
New Diterpenoids and Isocoumarin Derivatives from the Mangrove-Derived Fungus Hypoxylon sp.


 ,
,  and
and
Abstract
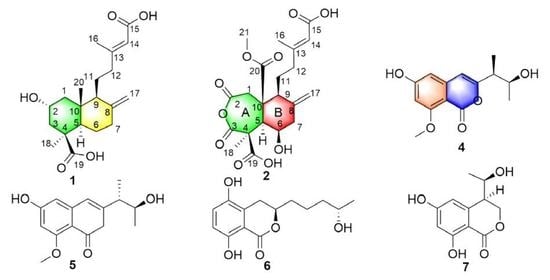
:
1. Introduction
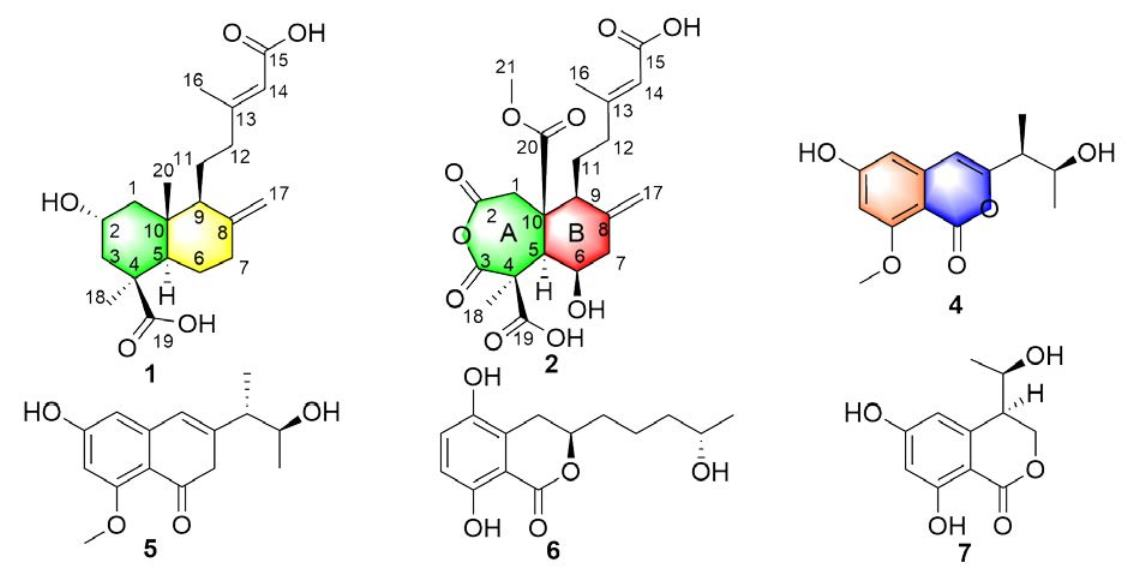
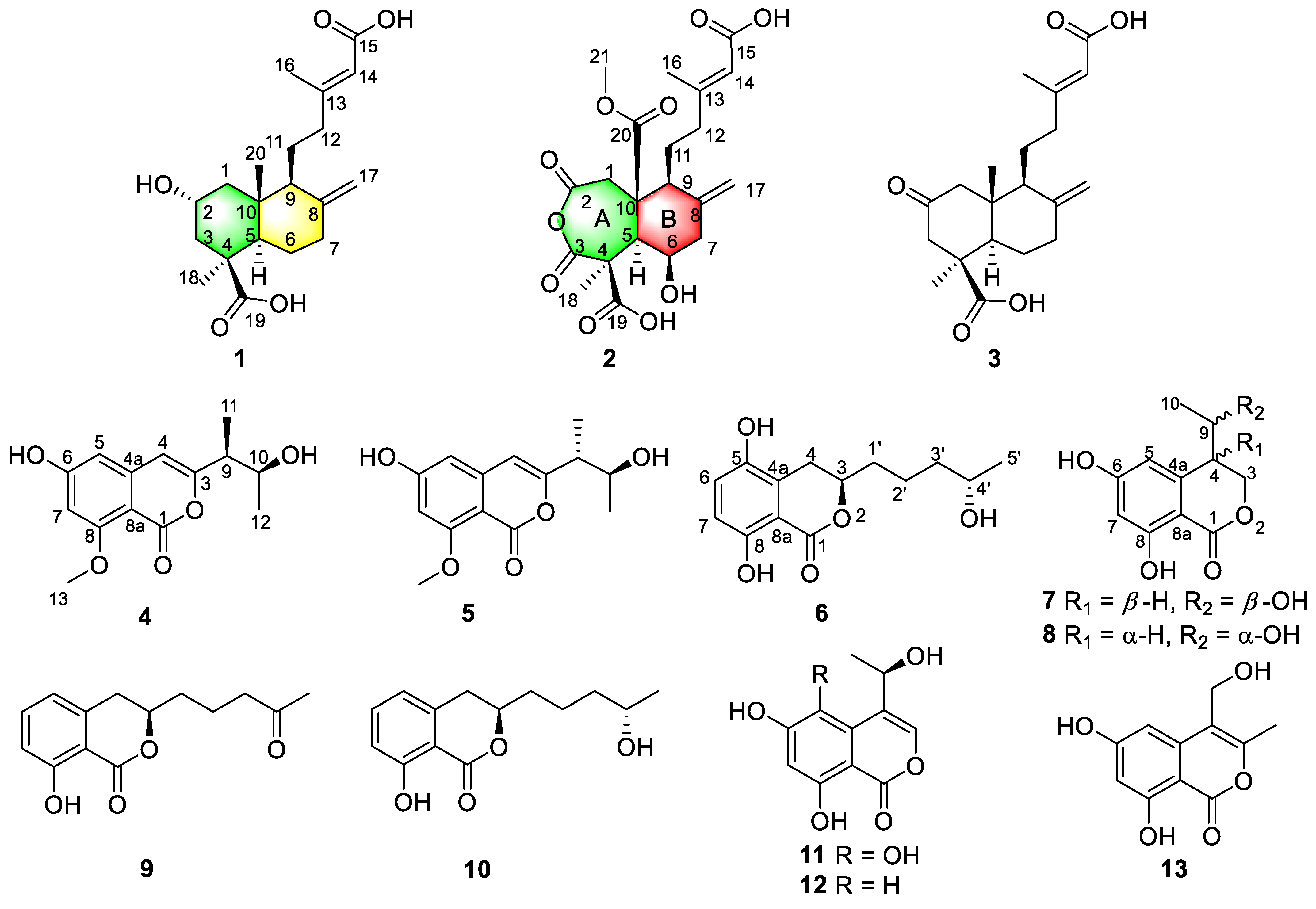
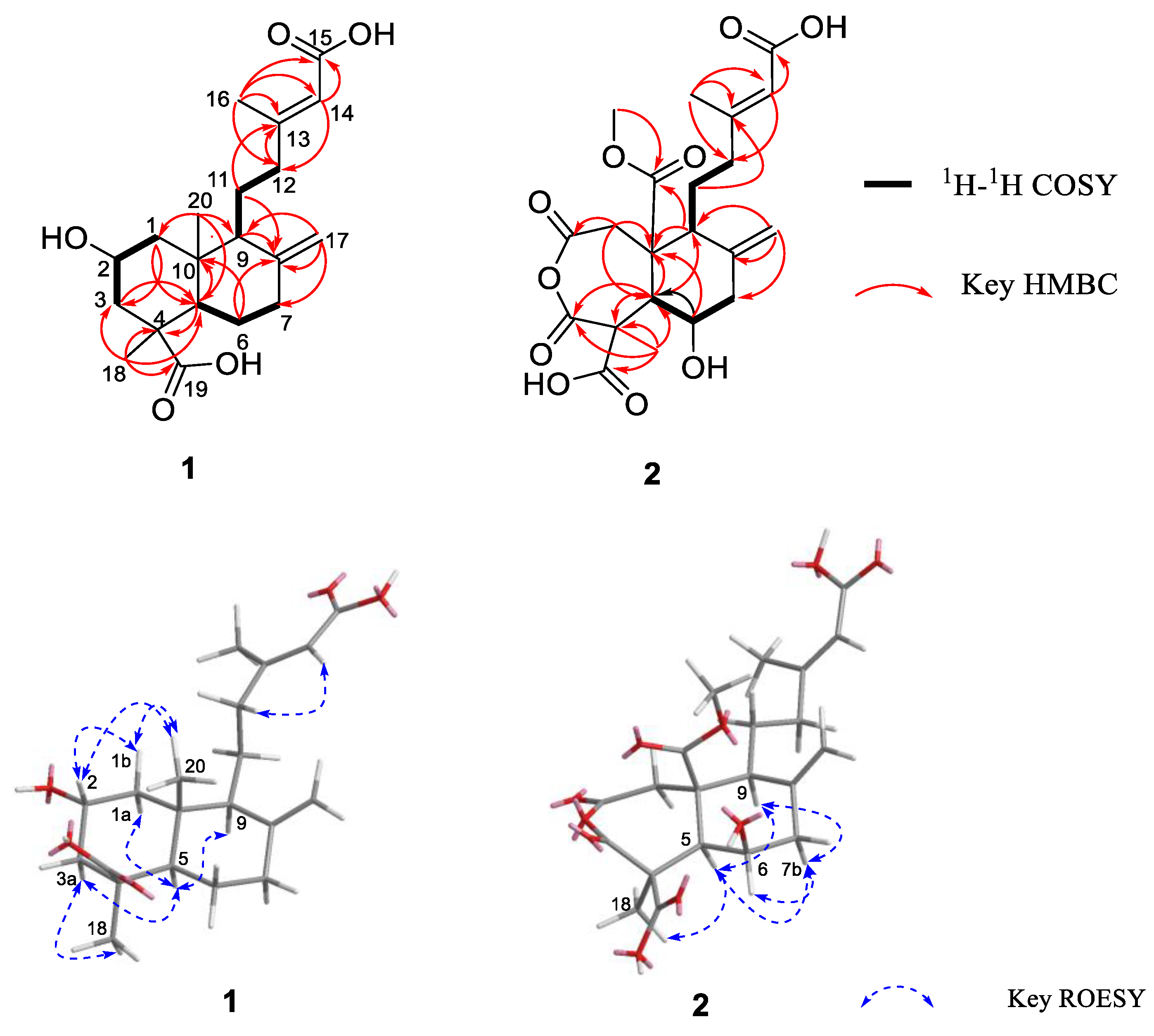
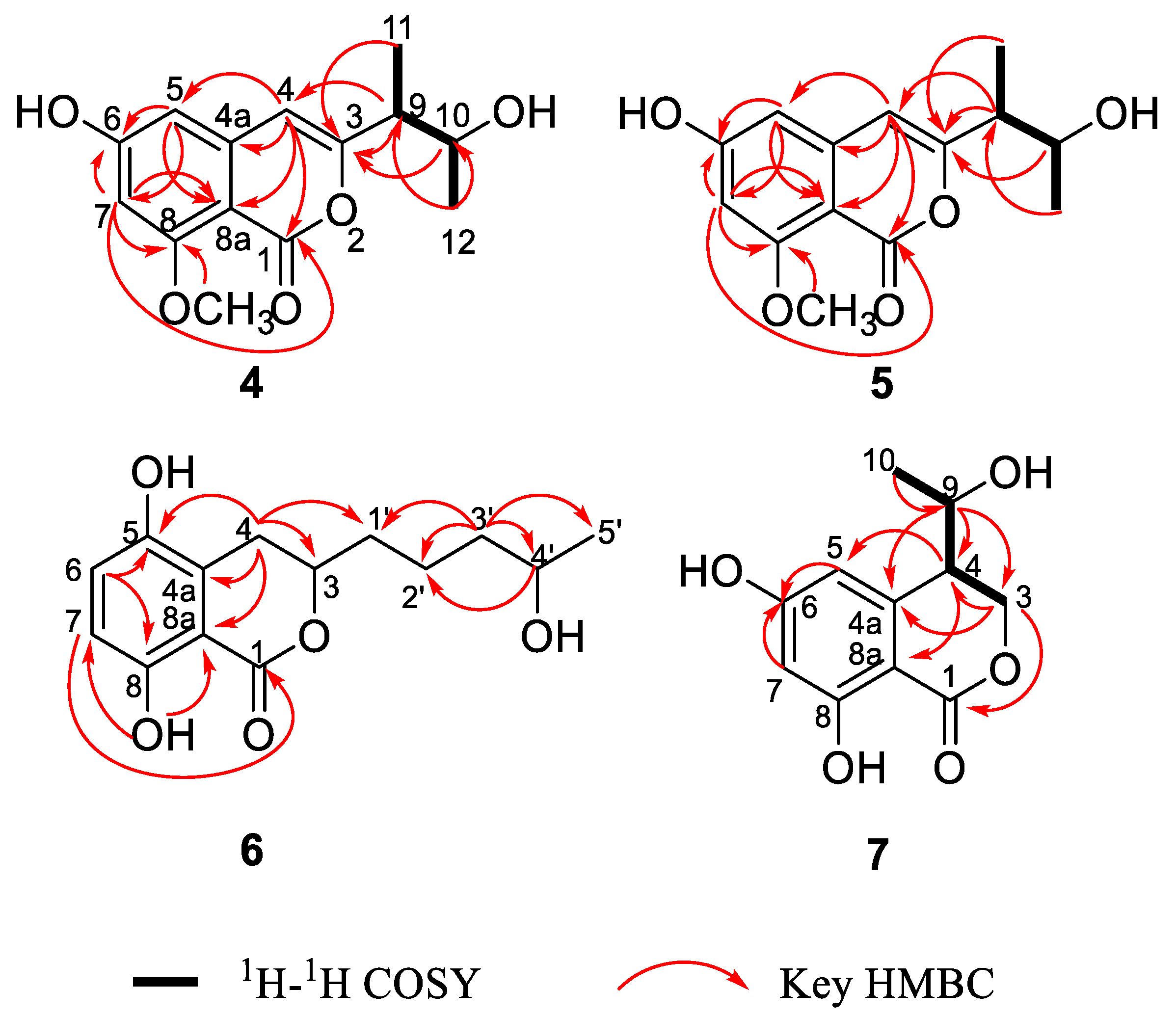
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedure
3.2. Strain and Fermentation
3.3. Extraction and Isolation
3.4. Antimicrobial Assay
3.5. DPPH Assay
3.6. α-Glucosidase Inhibitory Assay
3.7. Rh2(OCOCF3)4-Induced ECD Experiments of 6 and 7
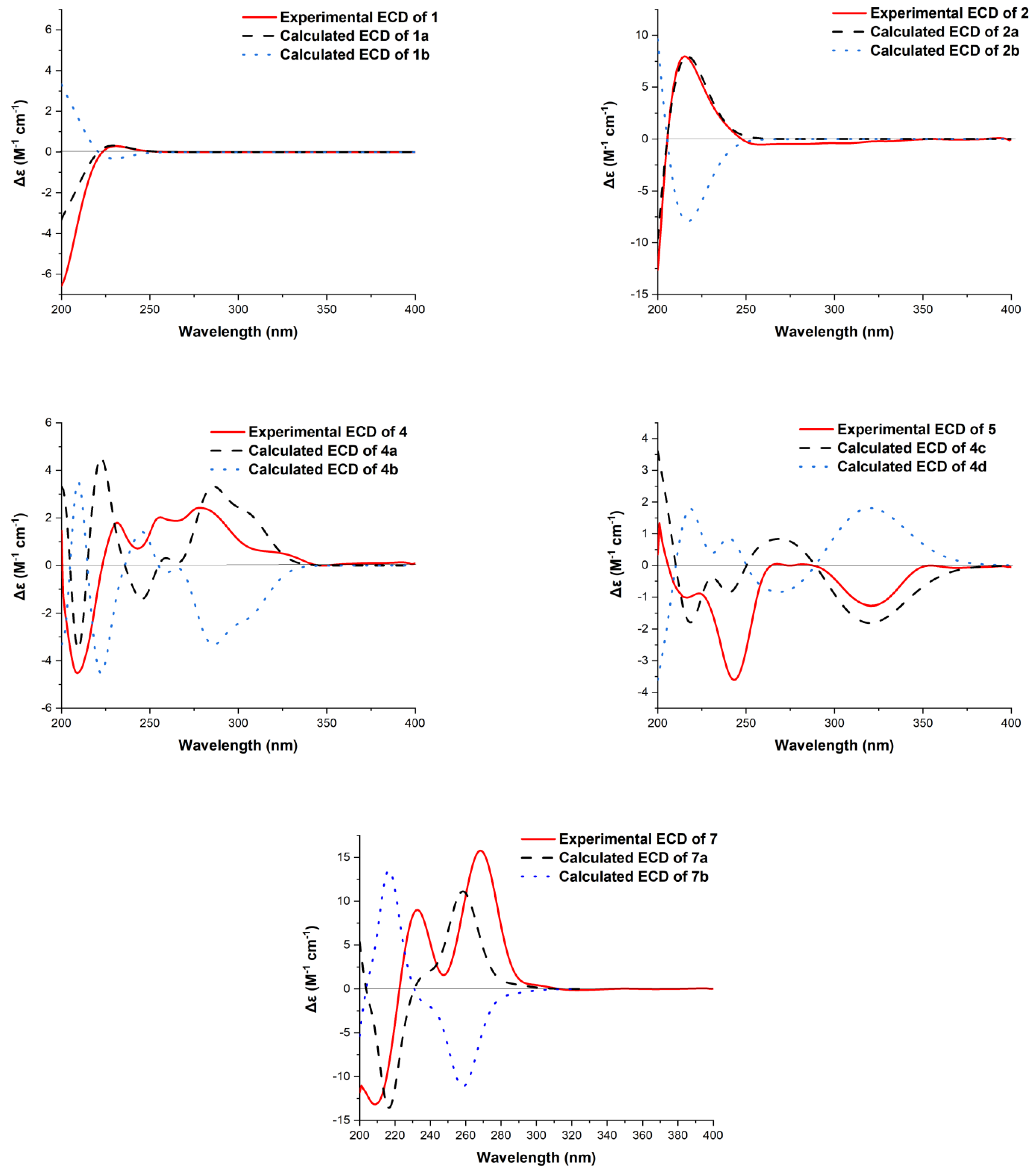
3.8. ECD Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H.W.; Bai, X.L.; Zhang, M.; Chen, J.W.; Wang, H. Bioactive natural products from endophytic microbes. Nat. Prod. J. 2018, 8, 86–108. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef]
- Xu, J. Bioactive natural products derived from mangrove-associated microbes. RSC Adv. 2015, 5, 841–892. [Google Scholar] [CrossRef]
- Guo, L.F.; Liu, G.R.; Liu, L. Caryophyllene-type sesquiterpenoids and α-furanones from the plant endophytic fungus Pestalotiopsis theae. Chin. J. Nat. Med. 2020, 18, 261–267. [Google Scholar] [CrossRef]
- Li, S.J.; Jiao, F.W.; Zhang, X.; Yan, W.; Jiao, R.H. Cytotoxic xanthone derivatives from the mangrove-derived endophytic fungus Peniophora incarnate Z4. J. Nat. Prod. 2020, 83, 2976–2982. [Google Scholar] [CrossRef]
