
Full-Length Transcriptome of Thalassiosira weissflogii as a Reference Resource and Mining of Chitin-Related Genes



Abstract
:
1. Introduction
2. Results and Discussion
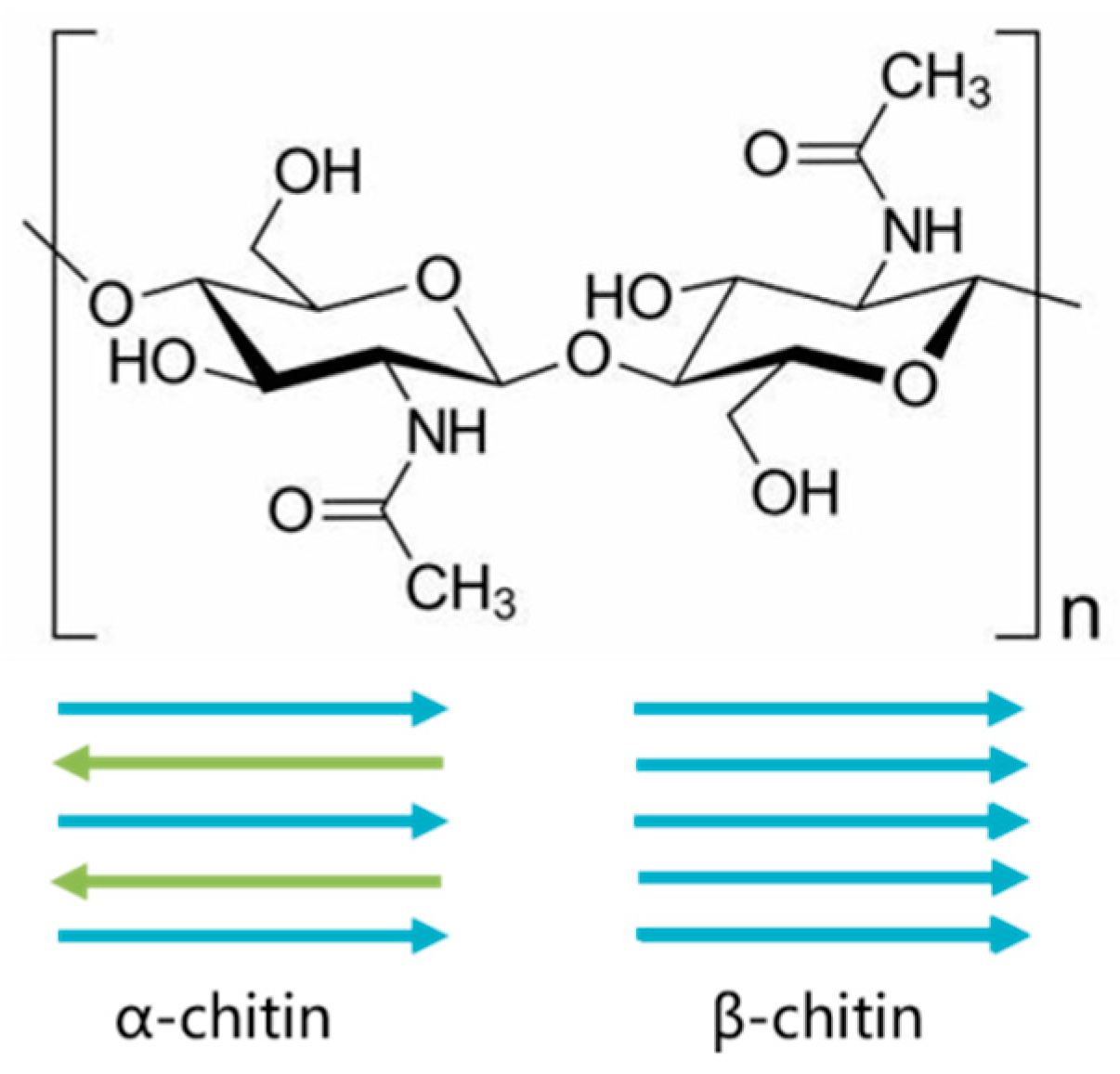
2.1. Glycosidic Linkage Analysis
