Cancer Biol. Ther. 2007, 6, 1409–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; He, Z.; Wu, J.; Lin, Y.; Zhu, X. A Novel Adriamycin Analogue Derived from Marine Microbes Induces Apoptosis by Blocking Akt Activation in Human Breast Cancer Cells. Mol. Med. Rep. 2011, 4, 261–265. [Google Scholar] [CrossRef]
- Zhou, X.-M.; Zheng, C.-J.; Chen, G.-Y.; Song, X.-P.; Han, C.-R.; Li, G.-N.; Fu, Y.-H.; Chen, W.-H.; Niu, Z.-G. Bioactive Anthraquinone Derivatives from the Mangrove-Derived Fungus Stemphylium Sp. 33231. J. Nat. Prod. 2014, 77, 2021–2028. [Google Scholar] [CrossRef]
- Li, H.-L.; Li, X.-M.; Li, X.; Wang, C.-Y.; Liu, H.; Kassack, M.U.; Meng, L.-H.; Wang, B.-G. Antioxidant Hydroanthraquinones from the Marine Algal-Derived Endophytic Fungus Talaromyces Islandicus EN-501. J. Nat. Prod. 2017, 80, 162–168. [Google Scholar] [CrossRef]
- Abdel-Wahab, N.; Scharf, S.; Özkaya, F.; Kurtán, T.; Mándi, A.; Fouad, M.; Kamel, M.; Müller, W.; Kalscheuer, R.; Lin, W.; et al. Induction of Secondary Metabolites from the Marine-Derived Fungus Aspergillus Versicolor through Co-Cultivation with Bacillus Subtilis. Planta Med. 2019, 85, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Trisuwan, K.; Khamthong, N.; Rukachaisirikul, V.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. Anthraquinone, Cyclopentanone, and Naphthoquinone Derivatives from the Sea Fan-Derived Fungi Fusarium Spp. PSU-F14 and PSU-F135. J. Nat. Prod. 2010, 73, 1507–1511. [Google Scholar] [CrossRef]
- Zheng, C.-J.; Shao, C.-L.; Guo, Z.-Y.; Chen, J.-F.; Deng, D.-S.; Yang, K.-L.; Chen, Y.-Y.; Fu, X.-M.; She, Z.-G.; Lin, Y.-C.; et al. Bioactive Hydroanthraquinones and Anthraquinone Dimers from a Soft Coral-Derived Alternaria Sp. Fungus. J. Nat. Prod. 2012, 75, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Pan, J.-H.; Chen, B.; Yu, M.; Huang, H.-B.; Zhu, X.; Lu, Y.-J.; She, Z.-G.; Lin, Y.-C. Three Bianthraquinone Derivatives from the Mangrove Endophytic Fungus Alternaria Sp. ZJ9-6B from the South China Sea. Mar. Drugs 2011, 9, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jin, H.; Song, B.; Zhu, X.; Zhao, H.; Cai, J.; Lu, Y.; Chen, B.; Lin, Y. The Cytotoxicity and Anticancer Mechanisms of Alterporriol L, a Marine Bianthraquinone, against MCF-7 Human Breast Cancer Cells. Appl. Microbiol. Biotechnol. 2012, 93, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, Y.-B.; Kurtán, T.; Liu, M.-X.; Tang, H.; Zhuang, C.-L.; Zhang, W. Anthraquinone Derivatives from a Coral Associated Fungus Stemphylium Lycopersici. Nat. Prod. Res. 2020, 34, 2116–2123. [Google Scholar] [CrossRef]
- Qiao, L.; Duan, Z.; Chen, Y.; Luan, Y.; Gu, Q.; Liu, Y.-K.; Li, D. Aspergiolides A and B: Core Structural Establishment and Synthesis of Structural Analogues. J. Org. Chem. 2019, 84, 4451–4457. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhu, T.; Fang, Y.; Liu, H.; Gu, Q.; Zhu, W. Aspergiolide A, a Novel Anthraquinone Derivative with Naphtho[1,2,3-de]Chromene-2,7-Dione Skeleton Isolated from a Marine-Derived Fungus Aspergillus Glaucus. Tetrahedron 2007, 63, 1085–1088. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Qi, X.; Li, D.; Zhu, T.; Mo, X. Anticancer Efficacy and Absorption, Distribution, Metabolism, and Toxicity Studies of Aspergiolide A in Early Drug Development. Drug Des. Devel. Ther. 2014, 1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Ai, J.; Li, D.; Zhu, T.; Wang, Y.; Knauer, M.; Bruhn, T.; Liu, H.; Geng, M.; Gu, Q.; et al. Aspergiolides C and D: Spirocyclic Aromatic Polyketides with Potent Protein Kinase c-Met Inhibitory Effects. Chem. Eur. J. 2011, 17, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Sun, C.; Feng, Y.; Wang, L.; Peng, J.; Che, Q.; Gu, Q.; Zhu, T.; Li, D.; Zhang, G. Anthraquinone Derivatives from a Marine-Derived Fungus Sporendonema Casei HDN16-802. Mar. Drugs 2019, 17, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Liu, W. Nidurufin as a New Cell Cycle Inhibitor from Marine-Derived Fungus Penicillium Flavidorsum SHK1-27. Arch. Pharm. Res. 2011, 34, 901–905. [Google Scholar] [CrossRef]
- Lee, Y.M.; Li, H.; Hong, J.; Cho, H.Y.; Bae, K.S.; Kim, M.A.; Kim, D.-K.; Jung, J.H. Bioactive Metabolites from the Sponge-Derived Fungus Aspergillus Versicolor. Arch. Pharm. Res. 2010, 33, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Fang, L.K.; Liu, J.W.; Cheng, W.Q.; Yun, M. Effect of Marine Fungal Metabolites from the South China Sea on Prostate Cancer Cell Line DU-145. J. Intern. Med. 2008, 35, 562–563. [Google Scholar]
- Chen, C.Q.; Fang, L.K.; Liu, J.W. Effects of Marine Fungal Metabolites 1386A from the South China Sea on Proliferation, Apoptosis and Mitochondrial Membrane Potential in Gastric Cancer Cell Line MCG-803. Chin. J. Pathophys 2010, 26, 1908–1912. [Google Scholar]
- Xia, X.; Li, Q.; Li, J.; Shao, C.; Zhang, J.; Zhang, Y.; Liu, X.; Lin, Y.; Liu, C.; She, Z. Two New Derivatives of Griseofulvin from the Mangrove Endophytic Fungus Nigrospora Sp. (Strain No. 1403) from Kandelia Candel (L.) Druce. Planta Med. 2011, 77, 1735–1738. [Google Scholar] [CrossRef]
- Buttachon, S.; May Zin, W.; Dethoup, T.; Gales, L.; Pereira, J.; Silva, A.; Kijjoa, A. Secondary Metabolites from the Culture of the Marine Sponge-Associated Fungi Talaromyces Tratensis and Sporidesmium Circinophorum. Planta Med. 2016, 82, 888–896. [Google Scholar] [CrossRef] [Green Version]
- Buttachon, S.; Ramos, A.A.; Inácio, Â.; Dethoup, T.; Gales, L.; Lee, M.; Costa, P.M.; Silva, A.M.S.; Sekeroglu, N.; Rocha, E. Bis-Indolyl Benzenoids, Hydroxypyrrolidine Derivatives and Other Constituents from Cultures of the Marine Sponge-Associated Fungus Aspergillus Candidus KUFA0062. Mar. Drugs 2018, 16, 119. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, H.; Rotinsulu, H.; Kaneko, T.; Murakami, K.; Fujiwara, H.; Ukai, K.; Namikoshi, M. A New Dibenz[b,e]Oxepine Derivative, 1-Hydroxy-10-Methoxy-Dibenz[b,e]Oxepin-6,11-Dione, from a Marine-Derived Fungus, Beauveria Bassiana TPU942. Mar. Drugs 2012, 10, 2691–2697. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tao, L.; Liang, Y.; Chen, L.; Mi, Y.; Zheng, L.; Wang, F.; She, Z.; Lin, Y.; To, K.K.W.; et al. Anthracenedione Derivatives as Anticancer Agents Isolated from Secondary Metabolites of the Mangrove Endophytic Fungi. Mar. Drugs 2010, 8, 1469–1481. [Google Scholar] [CrossRef]
- Tan, Q.-W.; Ouyang, M.-A.; Shen, S.; Li, W. Bioactive Metabolites from a Marine-Derived Strain of the Fungus Neosartorya Fischeri. Nat. Prod. Res. 2012, 26, 1402–1407. [Google Scholar] [CrossRef]
- Pang, X.; Cai, G.; Lin, X.; Salendra, L.; Zhou, X.; Yang, B.; Wang, J.; Wang, J.; Xu, S.; Liu, Y. New Alkaloids and Polyketides from the Marine Sponge-Derived Fungus Penicillium Sp. SCSIO41015. Mar. Drugs 2019, 17, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Cai, X.; Pan, J.; Gao, J.; Li, J.; Yuan, J.; Fu, L.; She, Z.; Lin, Y. Structure Elucidation and NMR Assignments for Three Anthraquinone Derivatives from the Marine Fungus Fusarium Sp. (No. ZH-210). Magn. Reson Chem. 2009, 47, 362–365. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, T.; Li, S.; Duan, J.; Ye, F.; Li, H.; She, Z.; Gao, G.; Yang, X. Anthraquinone G503 Induces Apoptosis in Gastric Cancer Cells through the Mitochondrial Pathway. PLoS ONE 2014, 9, e108286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.-H.; Liu, D.; Xu, Y.; Chen, J.-L.; Lin, W.-H. Antioxidant Xanthones and Anthraquinones Isolated from a Marine-Derived Fungus Aspergillus Versicolor. Chin. J. Nat. Med. 2018, 16, 219–224. [Google Scholar] [CrossRef]