- Gan, Q.; Lin, C.Y.; Lu, C.J.; Chang, Y.M.; Che, Q.; Zhang, G.J.; Zhu, T.J.; Gu, Q.Q.; Wu, Z.Q.; Li, M.Y.; et al. New furo [3,2-h] isochroman from the mangrove endophytic fungus Aspergillus sp. 085242. Chin. J. Nat. Med. 2020, 18, 855–859. [Google Scholar]
- Yu, X.Q.; Müller, W.E.G.; Meier, D.; Kalscheuer, R.; Guo, Z.; Zou, K.; Umeokoli, B.O.; Liu, Z.; Proksch, P. Polyketide derivatives from mangrove derived endophytic fungus Pseudopestalotiopsis theae. Mar. Drugs 2020, 18, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.X.; Shao, T.M.; Mei, R.Q.; Huang, G.L.; Zhou, X.M.; Zheng, C.J.; Wang, C.Y. Bioactive secondary metabolites from the culture of the mangrove-derived fungus Daldinia eschscholtzii HJ004. Mar. Drugs 2019, 17, 710. [Google Scholar] [CrossRef] [Green Version]
- Li, W.S.; Hu, H.B.; Huang, Z.H.; Yan, R.J.; Tian, L.W.; Wu, J. Phomopsols A and B from the mangrove endophytic fungus Phomopsis sp. xy21: Structures, neuroprotective effects, and biogenetic relationships. Org. Lett. 2019, 21, 7919–7922. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.R.; Niu, S.B.; Liu, L. Alterchromanone A, one new chromanone derivative from the mangrove endophytic fungus Alternaria longipes. J. Antibiot. 2020, 74, 152–155. [Google Scholar] [CrossRef]