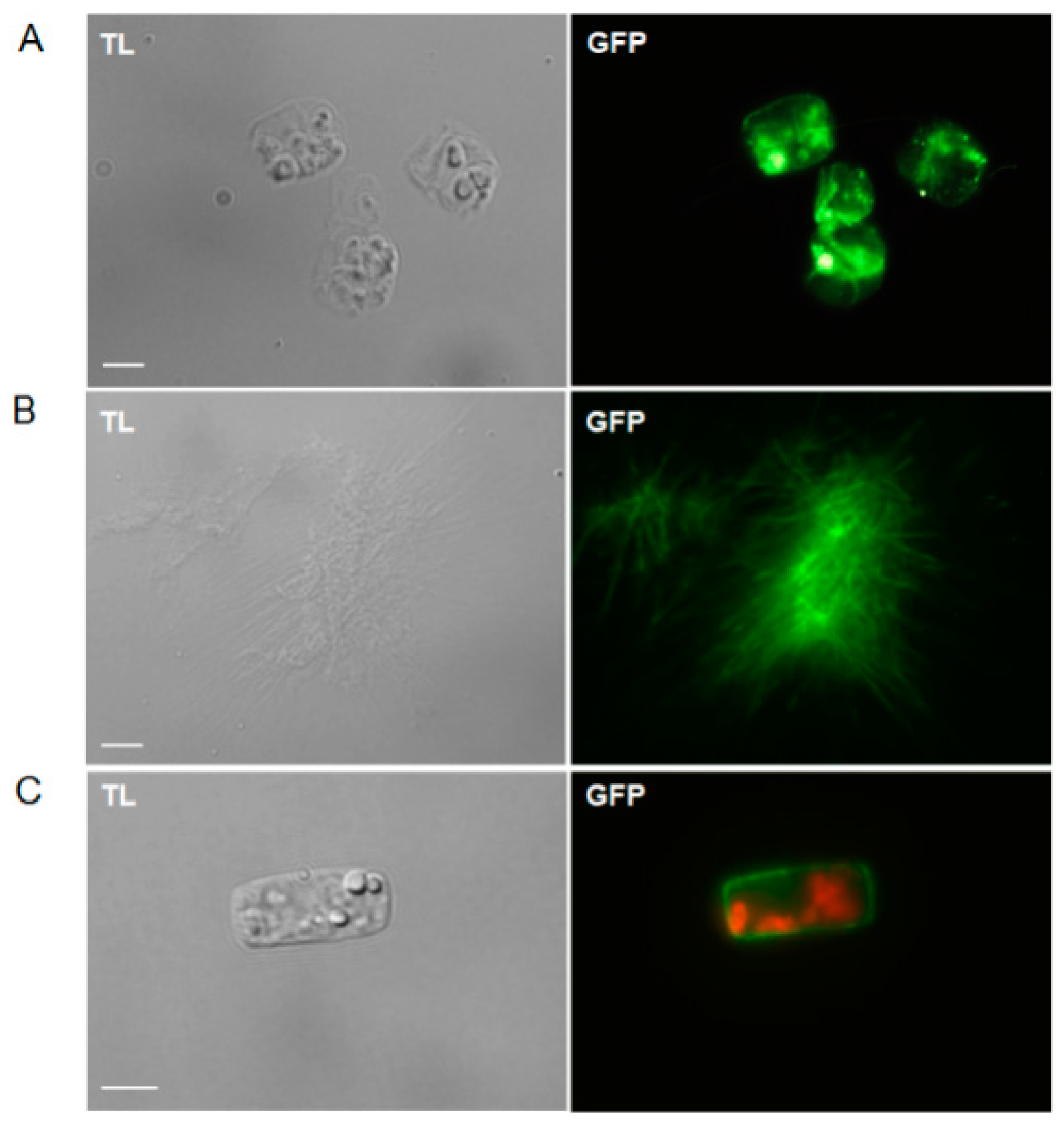
2.2. Staining of Chitin and Chitosan
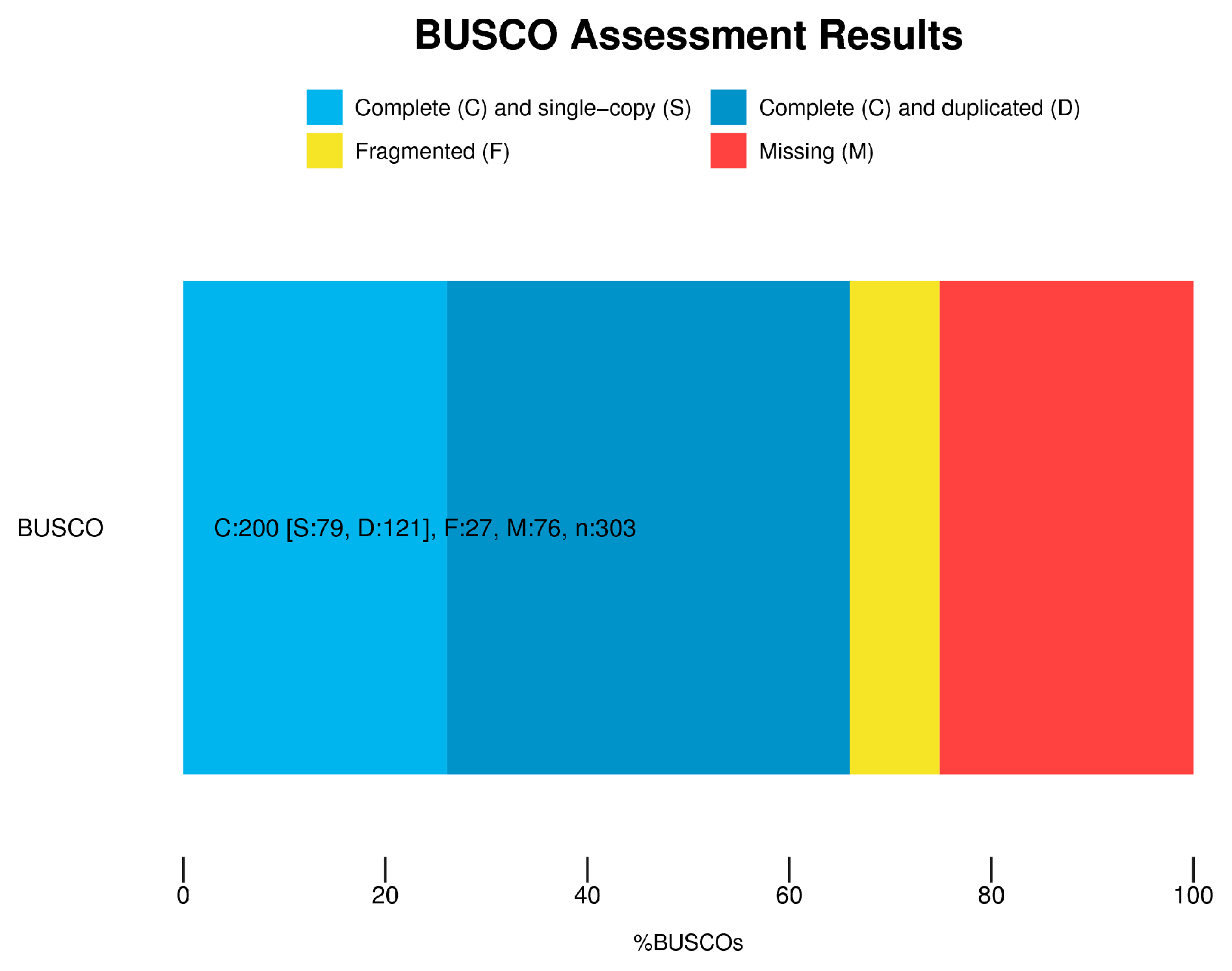