- Wang, C.C.C.; Chiang, Y.-M.; Kuo, P.-L.; Chang, J.-K.; Hsu, Y.-L. Norsolorinic Acid from Aspergillus Nidulans Inhibits the Proliferation of Human Breast Adenocarcinoma MCF-7 Cells via Fas-Mediated Pathway. Basic Clin. Pharmacol. Toxicol. 2008, 102, 491–497. [Google Scholar] [CrossRef]
- Khamthong, N.; Rukachaisirikul, V.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. Bioactive Polyketides from the Sea Fan-Derived Fungus Penicillium Citrinum PSU-F51. Tetrahedron 2012, 68, 8245–8250. [Google Scholar] [CrossRef]
- Wijesekara, I.; Zhang, C.; Van Ta, Q.; Vo, T.-S.; Li, Y.-X.; Kim, S.-K. Physcion from Marine-Derived Fungus Microsporum Sp. Induces Apoptosis in Human Cervical Carcinoma HeLa Cells. Microbiol. Res. 2014, 169, 255–261. [Google Scholar] [CrossRef]
- Wang, X.; Tan, T.; Mao, Z.-G.; Lei, N.; Wang, Z.-M.; Hu, B.; Chen, Z.-Y.; She, Z.-G.; Zhu, Y.-H.; Wang, H.-J. The Marine Metabolite SZ-685C Induces Apoptosis in Primary Human Nonfunctioning Pituitary Adenoma Cells by Ihibition of the Akt Pathway in Vitro. Mar. Drugs 2015, 13, 1569–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-H.; Xiao, W.-W.; Jiang, X.-B.; Wang, J.-W.; Mao, Z.-G.; Lei, N.; Fan, X.; Song, B.-B.; Liao, C.-X.; Wang, H.-J.; et al. A Novel Marine Drug, SZ-685C, Induces Apoptosis of MMQ Pituitary Tumor Cells by Downregulating MiR-200c. Curr. Med. Chem. 2013, 20, 2145–2154. [Google Scholar] [CrossRef]
- Wang, D.; Wang, S.; Liu, Q.; Wang, M.; Wang, C.; Yang, H. SZ-685C Exhibits Potent Anticancer Activity in Both Radiosensitive and Radioresistant NPC Cells through the MiR-205-PTEN-Akt Pathway. Oncol. Rep. 2013, 29, 2341–2347. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; He, Z.; Wu, J.; Yuan, J.; Wen, W.; Hu, Y.; Jiang, Y.; Lin, C.; Zhang, Q.; Lin, M.; et al. A Marine Anthraquinone SZ-685C Overrides Adriamycin-Resistance in Breast Cancer Cells through Suppressing Akt Signaling. Mar. Drugs 2012, 10, 694–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Zhu, X.; Li, Q.; Gu, M.; He, Z.; Wu, J.; Li, J.; Lin, Y.; Li, M.; She, Z.; et al. SZ-685C, a Marine Anthraquinone, Is a Potent Inducer of Apoptosis with Anticancer Activity by Suppression of the Akt/FOXO Pathway: SZ-685C Induces Apoptosis and Inhibits Tumour Growth. Br. J. Pharmacol. 2010, 159, 689–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuli, H.S.; Aggarwal, V.; Tuorkey, M.; Aggarwal, D.; Parashar, N.C.; Varol, M.; Savla, R.; Kaur, G.; Mittal, S.; Sak, K. Emodin: A Metabolite That Exhibits Anti-Neoplastic Activities by Modulating Multiple Oncogenic Targets. Toxicol. In Vitro 2021, 73, 105142. [Google Scholar] [CrossRef]
- Ye, F.; Chen, C.; Qin, J.; Liu, J.; Zheng, C. Genetic Profiling Reveals an Alarming Rate of Cross-Contamination among Human Cell Lines Used in China. FASEB J. 2015, 29, 4268–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, X.; Yang, Z.; Feng, H.; Sun, H.; Liu, Y. A Combination of Species Identification and STR Profiling Identifies Cross-Contaminated Cells from 482 Human Tumor Cell Lines. Sci. Rep. 2017, 7, 9774. [Google Scholar] [CrossRef] [Green Version]
- Kamiloglu, S.; Sari, G.; Ozdal, T.; Capanoglu, E. Guidelines for Cell Viability Assays. Food Front. 2020, 1, 332–349. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT Signaling Pathway and Cancer: An Updated Review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Greco, G.; Catanzaro, E.; Fimognari, C. Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer. Cancers 2021, 13, 304. [Google Scholar] [CrossRef]
- Zhou, J.; Li, G.; Han, G.; Feng, S.; Liu, Y.; Chen, J.; Liu, C.; Zhao, L.; Jin, F. Emodin Induced Necroptosis in the Glioma Cell Line U251 via the TNF-α/RIP1/RIP3 Pathway. Invest. New Drugs 2020, 38, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The Role and Mechanisms of Action of MicroRNAs in Cancer Drug Resistance. Clin. Epigenet 2019, 11, 25. [Google Scholar] [CrossRef]
- Li, N.; Wang, C.; Zhang, P.; You, S. Emodin Inhibits Pancreatic Cancer EMT and Invasion by Up-regulating MicroRNA-1271. Mol. Med. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-Z.; Xu, J.-B.; Ji, X.; Chen, H.; Xu, H.-T.; Hu, P.; Chen, L.; Guo, J.-Q.; Chen, M.-Y.; Lu, D.; et al. Emodin Inhibits Angiogenesis in Pancreatic Cancer by Regulating the Transforming Growth Factor-β/Drosophila Mothers against Decapentaplegic Pathway and Angiogenesis-Associated MicroRNAs. Mol. Med. Rep. 2015, 12, 5865–5871. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Tsai, S.-J. Effect of Emodin on Cooked-Food Mutagen Activation. Food Chem. Toxicol. 1991, 29, 765–770. [Google Scholar] [CrossRef]
- Su, H.-Y.; Cherng, S.-H.; Chen, C.-C.; Lee, H. Emodin Inhibits the Mutagenicity and DNA Adducts Induced by 1-Nitropyrene. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1995, 329, 205–212. [Google Scholar] [CrossRef]
- Wu, C.H.; Hsieh, C.L.; Song, T.Y.; Yen, G.C. Inhibitory Effects of Cassia Tora L. on Benzo[a]Pyrene-Mediated DNA Damage toward HepG2 Cells. J. Agric. Food Chem. 2001, 49, 2579–2586. [Google Scholar] [CrossRef]
- Słoczyńska, K.; Powroźnik, B.; Pękala, E.; Waszkielewicz, A.M. Antimutagenic Compounds and Their Possible Mechanisms of Action. J. Appl. Genet. 2014, 55, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Bhattachar, S. Natural Antimutagens: A Review. Res. J. Med. Plant. 2011, 5, 116–126. [Google Scholar] [CrossRef] [Green Version]
- AbdelHakem, A.M.; Abdelhafez, E.-S.M.N. Current trends and future perspectives of antimutagenic agents. In Genotoxicity and Mutagenicity-Mechanisms and Test Methods; Soloneski, S.L., Larramendy, M., Eds.; IntechOpen: London, UK, 2021; ISBN 978-1-83880-041-3. [Google Scholar]
- Sun, M.; Sakakibara, H.; Ashida, H.; Danno, G.; Kanazawa, K. Cytochrome P4501A1-Inhibitory Action of Antimutagenic Anthraquinones in Medicinal Plants and the Structure-Activity Relationship. Biosci. Biotechnol. Biochem. 2000, 64, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Sevcovicova, A.; Bodnarova, K.; Loderer, D.; Imreova, P.; Galova, E.; Miadokova, E. Dual Activities of Emodin—DNA Protectivity vs Mutagenicity. Neuro Endocrinol. Lett. 2014, 35, 149–154. [Google Scholar] [PubMed]
- Shah, M.A.; Adnan, M.; Rasul, A.; Hussain, G.; Sarfraz, I.; Nageen, B.; Riaz, A.; Khalid, R.; Asrar, M.; Selamoglu, Z.; et al. Physcion and Physcion 8-O-β-D-Glucopyranoside: Natural Anthraquinones with Potential Anti-Cancer Activities. Curr. Drug Targets 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Szatmári, Z. Autophagy-from Molecular Mechanisms to Clinical Relevance. Cell Biol. Toxicol. 2017, 33, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.-J.; Yang, Z.; Zhang, X.-L.; Liu, Z.-F.; Fan, J.; Zhang, H.-Y. Physcion, a Naturally Occurring Anthraquinone Derivative, Induces Apoptosis and Autophagy in Human Nasopharyngeal Carcinoma. Acta Pharmacol. Sin. 2016, 37, 1623–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, T.F.; Sorbo, J.M.; de Oliveira Souza, F.; Fernandes, B.C.; Ocampos, F.M.M.; de Oliveira Soares, D.M.; Arcaro, C.A.; Assis, R.P.; Barison, A.; Miguel, O.G.; et al. Emodin, Physcion, and Crude Extract of Rhamnus Sphaerosperma var. Pubescens Induce Mixed Cell Death, Increase in Oxidative Stress, DNA Damage, and Inhibition of AKT in Cervical and Oral Squamous Carcinoma Cell Lines. Oxid Med. Cell Longev. 2018, 2018, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Zhang, J.; Tong, Y.; Li, J.; Liu, B. Physcion 8-O-β-Glucopyranoside Induced Ferroptosis via Regulating MiR-103a-3p/GLS2 Axis in Gastric Cancer. Life Sci. 2019, 237, 116893. [Google Scholar] [CrossRef]