- Qiu, P.; Cai, R.L.; Li, L.; She, Z.G. Three new isocoumarin derivatives from the mangrove endophytic fungus Penicillium sp. YYSJ-3. Chin. J. Nat. Med. 2020, 18, 256–260. [Google Scholar] [CrossRef]
- Quang, D.N.; Hashimoto, T.; Stadler, M.; Asakawa, Y. New azaphilones from the inedible mushroom Hypoxylon rubiginosum. J. Nat. Prod. 2004, 67, 1152–1155. [Google Scholar] [CrossRef]
- Quang, D.N.; Stadler, M.; Fournier, J.; Asakawa, Y. Carneic acids A and B, chemotaxonomically significant antimicrobial agents from the xylariaceous ascomycete Hypoxylon carneum. J. Nat. Prod. 2006, 69, 1198–1202. [Google Scholar] [CrossRef]
- Leman-Loubière, C.; Le Goff, G.; Retailleau, P.; Debitus, C.; Ouazzani, J. Sporothriolide-related compounds from the fungus Hypoxylon monticulosum CLL-205 isolated from a Sphaerocladina sponge from the Tahiti coast. J. Nat. Prod. 2017, 80, 2850–2854. [Google Scholar] [CrossRef]
- Fukai, M.; Tsukada, M.; Miki, K.; Suzuki, T.; Sugita, T.; Kinoshita, K.; Takahashi, K.; Shiro, M.; Koyama, K. Hypoxylonols C−F, benzo[j]fluoranthenes from Hypoxylon truncatum. J. Nat. Prod. 2012, 75, 22–25. [Google Scholar] [CrossRef]
- Koyama, K.; Kuramochi, D.; Kinoshita, K.; Takahashi, K. Hypoxylonols A and B, novel reduced benzo[j]fluoranthene derivatives from the mushroom Hypoxylon truncatum. J. Nat. Prod. 2002, 65, 1489–1490. [Google Scholar] [CrossRef] [PubMed]