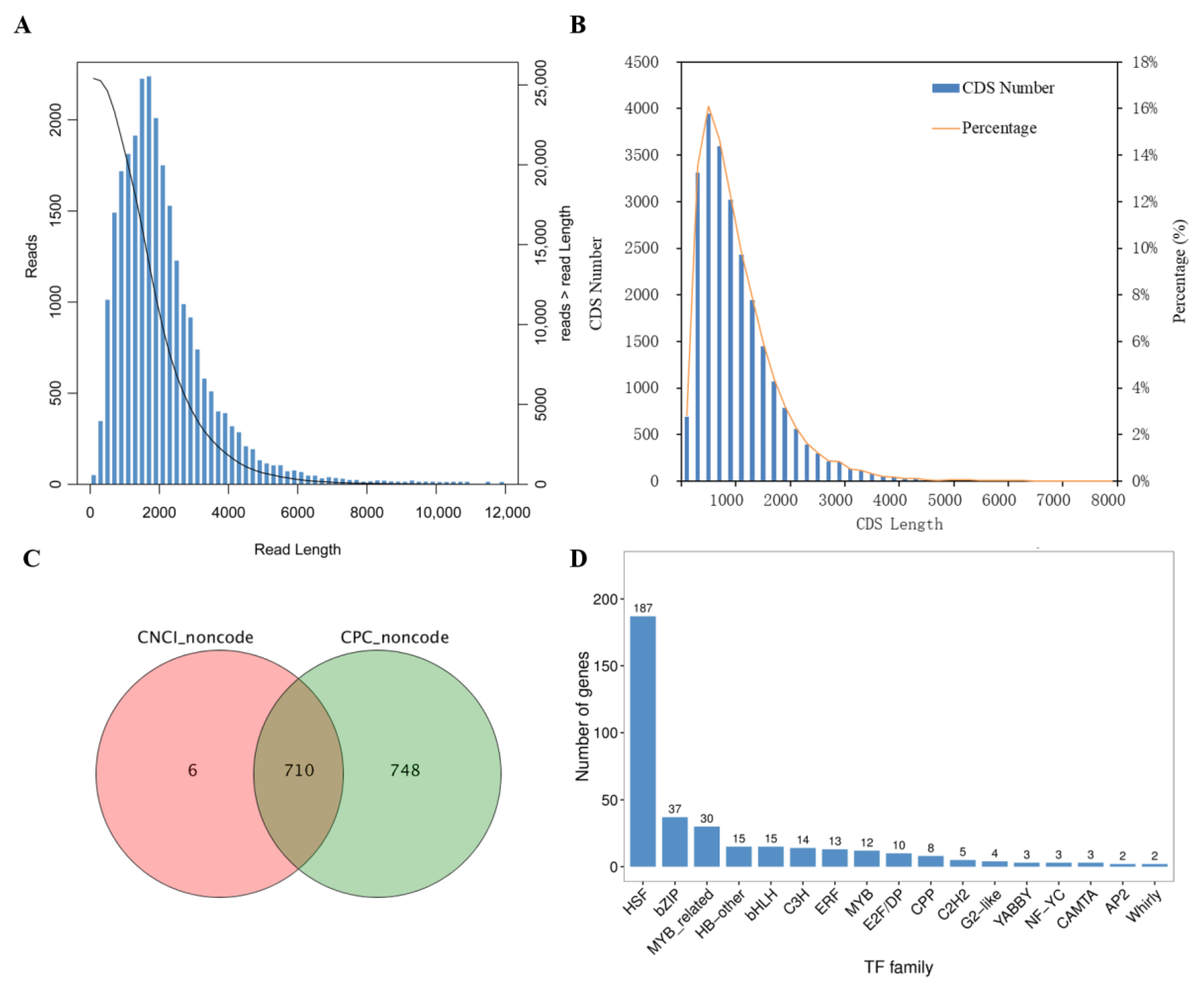
2.3. Sequencing and Data Processing
2.4. Analyses of Coding