- Durán, N.; Teixeira, M.F.S.; De Conti, R.; Esposito, E. Ecological-Friendly Pigments from Fungi. Crit. Rev. Food Sci. Nutr. 2002, 42, 53–66. [Google Scholar] [CrossRef]
- Mapari, S.A.; Nielsen, K.F.; Larsen, T.O.; Frisvad, J.C.; Meyer, A.S.; Thrane, U. Exploring Fungal Biodiversity for the Production of Water-Soluble Pigments as Potential Natural Food Colorants. Curr. Opin. Biotechnol. 2005, 16, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Frisvad, J.C.; Filtenborg, O. Terverticillate Penicillia: Chemotaxonomy and Mycotoxin Production. Mycologia 1989, 81, 837. [Google Scholar] [CrossRef]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. GammaH2AX: A Sensitive Molecular Marker of DNA Damage and Repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Solier, S.; Pommier, Y. The Nuclear γ-H2AX Apoptotic Ring: Implications for Cancers and Autoimmune Diseases. Cell Mol. Life Sci. 2014, 71, 2289–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.-S.; Hou, J.-N.; Guo, Y.-B.; Yang, H.-L.; Xie, C.-M.; Lin, Y.-C.; She, Z.-G. Bostrycin Inhibits Proliferation of Human Lung Carcinoma A549 Cells via Downregulation of the PI3K/Akt Pathway. J. Exp. Clin. Cancer Res. 2011, 30, 17. [Google Scholar] [CrossRef] [Green Version]
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of Regulatory Oncogenic or Tumor Suppressor MiRNAs of PI3K/AKT Signaling Axis in the Pathogenesis of Colorectal Cancer. Curr. Pharm. Des. 2019, 24, 4605–4610. [Google Scholar] [CrossRef] [PubMed]
- Gasparri, M.L.; Besharat, Z.M.; Farooqi, A.A.; Khalid, S.; Taghavi, K.; Besharat, R.A.; Sabato, C.; Papadia, A.; Panici, P.B.; Mueller, M.D.; et al. MiRNAs and Their Interplay with PI3K/AKT/MTOR Pathway in Ovarian Cancer Cells: A Potential Role in Platinum Resistance. J. Cancer Res. Clin. Oncol. 2018, 144, 2313–2318. [Google Scholar] [CrossRef]
- Rahmani, F.; Ziaeemehr, A.; Shahidsales, S.; Gharib, M.; Khazaei, M.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of Regulatory MiRNAs of the PI3K/AKT/MTOR Signaling in the Pathogenesis of Hepatocellular Carcinoma. J. Cell Physiol. 2020, 235, 4146–4152. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Pereira, D.M.; Valentão, P.; Correia-da-Silva, G.; Teixeira, N.; Andrade, P.B. Translating Endoplasmic Reticulum Biology into the Clinic: A Role for ER-Targeted Natural Products? Nat. Prod. Rep. 2015, 32, 705–722. [Google Scholar] [CrossRef]
- Schirrmacher, V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Gandellini, P. Unveiling the Ups and Downs of MiR-205 in Physiology and Cancer: Transcriptional and Post-Transcriptional Mechanisms. Cell Death Dis. 2020, 11, 980. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Wang, E.L.; Zhou, H.-M.; Yoshimoto, K.; Qian, Z.R. MicroRNAs in Human Pituitary Adenomas. Int. J. Endocrinol. 2014, 2014, 435171. [Google Scholar] [CrossRef]
- Los, M.; Maddika, S.; Erb, B.; Schulze-Osthoff, K. Switching Akt: From Survival Signaling to Deadly Response. Bioessays 2009, 31, 492–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and Regulation of Apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [Green Version]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the Act between Death Receptors and Mitochondria. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.; Nielson, J.; Tapiolas, D.; Motti, C.; Ovenden, S.P.; Kearns, P.; Liptrot, C. Detailed NMR, Including 1,1-ADEQUATE, and Anticancer Studies of Compounds from the Echinoderm Colobometra Perspinosa. Mar. Drugs 2009, 7, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Wätjen, W.; Ebada, S.S.; Bergermann, A.; Chovolou, Y.; Totzke, F.; Kubbutat, M.H.G.; Lin, W.; Proksch, P. Cytotoxic Effects of the Anthraquinone Derivatives 1′-Deoxyrhodoptilometrin and (S)-(−)-Rhodoptilometrin Isolated from the Marine Echinoderm Comanthus sp. Arch. Toxicol. 2017, 91, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Khokhar, S.; Pierens, G.K.; Hooper, J.N.A.; Ekins, M.G.; Feng, Y.; Davis, R.A. Rhodocomatulin-Type Anthraquinones from the Australian Marine Invertebrates Clathria Hirsuta and Comatula Rotalaria. J. Nat. Prod. 2016, 79, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Tian, L.; Fu, H.-W.; Hua, H.-M.; Pei, Y.-H. One New Anthraquinone from Marine Streptomyces Sp. FX-58. Nat. Prod. Res. 2006, 20, 1207–1210. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Cui, H.; Li, Z.; Xie, Z.; Pu, Y.; Li, F.; Qin, S. A New Anthracene Derivative from Marine Streptomyces Sp. W007 Exhibiting Highly and Selectively Cytotoxic Activities. Mar. Drugs 2011, 9, 1502–1509. [Google Scholar] [CrossRef]
- Lai, Z.; Yu, J.; Ling, H.; Song, Y.; Yuan, J.; Ju, J.; Tao, Y.; Huang, H. Grincamycins I–K, Cytotoxic Angucycline Glycosides Derived from Marine-Derived Actinomycete Streptomyces Lusitanus SCSIO LR32. Planta Med. 2018, 84, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Liu, G.; Li, J.; Huang, H.; Zhang, X.; Zhang, H.; Ju, J. Cytotoxic and Antibacterial Angucycline- and Prodigiosin- Analogues from the Deep-Sea Derived Streptomyces Sp. SCSIO 11594. Mar. Drugs 2015, 13, 1304–1316. [Google Scholar] [CrossRef]
- Qu, X.-Y.; Ren, J.-W.; Peng, A.-H.; Lin, S.-Q.; Lu, D.-D.; Du, Q.-Q.; Liu, L.; Li, X.; Li, E.-W.; Xie, W.-D. Cytotoxic, Anti-Migration, and Anti-Invasion Activities on Breast Cancer Cells of Angucycline Glycosides Isolated from a Marine-Derived Streptomyces sp. Mar. Drugs 2019, 17, 277. [Google Scholar] [CrossRef] [Green Version]
- Peng, A.; Qu, X.; Liu, F.; Li, X.; Li, E.; Xie, W. Angucycline Glycosides from an Intertidal Sediments Strain Streptomyces Sp. and Their Cytotoxic Activity against Hepatoma Carcinoma Cells. Mar. Drugs 2018, 16, 470. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Martinez, E.D.; MacMillan, J.B. Anthraquinones from a Marine-Derived Streptomyces Spinoverrucosus. J. Nat. Prod. 2012, 75, 1759–1764. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Yang, T.; Ren, X.; Liu, J.; Song, Y.; Sun, A.; Ma, J.; Wang, B.; Zhang, Y.; Huang, C.; et al. Cytotoxic Angucycline Class Glycosides from the Deep Sea Actinomycete Streptomyces Lusitanus SCSIO LR32. J. Nat. Prod. 2012, 75, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Duan, Y.; Cui, Z.; Wang, Z.; Li, Z.; Zhang, Y.; Ju, J.; Huang, H. Cytotoxic Rearranged Angucycline Glycosides from Deep Sea-Derived Streptomyces Lusitanus SCSIO LR32. J. Antibiot. 2017, 70, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Liu, B.; Wang, H.; Yang, S.; Zhang, H.; Wang, Y.; Ji, N.; Qin, S.; Laatsch, H. Kiamycin, a Unique Cytotoxic Angucyclinone Derivative from a Marine Streptomyces sp. Mar. Drugs 2012, 10, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.D.A.; Tan, L.T.; Jensen, P.R.; Dimayuga, R.E.; Fairchild, C.R.; Raventos-Suarez, C.; Fenical, W. Marmycins A and B, Cytotoxic Pentacyclic C-Glycosides from a Marine Sediment-Derived Actinomycete Related to the Genus Streptomyces. J. Nat. Prod. 2007, 70, 1406–1409. [Google Scholar] [CrossRef] [PubMed]
- Adinarayana, G.; Venkateshan, M.R.; Bapiraju, V.V.S.N.K.; Sujatha, P.; Premkumar, J.; Ellaiah, P.; Zeeck, A. Cytotoxic Compounds from the Marine Actinobacterium Streptomyces Corchorusii AUBN1/71. Russ. J. Bioorg. Chem. 2006, 32, 295–300. [Google Scholar] [CrossRef]
- Murphy, B.T.; Narender, T.; Kauffman, C.A.; Woolery, M.; Jensen, P.R.; Fenical, W. Saliniquinones A-F, New Members of the Highly Cytotoxic Anthraquinone-γ-Pyrones from the Marine Actinomycete Salinispora Arenicola. Aust. J. Chem. 2010, 63, 929. [Google Scholar] [CrossRef]