- Surup, F.; Kuhnert, E.; Lehmann, E.; Heitkämper, S.; Hyde, K.D.; Fournier, J.; Stadler, M. Sporothriolide derivatives as chemotaxonomic markers for Hypoxylon monticulosum. Mycology 2014, 5, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, B.W.; Jia, F.F.; Luo, Y.; Ding, N.; Li, S.N.; Shi, F.Y.; Hai, Y.; Wang, L.L.; Zhu, Z.X.; Liu, X.; et al. Two new diterpenoids from Penicillium chrysogenum MT-12, an endophytic fungus isolated from Huperzia serrata. Nat. Prod. Res. 2020, 25, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Shao, C.L.; Li, Z.Y.; Gan, L.S.; Fu, X.M.; Bian, W.T.; Zhao, H.Y.; Wang, C.Y. Isocoumarin derivatives and benzofurans from a sponge-derived Penicillium sp. Fungus. J. Nat. Prod. 2013, 76, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Wei, M.Y.; Chen, G.Y.; Lin, Y.C. Two new dihydroisocoumarins from the endophytic fungus Aspergillus sp. collected from the south China sea. Chem. Nat. Compd. 2012, 48, 371–373. [Google Scholar] [CrossRef]
- Findlay, J.A.; Li, G.Q.; Miller, J.D.; Womiloju, T.O. Insect toxins from spruce endophytes. Can. J. Chem. 2003, 81, 284–292. [Google Scholar] [CrossRef]
- Kimura, Y.; Nakajima, H.; Hamasaki, T. Sescandelin, a new root promoting substance produced by the fungus, Sesquicillium candelabrum. Agric. Biol. Chem. 1990, 54, 2477–2479. [Google Scholar] [CrossRef]
- Kimura, Y.; Nakadoi, M.; Nakajima, H.; Hamasaki, T.; Nagai, T.; Kohmoto, K.; Shimada, A. Structure of sescandelin-B, a new metabolite produced by the fungus, Sesquicillium candelabrum. Agric. Biol. Chem. 2006, 55, 1887–1888. [Google Scholar]