Sequence, Long Non-Coding RNAs and Transcription Factors
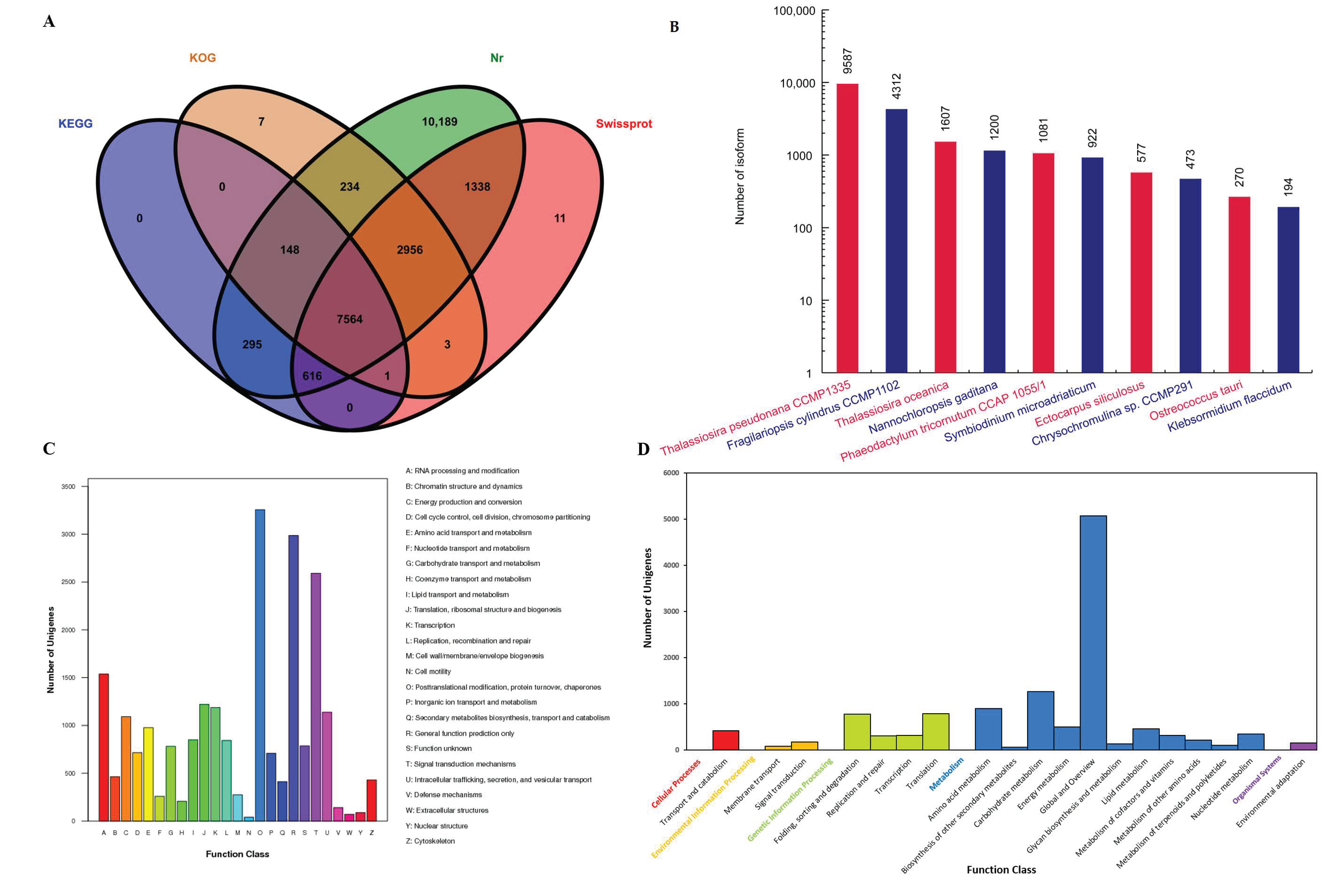
2.5. Annotation Analyses