- Lu, Y.; Xing, Y.; Chen, C.; Lu, J.; Ma, Y.; Xi, T. Anthraquinone Glycosides from Marine Streptomyces Sp. Strain. Phytochem. Lett. 2012, 5, 459–462. [Google Scholar] [CrossRef]
- Shrimali, D.; Shanmugam, M.K.; Kumar, A.P.; Zhang, J.; Tan, B.K.H.; Ahn, K.S.; Sethi, G. Targeted Abrogation of Diverse Signal Transduction Cascades by Emodin for the Treatment of Inflammatory Disorders and Cancer. Cancer Lett. 2013, 341, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.-T.; Lin, S.-Z.; Liu, D.-L.; Wang, Z.-H. The Distinct Mechanisms of the Antitumor Activity of Emodin in Different Types of Cancer. Oncol. Rep. 2013, 30, 2555–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelfattah, M.S.; Elmallah, M.I.Y.; Faraag, A.H.I.; Hebishy, A.M.S.; Ali, N.H. Heliomycin and Tetracinomycin D: Anthraquinone Derivatives with Histone Deacetylase Inhibitory Activity from Marine Sponge-Associated Streptomyces Sp. SP9. 3 Biotech 2018, 8, 282. [Google Scholar] [CrossRef]
- Qiao, X.; Gan, M.; Wang, C.; Liu, B.; Shang, Y.; Li, Y.; Chen, S. Tetracenomycin X Exerts Antitumour Activity in Lung Cancer Cells through the Downregulation of Cyclin D1. Mar. Drugs 2019, 17, 63. [Google Scholar] [CrossRef] [Green Version]
- Kelly, A.D.; Issa, J.-P.J. The Promise of Epigenetic Therapy: Reprogramming the Cancer Epigenome. Curr. Opin. Genet. Dev. 2017, 42, 68–77. [Google Scholar] [CrossRef]
- Peng, X.; Sun, Z.; Kuang, P.; Chen, J. Recent Progress on HDAC Inhibitors with Dual Targeting Capabilities for Cancer Treatment. Eur. J. Med. Chem. 2020, 208, 112831. [Google Scholar] [CrossRef]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules 2016, 21, 1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon Cancer Activity of Largazole, a Marine-Derived Tunable Histone Deacetylase Inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Wang, J.; Wang, X.; Liu, H.; Zhang, M.; Liu, Y.-C.; Zhang, C.; Su, Y.; Shen, Y.; Guo, Y.; et al. Marine-Derived Chromopeptide A, a Novel Class I HDAC Inhibitor, Suppresses Human Prostate Cancer Cell Proliferation and Migration. Acta Pharmacol. Sin. 2017, 38, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohr, J.; Thiericke, R. Angucycline Group Antibiotics. Nat. Prod. Rep. 1992, 9, 103. [Google Scholar] [CrossRef]
- Kharel, M.K.; Pahari, P.; Shepherd, M.D.; Tibrewal, N.; Nybo, S.E.; Shaaban, K.A.; Rohr, J. Angucyclines: Biosynthesis, Mode-of-Action, New Natural Products, and Synthesis. Nat. Prod. Rep. 2012, 29, 264–325. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H.; Arlt, V.M. Genotoxicity: Damage to DNA and Its Consequences. EXS 2009, 99, 87–110. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, S.R.; Loeb, L.A.; Herr, A.J. Somatic Mutations in Aging, Cancer and Neurodegeneration. Mech. Ageing Dev. 2012, 133, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Weakley, S.M.; Jiang, J.; Kougias, P.; Lin, P.H.; Yao, Q.; Brunicardi, F.C.; Gibbs, R.A.; Chen, C. Role of Somatic Mutations in Vascular Disease Formation. Expert Rev. Mol. Diagn. 2010, 10, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Review 2012-REACH-Chemicals-Environment-European Commission. Available online: https://ec.europa.eu/environment/chemicals/reach/review_2012_en.htm (accessed on 10 April 2021).
- EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS); Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Filipič, M.; Frutos, M.J.; Galtier, P.; Gott, D.; Gundert-Remy, U. Safety of Hydroxyanthracene Derivatives for Use in Food. EFSA J. 2018, 16. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, N.; Bettiol, A.; Crescioli, G.; Maggini, V.; Gallo, E.; Sivelli, F.; Sofi, F.; Gensini, G.F.; Vannacci, A.; Firenzuoli, F. Association between Anthraquinone Laxatives and Colorectal Cancer: Protocol for a Systematic Review and Meta-Analysis. Syst. Rev. 2020, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.T.; Silva, G.; Pungartnik, C.; Brendel, M. Study of DNA-Emodin Interaction by FTIR and UV-Vis Spectroscopy. J. Photochem. Photobiol. B 2012, 111, 59–63. [Google Scholar] [CrossRef]
- Müller, S.O.; Eckert, I.; Lutz, W.K.; Stopper, H. Genotoxicity of the Laxative Drug Components Emodin, Aloe-Emodin and Danthron in Mammalian Cells: Topoisomerase II Mediated? Mutat. Res. 1996, 371, 165–173. [Google Scholar] [CrossRef]
- Li, Y.; Luan, Y.; Qi, X.; Li, M.; Gong, L.; Xue, X.; Wu, X.; Wu, Y.; Chen, M.; Xing, G.; et al. Emodin Triggers DNA Double-Strand Breaks by Stabilizing Topoisomerase II-DNA Cleavage Complexes and by Inhibiting ATP Hydrolysis of Topoisomerase II. Toxicol. Sci. 2010, 118, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amacher, D.E. The L5178Y/TK Gene Mutation Assay System. In Chemical Mutagens; de Serres, F.J., Ed.; Springer US: Boston, MA, USA, 1984; pp. 183–212. ISBN 978-1-4612-9267-8. [Google Scholar]
- Chen, Y.-Y.; Chiang, S.-Y.; Lin, J.-G.; Yang, J.-S.; Ma, Y.-S.; Liao, C.-L.; Lai, T.-Y.; Tang, N.-Y.; Chung, J.-G. Emodin, Aloe-Emodin and Rhein Induced DNA Damage and Inhibited DNA Repair Gene Expression in SCC-4 Human Tongue Cancer Cells. Anticancer Res. 2010, 7. [Google Scholar]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single Cell Gel/Comet Assay: Guidelines for in Vitro and in Vivo Genetic Toxicology Testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef]
- Mueller, S.O.; Stopper, H.; Dekant, W. Biotransformation of the Anthraquinones Emodin and Chrysophanol by Cytochrome P450 Enzymes. Bioactivation to Genotoxic Metabolites. Drug Metab. Dispos. 1998, 26, 540–546. [Google Scholar] [PubMed]
- Stark, A.A.; Townsend, J.M.; Wogan, G.N.; Demain, A.L.; Manmade, A.; Ghosh, A.C. Mutagenicity and Antibacterial Activity of Mycotoxins Produced by Penicillium Islandicum Sopp and Penicillium Rugulosum. J. Environ. Pathol. Toxicol. 1978, 2, 313–324. [Google Scholar]
- van der Hoeven, J.C. Occurrence and Detection of Natural Mutagens and Modifying Factors in Food Products. Princess Takamatsu Symp. 1985, 16, 119–137. [Google Scholar]
- Bruggeman, I.M.; van der Hoeven, J.C.M. Lack of Activity of the Bacterial Mutagen Emodin in HGPRT and SCE Assay with V79 Chinese Hamster Cells. Mutat. Res. Genet. Toxicol. 1984, 138, 219–224. [Google Scholar] [CrossRef]
- Kevekordes, S.; Spielberger, J.; Burghaus, C.M.; Birkenkamp, P.; Zietz, B.; Paufler, P.; Diez, M.; Bolten, C.; Dunkelberg, H. Micronucleus Formation in Human Lymphocytes and in the Metabolically Competent Human Hepatoma Cell Line Hep-G2: Results with 15 Naturally Occurring Substances. Anticancer Res. 2001, 21, 461–469. [Google Scholar]
- Mengs, U.; Krumbiegel, G.; Völkner, W. Lack of Emodin Genotoxicity in the Mouse Micronucleus Assay. Mutat. Res. 1997, 393, 289–293. [Google Scholar] [CrossRef]
- Dong, X.; Fu, J.; Yin, X.; Cao, S.; Li, X.; Lin, L.; Huyiligeqi, H.; Ni, J. Emodin: A Review of Its Pharmacology, Toxicity and Pharmacokinetics. Phytother. Res. 2016, 30, 1207–1218. [Google Scholar] [CrossRef]
- Lewis, D.F.; Ioannides, C.; Parke, D.V. COMPACT and Molecular Structure in Toxicity Assessment: A Prospective Evaluation of 30 Chemicals Currently Being Tested for Rodent Carcinogenicity by the NCI/NTP. Environ. Health Perspect. 1996, 104, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program NTP. Toxicology and Carcinogenesis Studies of EMODIN (CAS NO. 518-82-1) Feed Studies in F344/N Rats and B6C3F1 Mice. Natl. Toxicol. Program. Tech. Rep. Ser. 2001, 493, 1–278. [Google Scholar]
- Dantron (Chrysazin; 1,8-Dihydroxyanthraquinone). IARC. Monogr. Eval Carcinog. Risks Hum. 1990, 50, 265–275.