- Zhou, K.; Zhao, X.L.; Han, L.P.; Cao, M.M.; Chen, C.; Shi, B.Z.; Luo, D.Q. Paecilomycines A and B, novel diterpenoids, isolated from insect-pathogenic fungi Paecilomyces sp. ACCC 37762. Helv. Chim. Acta 2015, 98, 642–649. [Google Scholar] [CrossRef]
- Appendino, G.; Prosperini, S.; Valdivia, C.; Ballero, M.; Colombano, G.; Billington, R.A.; Genazzani, A.A.; Sterner, O. Serca-inhibiting activity of C-19 terpenolides from Thapsia garganica and their possible biogenesis. J. Nat. Prod. 2005, 68, 1213–1217. [Google Scholar] [CrossRef]
- Chen, X.; Ding, J.; Ye, Y.M.; Zhang, J.S. Bioactive abietane and seco-abietane diterpenoids from Salvia prionitis. J. Nat. Prod. 2002, 65, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.H.; Chen, C.H.; Huang, S.L. New diterpenes from the heartwood of Chamaecyparis obtusa var. formosana. J. Nat. Prod. 1998, 61, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Frelek, J.; Szczepek, W.J. [Rh2(OCOCF3)4] as an auxiliary chromophore in chiroptical studies on steroidal alcohols. Tetrahedron: Asymmetry 1999, 10, 1507–1520. [Google Scholar] [CrossRef]
- Chen, S.H.; Liu, Y.Y.; Liu, Z.M.; Cai, R.L.; Lu, Y.J.; Huang, X.S.; She, Z.G. Isocoumarins and benzofurans from the mangrove endophytic fungus Talaromyces amestolkiae possess α-glucosidase inhibitory and antibacterial activitie. RSC Adv. 2016, 6, 26412–26420. [Google Scholar] [CrossRef]
- Fan, W.W.; Li, E.W.; Ren, J.W.; Wang, W.Z.; Liu, X.Z.; Zhang, Y.J. Cordycepamides A−E and cordyglycoside A, new alkaloidal and glycoside metabolites from the entomopathogenic fungus Cordyceps sp. Fitoterapia 2020, 142, 104525. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1a | 48.0, CH2 | 0.87, td (12, 3) | 35.8, CH2 | 2.72, d (18.3) |
| 1b | 1.96, m | 3.11, d (18.3) | ||
| 2 | 62.9, CH | 3.88, tt (4.2, 12) | 172.8, C | |
| 3a | 46.9, CH2 | 0.87, td (12, 3) | 173.5, C | |
| 3b | 2.19, m | |||
| 4 | 44.2, qC | 55.0, C | ||
| 5 | 54.5, CH | 1.26, m | 54.7, CH | 3.50, d (11.3) |
| 6a | 25.5, CH2 | 1.70, m | 77.7, CH | 4.93, m |
| 6b | 1.88, m | |||
| 7a | 38.0, CH2 | 1.84, m | 42.4, CH2 | 2.38, m |
| 7b | 2.34, m | 3.05, dd (4.5, 11.3) | ||
| 8 | 147.7, qC | 142.7, C | ||
| 9 | 54.6, CH | 1.60, m | 47.4, CH | 3.18, m |
| 10 | 40.9, C | 51.8, C | ||
| 11a | 21.5, CH2 | 1.44, m | 24.1, CH2 | 1.47, m |
| 11b | 1.62, m | 1.77, m | ||
| 12a | 38.9, CH2 | 2.21, m | 40.1, CH2 | 2.10, m |
| 12b | 1.94, m | 2.34, m | ||
| 13 | 158.6, C | 160.1, C | ||
| 14 | 116.8, CH | 5.54, s | 116.5, CH | 5.65, s |
| 15 | 167.6, C | 167.7, C | ||
| 16 | 18.3, CH3 | 2.07, s | 18.7, CH3 | 2.13, s |
| 17a | 106.8, CH2 | 4.51, s | 113.7, CH2 | 4.91, m |
| 17b | 4.88, s | 5.21, s | ||
| 18 | 28.7, CH3 | 1.15, s | 13.3, CH3 | 1.25, s |