- Zhang, Z.; Fu, J.; Yao, B.; Zhang, X.; Zhao, P.; Zhou, Z. In Vitro Genotoxicity of Danthron and Its Potential Mechanism. Mutat. Res. 2011, 722, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Krivobok, S.; Seigle-Murandi, F.; Steiman, R.; Marzin, D.R.; Betina, V. Mutagenicity of Substituted Anthraquinones in the Ames/Salmonella Microsome System. Mutat. Res. 1992, 279, 1–8. [Google Scholar] [CrossRef]
- Ryden, E. Comparison of the Sensitivities of Salmonella Typhimurium Strains TA102 and TA2638A to 16 Mutagens. Mutagenesis 2000, 15, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Simi, S.; Monrelli, S.; Gervasi, P.G.; Rainaldi, G. Clastogenicity of Anthraquinones in V79 and in Three Derived Cell Lines Expressing P450 Enzymes. Mutat. Res. Lett. 1995, 347, 151–156. [Google Scholar] [CrossRef]
- Kawai, K.; Mori, H.; Sugie, S.; Yoshimi, N.; Inoue, T.; Nakamaru, T.; Nozawa, Y.; Matsushima, T. Genotoxicity in the Hepatocyte/DNA Repair Test and Toxicity to Liver Mitochondria of 1-Hydroxyanthraquinone and Several Dihydroxyanthraquinones. Cell Biol. Toxicol. 1986, 2, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.O.; Stopper, H. Characterization of the Genotoxicity of Anthraquinones in Mammalian Cells. Biochim. Biophys. Acta 1999, 1428, 406–414. [Google Scholar] [CrossRef]
- Mori, H.; Sugie, S.; Niwa, K.; Takahashi, M.; Kawai, K. Induction of Intestinal Tumours in Rats by Chrysazin. Br. J. Cancer 1985, 52, 781–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, H.; Sugie, S.; Niwa, K.; Yoshimi, N.; Tanaka, T.; Hirono, I. Carcinogenicity of Chrysazin in Large Intestine and Liver of Mice. Jpn. J. Cancer Res. 1986, 77, 871–876. [Google Scholar]
- EFSA Scientific Committee Guidance on Selected Default Values to Be Used by the EFSA Scientific Committee, Scientific Panels and Units in the Absence of Actual Measured Data. EFSA J. 2012, 10. [CrossRef]
- Xie, L.; Tang, H.; Song, J.; Long, J.; Zhang, L.; Li, X. Chrysophanol: A Review of Its Pharmacology, Toxicity and Pharmacokinetics. J. Pharm Pharmacol. 2019, 71, 1475–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.M.; Li, J.S.; Huang, G.X.; Li, Q.Q.; Yan, L.J. Study on Potential Toxic Mechanism of Chrysophanol Binding DNA by Saturation Value Binding DNA. Asian J. Chem. 2012, 24, 551–557. [Google Scholar]
- Yang, X.M.; Li, J.S.; Li, Q.Q.; Huang, G.X.; Yan, L.J. Evaluation of the Potential Toxicity of Anthraquinone Derivatives in Chinese Herbal Medicines by the Resonance Light Scattering Spectrum. Asian J. Chem. 2011, 23, 3631–3634. [Google Scholar]
- Tikkanen, L.; Matsushima, T.; Natori, S. Mutagenicity of Anthraquinones in the Salmonella Preincubation Test. Mutat. Res. 1983, 116, 297–304. [Google Scholar] [CrossRef]
- Mengs, U.; Schuler, D.; Marshall, R.R. No Induction of Chromosomal Aberrations in Chinese Hamster Ovary Cells by Chrysophanol. Mutat. Res. 2001, 492, 69–72. [Google Scholar] [CrossRef]
- Heidemann, A.; Völkner, W.; Mengs, U. Genotoxicity of Aloeemodin in Vitro and in Vivo. Mutat. Res. 1996, 367, 123–133. [Google Scholar] [CrossRef]
- Özenver, N.; Saeed, M.; Demirezer, L.Ö.; Efferth, T. Aloe-Emodin as Drug Candidate for Cancer Therapy. Oncotarget 2018, 9, 17770–17796. [Google Scholar] [CrossRef] [Green Version]
- Nesslany, F.; Simar-Meintières, S.; Ficheux, H.; Marzin, D. Aloe-Emodin-Induced DNA Fragmentation in the Mouse in Vivo Comet Assay. Mutat. Res. 2009, 678, 13–19. [Google Scholar] [CrossRef]
- Yu, C.-P.; Shia, C.-S.; Lin, H.-J.; Hsieh, Y.-W.; Lin, S.-P.; Hou, Y.-C. Analysis of the Pharmacokinetics and Metabolism of Aloe-Emodin Following Intravenous and Oral Administrations in Rats: Pharmacokinetics and Metabolism of Aloe-Emodin. Biomed. Chromatogr. 2016, 30, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zeng, Y.; Liu, Y.; You, L.; Yin, X.; Fu, J.; Ni, J. Aloe-emodin: A Review of Its Pharmacology, Toxicity, and Pharmacokinetics. Phytother Res. 2020, 34, 270–281. [Google Scholar] [CrossRef]
- Westendorf, J.; Marquardt, B.; Poginski, M.; Dominiak, J.; Schmidt, H.; Marquardt, H. Genotoxicity of Naturally Occurring Hydroxyanthraquinones. Mutat. Res. 1990, 1–12. [Google Scholar] [CrossRef]
- Mengs, U.; Heidemann, A. Genotoxicity of Sennosides and Rhein in Vitro and in Vivo. Med. Sci. 1993, 749–750. [Google Scholar]
- Nohmi, T. Thresholds of Genotoxic and Non-Genotoxic Carcinogens. Toxicol. Res. 2018, 34, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gao, J.; Wang, T.-S.; Guo, C.; Yan, Y.-J.; Mao, C.-Y.; Gu, L.-W.; Yang, Y.; Li, Z.-F.; Liu, A. NMR-Based Metabolomic Techniques Identify the Toxicity of Emodin in HepG2 Cells. Sci. Rep. 2018, 8, 9379. [Google Scholar] [CrossRef] [PubMed]
- Anthraquinone-Registration Dossier-ECHA. Available online: https://echa.europa.eu/registration-dossier/-/registered-dossier/5769/7/7/3 (accessed on 23 March 2021).
- EUR-Lex-Ares(2020)1357432-EN-EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=pi_com%3AAres%282020%291357432 (accessed on 25 April 2021).


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Fungus Species | Source of Isolation | Cell Line(s) | IC50 a/ Cell Growth Inhibition Rate | Time (Where Indicated) | Reference |
|---|---|---|---|---|---|---|
| (1’S)-6,1′-O,O-dimethylaverantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM >50 μM >50 μM | 72 h 72 h 72 h | [26] |
| (1′S)-7-chloroaverantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM 36.41 μM >50 μM | 72 h 72 h 72 h | [26] |
| (cis)-emodin-physcion bianthrone | Aspergillus glaucus | Marine sediment | HL-60 A549 | 44 μM 14.2 μM | / | [27] |
| (trans)-emodin-physcion bianthrone | Aspergillus glaucus | Marine sediment | HL-60 A549 | 7.8 μM 9.2 μM | / | [27] |
| 1,2,3-trimethoxy-7-hydroxymethylanthracene-9,10-dione | Aspergillus tritici (strain SP2-8-1) | Coral Galaxea fascicularis | HeLa A549 HepG2 | >50 μM >50 μM 42.07 μM | 48 h 48 h 48 h | [28] |
| 1,4,6- trihydroxy-2-methoxy-7-methylanthracene-9,10-dione | Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | KB KBv200 | >50 μg/mL >50 μg/mL | 72 h 72 h | [29] |
| 1-hydroxy-6-methyl-8-hydroxymethylxanthone | Chaetomium sp. (strain YMF 1.02105) | Submerged woody substrate | A549 Raji HepG2 MCF-7 HL-60 | >100 μg/mL >100 μg/mL >100 μg/mL >100 μg/mL >100 μg/mL | / | [30] |
| 1′-O-methyl-7-chloroaverantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | 34.06 μM 26.09 μM 37.19 μM | 72 h 72 h 72 h | [26] |
| 1′-O-methylaverantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | 33.59 μM 35.31 μM 44.22 μM | 72 h 72 h 72 h | [26] |
| 1403P-3 | Endophytic fungus No. 1403 (strain 1403) | Mangrove (specie was not indicated) | KB KBv200 | 19.66 μM 19.27 μM | 72 h 72 h | [31] |
| MCF-7 MDA-MB-435 | 9.7 μM 7.6 μM | / | [32] | |||
| 2-O-Acetylaltersolanol B | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| 2-O-Acetylaltersolanol L | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| 3-hydroxy-1,2,5,6-tetramethoxyanthracene-9,10-dione | Aspergillus tritici (strain SP2-8-1) | Coral Galaxea fascicularis | HeLa A549 HepG2 | >50 μM >50 μM >50 μM | 48 h 48 h 48 h | [28] |
| 4R,8-dihydroxyconiothyrione B | Talaromyces islandicus (strain EN-501) | Red alga Laurencia okamurai | A2780 A2780 CisR | <10 μM <10 μM | / | [34] |
| 4S,8-dihydroxyconiothyrinone B | Talaromyces islandicus (strain EN-501) | Red alga Laurencia okamurai | A2780 A2780 CisR | <10 μM <10 μM | / | [34] |
| 4S,8-dihydroxy-10-O-methyldendryol E | Talaromyces islandicus (strain EN-501) | Red alga Laurencia okamurai | A2780 A2780 CisR | <10 μM <10 μM | / | [34] |