| 19 | 178.3, C | 175.9, C | ||
| 20 | 13.5, CH3 | 0.54, s | 172.6, C | |
| 21 | 52.0, CH3 | 3.67, s | ||
| Position | 4 | 5 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 158.6, C | 158.6, C | ||
| 3 | 161.6, C | 161.3, C | ||
| 4 | 103.5, CH | 6.22, s | 103.8, CH | 6.20, s |
| 4a | 143.2, C | 143.3, C | ||
| 5 | 103.6, CH | 6.43, s | 103.5, CH | 6.42, s |
| 6 | 164.6, C | 164.6, C | ||
| 7 | 99.4, CH | 6.51, s | 99.4, CH | 6.51, s |
| 8 | 164.7 C | 164.7, C | ||
| 8a | 102.8, C | 102.9, C | ||
| 9 | 46.4, CH | 2.45, m | 46.7, CH | 2.53, m |
| 10 | 69.1, CH | 3.98, m | 69.3, CH | 3.98, m |
| 11 | 13.7, CH3 | 1.26, d (7.0) | 14.2, CH3 | 1.19, d (7.0) |
| 12 | 22.0, CH3 | 1.16, d (6.3) | 20.7, CH3 | 1.17, d (6.2) |
| 13 | 56.2, CH3 | 3.87, s | 56.3, CH3 | 3.87, s |
| OH-6 | 9.63, br s | 9.59, br s | ||
| Position | 6 | Position | 7 | ||
|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | ||
| 1 | 170.8, C | 1 | 169.1, C | ||
| 3 | 80.5, CH | 4.63, m | 3 | 68.3, CH2 | 4.51, dd (4.1, 11.6) 4.61, dd (1.8, 11.6) |
| 4 | 27.4, CH2 | 2.68, dd (3.4, 16.8) 3.19, dd (10.6, 16.8) | 4 | 43.2, CH | 2.79, m |
| 4a | 125.6, C | 4a | 143.0, C | ||
| 5 | 146.3, C | 5 | 108.2, CH | 6.26, d (2.2) | |
| 6 | 124.7, CH | 7.12, d (8.8) | 6 | 164.5, C | |
| 7 | 116.1, CH | 6.71, d (8.8) | 7 | 101.2, CH | 6.19, d (2.2) |
| 8 | 156.2, C | 8 | 163.1, C | ||
| 8a | 109.3, C | 8a | 100.1, C | ||