| 6-O-methyl-averantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM >50 μM >50 μM | 72 h 72 h 72 h | [26] |
| 6-O-methyl-7-bromoaverantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | 24.69 μM 25.62 μM 18.91 μM | 72 h 72 h 72 h | [26] |
| 6-O-methyl-7-chloroaverantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | 7.11 μM 6.64 μM 7.42 μM | 72 h 72 h 72 h | [26] |
| 6-O-methyl-7-chloroaverythrin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM 24.38 μM >50 μM | 72 h 72 h 72 h | [26] |
| 6,1′-O,O-dimethyl-7-bromoaverantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM >50 μM >50 μM | 72 h 72 h 72 h | [26] |
| 6,1′-O,O-dimethyl-7-chloroaverantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM >50 μM >50 μM | 72 h 72 h 72 h | [26] |
| 7-chloroaverantin-1′-butyl ether | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM 49.53 μM >50 μM | 72 h 72 h 72 h | [26] |
| 7-chloroaverythrin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM >50 μM >50 μM | 72 h 72 h 72 h | [26] |
| 8- hydroxyconiothyrinone B | Talaromyces islandicus (strain EN-501) | Red alga Laurencia okamurai | A2780 A2780 CisR | <10 μM <10 μM | / | [34] |
| 8-O-Methylversicolorin B | Aspergillus versicolor co-cultured with Bacillus subtilis | Sponge Agelas oroides | L5178Y | 21.2 μM | 72 h | [35] |
| 8,11-Dihydroxyconiothyrinone B | Talaromyces islandicus (strain EN-501) | Red alga Laurencia okamurai | A2780 A2780 CisR | <10 μM <10 μM | / | [34] |
| 9R-hydroxydihydrodesoxybostrycin | Fusarium spp. (strains PSU-F14 and PSU-F135) | Gorgonian sea fan (Annella sp.) | KB MCF-7 Vero b | 19 μM 15 μM 57 μM | / | [36] |
| 9R-hydroxyhalorosellinia A | Fusarium spp. (strains PSU-F14 and PSU-F135) | Gorgonian sea fan (Annella sp.) | KB MCF-7 Vero b | 49 μM 6.2 μM 54 μM | / | [36] |
| Alterporriol A | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alterporriol B | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alterporriol C | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | 24 μM 27 μM 53 μM 51 μM 98 μM | / | [37] | |
| Alterporriol D | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alterporriol E | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alterporriol K | Alternaria sp. (strain ZJ9-6B) | Fruit of mangrove Aegiceras corniculatum | MDA-MB-435 MCF-7 | 26.97 μM 29.11 μM | / | [38] |
| Alterporriol L | Alternaria sp. (strain ZJ9-6B) | Fruit of mangrove Aegiceras corniculatum | MDA-MB-435 MCF-7 | 13.11 μM 20.04 μM | / | [38] |
| Alternaria sp. (strain ZJ9-6B) | Mangrove Aegiceras corniculatum | MCF-7 MDA-MB-435 | 20.04 μM 13.11 μM | 48 h 48 h | [39] | |
| Alterporriol N | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Alterporriol O | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Alterporriol P | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | 8.6 μM 6.4 μM 20 μM 21 μM 23 μM | / | [37] |
| Alterporriol R | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alterporriol U | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alterporriol V | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alterporriol W | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Alterporriol Y | Stemphylium lycopersici | Coral Dichotella gemmacea | HCT-116 MCF- Hu7 | >50 μM >50 μM >50 μM | / | [40] |
| Altersolanol A | Stemphylium lycopersici | Coral Dichotella gemmacea | HCT-116 MCF-7 Hu7 | 1.3 μM 7.2 μM 38 μM | / | [40] |
| Altersolanol B | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Stemphylium lycopersici | Coral Dichotella gemmacea | HCT-116 MCF-7 Hu7 | 3.5 μM 9 μM >50 μM | / | [40] | |
| Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] | |
| Altersolanol C | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | 2.2 μM 7.6 μM 8.9 μM 8.2 μM 3.2 μM | / | [37] |
| Altersolanol L | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] | |
| Ampelanol | Alternaria sp. (strain ZJ-2008003) | Soft coral )Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Stemphylium lycopersici | Coral Dichotella gemmacea | HCT-116 MCF-7 Hu7 | >50 μM >50 μM >50 μM | / | [40] | |
| Aspergiolide A | Aspergillus glaucus (strain HB1-19) | Marine sediment | HeLa A549 K562 HL-60 HCT-116 | 3.1 μM 0.1 μM 7.1 μM 0.3 μM 4.4 μM | / | [41] |
| P388 HL-60 BEL-7402 A549 | 35 μM 0.28 μM 7.5 μM 0.13 μM | / | [42] | |||
| HeLa SMMC-7721 SGC-7901 MCF-7 MDA-MB-468 U251 A431 SK-OV-3 BxPC3 786-O Bel-7402 | Range between 2.37 μM and 7.07 μM c | 72 h | [43] | |||
| Aspergiolide B | Aspergillus glaucus | Marine sediment | HL-60 A549 | 0.51 μM 0.24 μM | 72 h 24 h | [27] |
| Aspergiolide C | Aspergillus glaucus (strain HB 1–19) | Marine sediment | P388 HL-60 BEL-7402 A549 | >50 μM >50 μM >50 μM >50 μM | / | [44] |
| Aspergiolide D | Aspergillus glaucus (strain HB 1–19) | Marine sediment | P388 HL-60 BEL-7402 A549 | >50 μM >50 μM >50 μM >50 μM | / | [44] |
| Aspetritone A (or 3, 9-deoxy-7-methoxybostrycin) | Aspergillus tritici (strain SP2-8-1) | Coral Galaxea fascicularis | HeLa A549 HepG2 | 2.67 μM 3.13 μM 3.87 μM | 48 h 48 h 48 h | [28] |
| Aspetritone B (or 1,2,3,4-tetrahydro-2,3,5-trihydroxy-3-methyl-6,7-dimethoxyanthracene-9,10-dione) | Aspergillus tritici (strain SP2-8-1) | Coral Galaxea fascicularis | HeLa A549 HepG2 | 10.57 μM 4.67 μM 8.57 μM | 48 h 48 h 48 h | [28] |
| Austrocortirubin | Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | KB KBv200 | >50 μg/mL >50 μg/mL | 72 h 72 h | [29] |
| Fusarium spp. (strain PSU- F14 and PSU-F135) | Sea fan Annella sp. | MCF-7 | 6.3 μM | / | [36] | |
| Auxarthrol C | Stemphylium lycopersici | Coral Dichotella gemmacea | HCT-116 MCF-7 Hu7 | >50 μM >50 μM >50 μM | / | [40] |
| Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] | |
| Auxarthrol D | Sporendonema casei (strain HDN16-802) | Marine sediment | HL-60 K562 HeLa HCT-116 MGC-803 HOS8910 MDA-MB-231 SH-SY5Y PC-3 BEL-7402 L-02 b | 7.5 μM >50 μM >50 μM 14.5 μM 21.8 μM >50 μM 19.1 μM 22.9 μM 21.9 μM 16.6 μM >50 μM | 72 h 72 h 72 h 72 h 72 h 72 h 72 h 72 h 72 h 72 h 72 h | [45] |
| Auxarthrol F | Sporendonema casei (strain HDN16-802) | Marine sediment | HL-60 K562 HeLa HCT-116 MGC-803 HOS8910 MDA-MB-231 SH-SY5Y PC-3 BEL-7402 L-02 b | 4.5 μM 16.5 μM 10.7 μM 7.8 μM 17.7 μM 18.7 μM 10.1 μM 17.2 μM 20 μM 21.3 μM 22.2 μM | 72 h 72 h 72 h 72 h 72 h 72 h 72 h 72 h 72 h 72 h 72 h | [45] |
| Averantin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM 45.47 μM >50 μM | 72 h 72 h 72 h | [26] |
| Penicillium flavidorsum (strain SHK1-27) | Marine sediment | K562 | 27.7 μM | / | [46] | |
| Aspergillus versicolor | Sponge Petrosia sp. | A549 SKOV-3 SK-MEL-2 XF-498 HCT-15 | 3.15 μg/mL 3.88 μg/mL 3.57 μg/mL 3.04 μg/mL 3.13 μg/mL | 72 h 72 h 72 h 72 h 72 h | [47] | |
| Averantin-1′-butyl ether | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | 47.19 μM 40.47 μM >50 μM | 72 h 72 h 72 h | [26] |
| Averufin | Aspergillus versicolor | Sponge Petrosia sp. | A549 SKOV-3 SK-MEL-2 XF-498 HCT-15 | 14.92 μg/mL 14.07 μg/mL 14.56 μg/mL 12.04 μg/mL 11.97 μg/mL | 72 h 72 h 72 h 72 h 72 h | [47] |
| Averythrin | Aspergillus sp. (strain SCSIO F063) | Marine sediment | SPF-268 MCF-7 NCI-H460 | >50 μM 29.69 μM >50 μM | 72 h 72 h 72 h | [26] |
| Bostrycin | Not indicated | Not indicated | DU-145 | 25.31 μM 8.62 μM 4.79 μM | 24 h 48 h 72 h | [48] |
| Not indicated | Not indicated | MGC-803 | 29.7 μM 14.07 μM 13.47 μM | 24 h 48 h 72 h | [49] | |
| Nigrospora sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | A549 Hep-2 HepG2 KB MCF-7 MCF-7/ADR | 2.64 μg/mL 5.39 μg/mL 5.90 μg/mL 4.19 μg/mL 6.13 μg/mL 6.68 μg/mL | / | [50] | |
| Fusarium spp. (strain PSU- F14 and PSU-F135) | Sea fan Annella sp. | KB MCF-7 Vero b | 0.9 μM 2.7 μM 4.2 μM | / | [36] | |
| Catenarin | Sporidesmium circinophorum (strain KUFA 0043) | Sponge Petrosia sp. | MCF-7 A375-C5 NCI-H460 | >150 mM c | / | [51] |
| Chrisophanic acid (or chrysophanol) | Aspergillus candidus (strain KUFA0062) | Sponge Epipolasis sp. | HepG2 HT-29 HCT-116 A549 A375 MCF-7 U251 T98G | 88.9% (100 μM) d 98.7% (100 μM) d 101.7% (100 μM) d 98.6% (100 μM) d 89.5% (100 μM) d 108.1% (100 μM) d 102.7% (100 μM) d 112% (100 μM) d | 48 h 48 h 48 h 48 h 48 h 48 h 48 h 48 h | [52] |
| Chrysazin (or danthron) | Beauveria bassiana (strain TPU942) | Unidentified sponge | HCT-15 Jurkat | >30 μM >30 μM | 48 h 48 h | [53] |
| Compound 6 | Halorosellinia sp. (strain 1403) and Guignardia sp. (strain 4382) | Mangrove (specie was not indicated) | KB KBv200 | 3.17 μM 3.21 μM | 72 h 72 h | [54] |
| Demethoxyaustrocortirubin | Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | KB KBv200 | >50 μg/mL >50 μg/mL | 72 h 72 h | [29] |
| Deoxybostrycin | Nigrospora sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | A549 Hep-2 HepG2 KB MCF-7 MCF-7/ADR | 2.44 μg/mL 3.15 μg/mL 4.41 μg/mL 3.15 μg/mL 4.76 μg/mL 5.46 μg/mL | / | [50] |
| Dihydroaltersolanol A | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Emodin | Aspergillus tritici (strain SP2-8-1) | Coral Galaxea fascicularis | HeLa A549 HepG2 | 25.07 μM 22.17 μM 30.20 μM | 48 h 48 h 48 h | [28] |
| Neosartorya fischeri (strain 1008F1) | Marine-derived (source was not specified) | SGC7901 BEL-7404 | 32.7% (200 μg/mL) d 11.3% (200 μg/mL) d | 72 h 72 h | [55] | |
| Penicillium sp. (strain SCSIO41015) | Sponge Callyspongia sp. | MGC803 | 5.19 μM | / | [56] | |
| Fusaquinon A | Fusarium sp. (strain ZH-210) | Mangrove sediment | KB KBv200 MCF-7 | >50 μg/mL >50 μg/mL >50 μg/mL | / | [57] |
| Fusaquinon B | Fusarium sp. (strain ZH-210) | Mangrove sediment | KB KBv200 MCF-7 | >50 μg/mL >50 μg/mL >50 μg/mL | / | [57] |
| Fusaquinon C | Fusarium sp. (strain ZH-210) | Mangrove sediment | KB KBv200 MCF-7 | >50 μg/mL >50 μg/mL >50 μg/mL | / | [57] |
| Fusarnaphthoquinone A | Fusarium spp. (strain PSU- F14 and PSU-F135) | Sea fan Annella sp. | KB MCF-7 | 130 μM 22 μM | / | [36] |
| G503 | Nigrospora sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | HONE-1 CNE2 5–8F A549 HepG2 B7402 Rb PC-3 SGC7901 HUVEC b Chang liver cells b | 35.7 μM 35.6 μM 31.7 μM 20 μM 13.3 μM 19.8 μM 44 μM 21.1 μM 10.24 μM 22.4 μM 17.5 μM | 48 h 48 h 48 h 48 h 48 h 48 h 48 h 48 h 48 h 48 h 48 h 48 h | [58] |
| Halorosellinia A (1,4,5,6,7,9-hexahydroxy-2-methoxy-7α-methyl-5β,9β,8αβ,6α,10aα- hexahydroanthracen-10(10aH)-one) | Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | KB KBv200 | >50 μg/mL >50 μg/mL | 72 h 72 h | [29] |
| Hydroxy-9,10-anthraquinone | Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | KB KBv200 | 1.40 μg/mL 2.58 μg/mL | 72 h 72 h | [29] |
| Macrosporin | Stemphylium lycopersici | Coral Dichotella gemmacea | HCT-116 MCF-7 Hu7 | >50 μM >50 μM >50 μM | / | [40] |
| Macrosporin 2-O-(6′-acetyl)-a-D-glucopyranoside | Stemphylium sp. (strain 33231) | Mangrove Burguiera sexangula var. rhynchopetala | B16F10 A549 | >10 μM >10 μM | / | [33] |
| Macrosporin 2-O-a-D-glucopyranoside | Stemphylium lycopersici | Coral Dichotella gemmacea | HCT-116 MCF-7 Hu7 | >50 μM >50 μM >50 μM | / | [40] |
| Methyl-averantin | Aspergillus versicolor | Sponge Petrosia sp. | A549 SKOV-3 SK-MEL-2 XF-498 HCT-15 | 0.64 μg/mL 1.17 μg/mL 1.10 μg/mL 0.41 μg/mL 0.49 μg/mL | 72 h 72 h 72 h 72 h 72 h | [47] |
| Nidurufin | Aspergillus versicolor | Sponge Petrosia sp. | A549 SKOV-3 SK-MEL-2 XF-498 HCT-15 | 1.83 μg/mL 3.39 μg/mL 3.16 μg/mL 1.78 μg/mL 2.20 μg/mL | 72 h 72 h 72 h 72 h 72 h | [47] |
| Aspergillus versicolor (strain A-21-2-7) | Marine sediment | A549 | 25.97 μM | / | [59] | |
| Penicillium flavidorsum (strain SHK1-27) | Marine sediment | K562 | 12.6 μM | 24 h | [46] | |
| Nigrosporin B | Fusarium spp. (strain PSU- F14 and PSU-F135) | Sea fan Annella sp. | KB MCF-7 Vero b | 88 μM 5.4 μM 29 μM | / | [36] |
| Norsolorinic acid | Aspergillus nidulans | Not specified | MCF-7 | 12.7 μM | 48 h | [60] |
| Penicillanthramin A | Penicillium citrinum (strain PSU-F51) | Sea fan Annella sp. | KB | 30 μg/mL | / | [61] |
| Physcion | Sporidesmium circinophorum (strain KUFA 0043) | Sponge Petrosia sp. | MCF-7 A375-C5 NCI-H460 | >150 mM (GI50 e) | / | [51] |
| Microsporum sp. (Strain MFS-YL) | Red alga Lomentaria catenata | HeLa | / | [62] | ||
| SZ-685C | Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | NFPA MMQ RPCs b | 18.76 μM 14.51 μM 56.09 μM | 24 h 24 h 24 h | [63] |
| Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | MMQ RPCs b | 13.2 μM 49.1 μM | 48 h 48 h | [64] | |
| Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | CNE2 | 46.89 μM 14.13 μM 8.97 μM | 24 h 48 h 72 h | [65] | |
| CNE2R | 69.11 μM 17.86 μM 8.94 μM | 24 h 48 h 72 h | ||||
| Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | MCF-7 MCF-7/ADR MCF-7/Akt K562 K562/ADR HL-60 HL-60/ADR | 7.38 μM 4.17 μM 3.36 μM 1.09 μM 1.35 μM 1.94 μM 1.76 μM | 48 h 48 h 48 h 48 h 48 h 48 h 48 h | [66] | |
| Halorosellinia sp. (strain 1403) | Mangrove Kandelia candel (L.) Druce | MCF-7 MDA-MB-435 | 7.5 μM 3 μM | 48 h 48 h | [67] | |
| Tetrahydroaltersolanol B | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Tetrahydroaltersolanol C | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Tetrahydroaltersolanol D | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Tetrahydroaltersolanol E | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Tetrahydroaltersolanol F | Alternaria sp. (strain ZJ-2008003) | Soft coral Sarcophyton sp. | HCT-116 PC-3 HepG2 Hep3B MCF-7/ADR | >100 μM >100 μM >100 μM >100 μM >100 μM | / | [37] |
| Versicolorin B | Aspergillus versicolor (strain A-21-2-7) | Marine sediment | A549 A2780 | 25.60 μM 38.76 μM | / | [59] |
| Versiconol | Aspergillus versicolor | Sponge Petrosia sp. | A549 SKOV-3 SK-MEL-2 XF-498 HCT-15 | 20.45 μg/mL 15.29 μg/mL 15.86 μg/mL 23.73 μg/mL 19.02 μg/mL | 72 h 72 h 72 h 72 h 72 h | [47] |
| Compound | Chemical Class | Species | Source of Isolation | Cell Line(s) | IC50 a/ Cell Growth Inhibition Rate | Time (Where Indicated) | Reference |
|---|---|---|---|---|---|---|---|
| 1,8-dihydroxy-2-ethyl-3-methylanthraquinone | Anthraquinone | Streptomyces sp. (strain FX-58) | Marine plant Salicornia herbacea | HL-60 BGC-823 MDA-MB-435 | 6.83 μg/mL 82.2 μg/mL 56.59 μg/mL | / | [115] |
| 3-hydroxy-1-keto-3-methyl-8- methoxy-1,2,3,4-tetrahydro-benz[α]anthracene | Anthraquinone | Streptomyces sp. (strain W007) | Marine sediment |
BEL-7402 A549 | 37.5% (100 μM) b 65.5% (100 μM) b | 72 h 72 h | [116] |
| A-7884 | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment |
MDA-MB-435 MDA-MB-231 NCI-H460 HCT-116 HepG2 MCF10A c |
2.14 μM 4.80 μM 6.90 μM 0.48 μM 4.57 μM 2.68 μM | 48 h | [117] |
| Dehydroxyaquayamycin | Angucycline glycoside | Streptomyces sp. (strain SCSIO11594) | Deep sea sediment | A549 CNE2 MCF-7 HepG2 HL7702 c | 16.40 μM 22.27 μM 23.65 μM 18.81 μM 49.34 μM | / | [118] |
| Deoxyrhodoptilometrin | Anthraquinone | / | Crinoid Colobometra perspinosa | SF-268 MCF-7 H460 | 72 μM 20 μM 25 μM | / | [112] |