| 1′ | 35.7, CH2 | 1.79, m 1.89, m | 9 | 68.0, CH | 3.85, m |
| 2′ | 22.0, CH2 | 1.62, m | 10 | 19.9, CH3 | 1.04, d (6.2) |
| 3′ | 39.3, CH2 | 1.48, m | 8-OH | 11.16, s | |
| 4′ | 67.4, CH | 3.77, m | |||
| 5′ | 24.1, CH3 | 1.14, d (6.2) | |||
| 8-OH | 10.6, s | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, B.; Liu, S.; Huo, R.; Li, Y.; Ren, J.; Wang, W.; Wei, T.; Jiang, X.; Yin, W.; Liu, H.; et al. New Diterpenoids and Isocoumarin Derivatives from the Mangrove-Derived Fungus Hypoxylon sp. Mar. Drugs 2021, 19, 362. https://doi.org/10.3390/md19070362
Hou B, Liu S, Huo R, Li Y, Ren J, Wang W, Wei T, Jiang X, Yin W, Liu H, et al. New Diterpenoids and Isocoumarin Derivatives from the Mangrove-Derived Fungus Hypoxylon sp. Marine Drugs. 2021; 19(7):362. https://doi.org/10.3390/md19070362
Chicago/Turabian StyleHou, Bolin, Sushi Liu, Ruiyun Huo, Yueqian Li, Jinwei Ren, Wenzhao Wang, Tao Wei, Xuejun Jiang, Wenbing Yin, Hongwei Liu, and et al. 2021. "New Diterpenoids and Isocoumarin Derivatives from the Mangrove-Derived Fungus Hypoxylon sp." Marine Drugs 19, no. 7: 362. https://doi.org/10.3390/md19070362
APA StyleHou, B., Liu, S., Huo, R., Li, Y., Ren, J., Wang, W., Wei, T., Jiang, X., Yin, W., Liu, H., Liu, L., & Li, E. (2021). New Diterpenoids and Isocoumarin Derivatives from the Mangrove-Derived Fungus Hypoxylon sp. Marine Drugs, 19(7), 362. https://doi.org/10.3390/md19070362