| Crinoid Comanthus sp. | C6 HCT-116 | 23.2 μM 13.1 μM | 24 h 24 h | [113] | |||
| Fridamycin D | Angucycline glycoside | Streptomyces sp. (strain OC1610.4) | Deep sea sediment | MCF-7 MDA-MB-231 BT-474 | 7.58 μM 8.01 μM 6.46 μM | / | [119] |
| Galtamycin C | Angucycline glycoside | Streptomyces sp. (strain OC1610.4) | Deep sea sediment | L-O2 c HepG2 SMMC-7721 Plc-prf-5 | >40 μM >40 μM >40 μM >40 μM | / | [120] |
| Galvaquinone A | Anthraquinone | Streptomyces spinoverrucosus | Marine sediment | Calu-3 H2887 | >50 μM >50 μM | 96 h 96 h | [121] |
| Galvaquinone B | Anthraquinone | Streptomyces spinoverrucosus | Marine sediment | Calu-3 H2887 | 12.2 μM 5 μM | 96 h 96 h | [121] |
| Galvaquinone C | Anthraquinone | Streptomyces spinoverrucosus | Marine sediment | Calu-3 H2887 | >50 μM >50 μM | 96 h 96 h | [121] |
| Grincamycin | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | B16 HepG2 SW-1990 HeLa NCI-H460 MCF-7 | 1.1 μM 5.3 μM 6.4 μM 5.3 μM 11 μM 2.1 μM | 48 h 48 h 48 h 48 h 48 h 48 h | [122] |
| Grincamycin B | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | B16 HepG2 SW-1990 HeLa NCI-H460 MCF-7 | 2.1 μM 8.5 μM 11 μM 6.4 μM >100 μM 12 μM | 48 h 48 h 48 h 48 h 48 h 48 h | [122] |
| Grincamycin C | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | HepG2 SW-1990 MCF-7 | 31 μM 31 μM 11 μM | 48 h 48 h 48 h | [122] |
| Grincamycin D | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | B16 HepG2 SW-1990 HeLa NCI-H460 MCF-7 | 9.7 μM 9.7 μM 22 μM 12 μM 30 μM 6.1 μM | 48 h 48 h 48 h 48 h 48 h 48 h | [122] |
| Grincamycin E | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | B16 HepG2 SW-1990 HeLa MCF-7 | 5.4 μM 11 μM 16 μM 11 μM 8.7 μM | 48 h 48 h 48 h 48 h 48 h | [122] |
| Grincamycin F | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | MCF-7 | 19 μM | 48 h | [122] |
| Grincamycin G | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | Jurkat | 0.3 μM | 72 h | [123] |
| Grincamycin H | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | Jurkat | >20 μM | 72 h | [123] |
| Grincamycin I | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment |
MDA-MB-435 MDA-MB-231 NCI-H460 HCT-116 HepG2 MCF10A c | 10.20 μM 25.87 μM 11.87 μM 8.97 μM 9.41 μM 2.90 μM | 48 h 48 h 48 h 48 h 48 h 48 h | [117] |
| Grincamycin J | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment |
MDA-MB-435 MDA-MB-231 NCI-H460 HCT-116 HepG2 MCF10A c | 2.63 μM 4.68 μM 5.40 μM 2.63 μM 4.80 μM 2.43 μM | 48 h 48 h 48 h 48 h 48 h 48 h | [117] |
| Grincamycin K | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | MDA-MB-435 MDA-MB-231 NCI-H460 HCT-116 HepG2 MCF10A c | >50 μM >50 μM >50 μM >50 μM >50 μM >50 μM | 48 h 48 h 48 h 48 h 48 h 48 h | [117] |
| Grincamycin L | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | MCF-7 MDA-MB-231 BT-474 | >20 μM >20 μM >20 μM | / | [119]. |
| Islandicin | Anthraquinone | Streptomyces spinoverrucosus | Marine sediment | Calu-3 H2887 | >50 μM >50 μM | 96 h 96 h | [121] |
| Kyamycin | Angucyclinone | Streptomyces sp. (strain M268) | Marine sediment | HL-60 A549 BEL-7402 | 68.8% (100 μM) 55.9% (100 μM) 31.7% (100 μM) | 72 h 72 h 72 h | [124] |
| Landomycin N | Angucycline glycoside | Streptomyces sp. (strain OC1610.4) | Deep sea sediment | L-02 c HepG2 SMMC-7721 Plc-prf-5 | >40 μM >40 μM >40 μM >40 μM | / | [120] |
| Lupinacidin A | Anthraquinone | Streptomyces spinoverrucosus | Marine sediment | Calu-3 H2887 | 3.1 μM 8.8 μM | 96 h 96 h | [121] |
| Marangucycline A | Angucycline glycoside | Streptomyces sp. (strain SCSIO11594) | Deep sea sediment | A549 CNE2 MCF-7 HepG2 HL7702 c | >50 μM >50 μM >50 μM >50 μM >50 μM | / | [118] |
| Marangucycline B | Angucycline glycoside | Streptomyces sp. (strain SCSIO11594) | Deep sea sediment | A549 CNE2 MCF-7 HepG2 HL7702 c | 0.45 μM 0.56 μM 0.24 μM 0.43 μM 3.67 μM | / | [118] |
| Marmycin A | Angucycline glycoside | Streptomyces sp. (strain CHN990) | Marine sediment | HCT-116 | 60.5 nM | 72 h | [125] |
| Marmycin B | Angucycline glycoside | Streptomyces sp. (strain CHN990) | Marine sediment | HCT-116 | 1.09 μM | 72 h | [125] |
| Moromycin B | Angucycline glycoside | Streptomyces sp. (strain OC1610.4) | Deep sea sediment | MCF-7 MDA-MB-231 BT-474 | 0.42 μM 0.35 μM 0.67 μM | / | [119] |
| Resistomycin (or heliomycin) | Anthraquinone | Actinobacterium Streptomyces (strain AUBN1/7) | Marine sediment | HMO2 HepG2 | 0.005 μg/mL 0.008 μg/mL | 48 h 48 h | [126] |
| Rhodocomatulin 5,7-dimethyl ether | Anthraquinone | / | -Sponge Clathria (Thalysias) hirsuta Hooper and Levi -Crinoid Comatula (Validia) rotalaria Lamarck | MCF-7 | 9% (10 μM) b | 72 h | [114] |
| Rhodoptilometrin | Anthraquinone | / | Crinoid Colobometra perspinosa | SF-268 MCF-7 H460 | 41 μM 21 μM 25 μM | / | [112] |
| Crinoid Chomanthus sp. | C6 HCT-116 | 30 μM 40.1 μM | 24 h 24 h | [113] | |||
| Saliniquinone A | Anthraquinone | Actinomycete Salinispora arenicola (Strain CNS-325) | Marine sediment | HCT-116 | 9.9 nM | 72 h | [127] |
| Saquayamycin B | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | Jurkat | 37 nM | 72 h | [123] |
| Streptomyces sp. (strain OC1610.4) | Deep sea sediment | MCF-7 MDA-MB-231 BT-474 | 0.40 μM 0.38 μM 0.41 μM | / | [119]. | ||
| Streptomyces sp. (strain OC1610.4) | Deep sea sediment | L-02 c HepG2 SMMC-7721 Plc-prf-5 | 0.34 μM 0.14 μM 0.03 μM 0.24 μM | / | [120] | ||
| Saquayamycin B1 | Angucycline glycoside | Streptomyces sp. (strain OC1610.4) | Deep sea sediment | MCF-7 MDA-MB-231 BT-474 | 0.24 μM 0.16 μM 0.28 μM | / | [119] |
| Strepnoneside A | Anthraquinone glycoside | Streptomyces coelicolor (strain WBF-16) | Marine sediment | HCT116 | 30.2 μM | 48 h | [128] |
| Strepnoneside B | Anthraquinone glycoside | Streptomyces coelicolor (strain WBF-16) | Marine sediment | HCT116 | 40.2 μM | 48 h | [128] |
| Tetracenomycin D | Anthraquinone | Streptomyces corchorusii (strain AUBN1/7) | Marine sediment | HMO2 HepG2 | 0.009 μg/mL 0.013 μg/mL | 48 h 48 h | [126] |
| Vineomycin A1 | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | Jurkat | 11 nM | 72 h | [123] |
| Vineomycin B2 | Angucycline glycoside | Streptomyces lusitanus (SCSIO LR32) | Deep sea sediment | Jurkat | 0.3 μM | 72 h | [123] |
| Vineomycin E | Angucycline glycoside | Streptomyces sp. (strain OC1610.4) | Deep sea sediment | MCF-7 MDA-MB-231 BT-474 | 6.07 μM 7.72 μM 4.27 μM | / | [119] |
| Vineomycin F | Angucycline glycoside | Streptomyces sp. (strain OC1610.4) | Deep sea sediment | MCF-7 MDA-MB-231 BT-474 | >20 μM >20 μM >20 μM | / | [119] |
| Vineomycinone B2 | Angucycline glycoside | Streptomyces sp. (strain OC1610.4) | Deep sea sediment | MCF-7 MDA-MB-231 BT-474 | >20 μM >20 μM >20 μM | / | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, G.; Turrini, E.; Catanzaro, E.; Fimognari, C. Marine Anthraquinones: Pharmacological and Toxicological Issues. Mar. Drugs 2021, 19, 272. https://doi.org/10.3390/md19050272
Greco G, Turrini E, Catanzaro E, Fimognari C. Marine Anthraquinones: Pharmacological and Toxicological Issues. Marine Drugs. 2021; 19(5):272. https://doi.org/10.3390/md19050272
Chicago/Turabian StyleGreco, Giulia, Eleonora Turrini, Elena Catanzaro, and Carmela Fimognari. 2021. "Marine Anthraquinones: Pharmacological and Toxicological Issues" Marine Drugs 19, no. 5: 272. https://doi.org/10.3390/md19050272
APA StyleGreco, G., Turrini, E., Catanzaro, E., & Fimognari, C. (2021). Marine Anthraquinones: Pharmacological and Toxicological Issues. Marine Drugs, 19(5), 272. https://doi.org/10.3390/md19050272