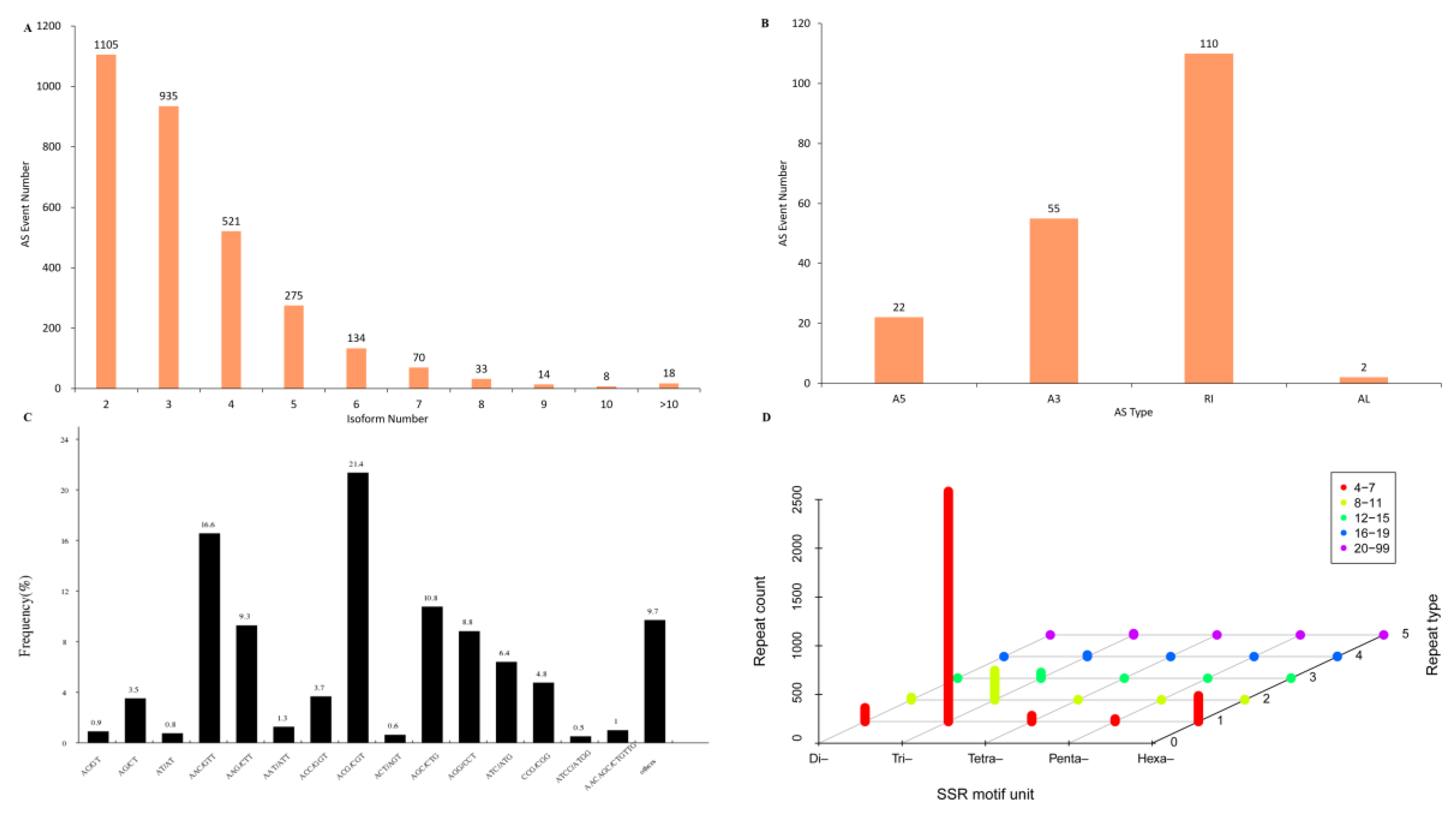
2.6. Analyses of Alternative Splicing and Simple Sequence Repeats
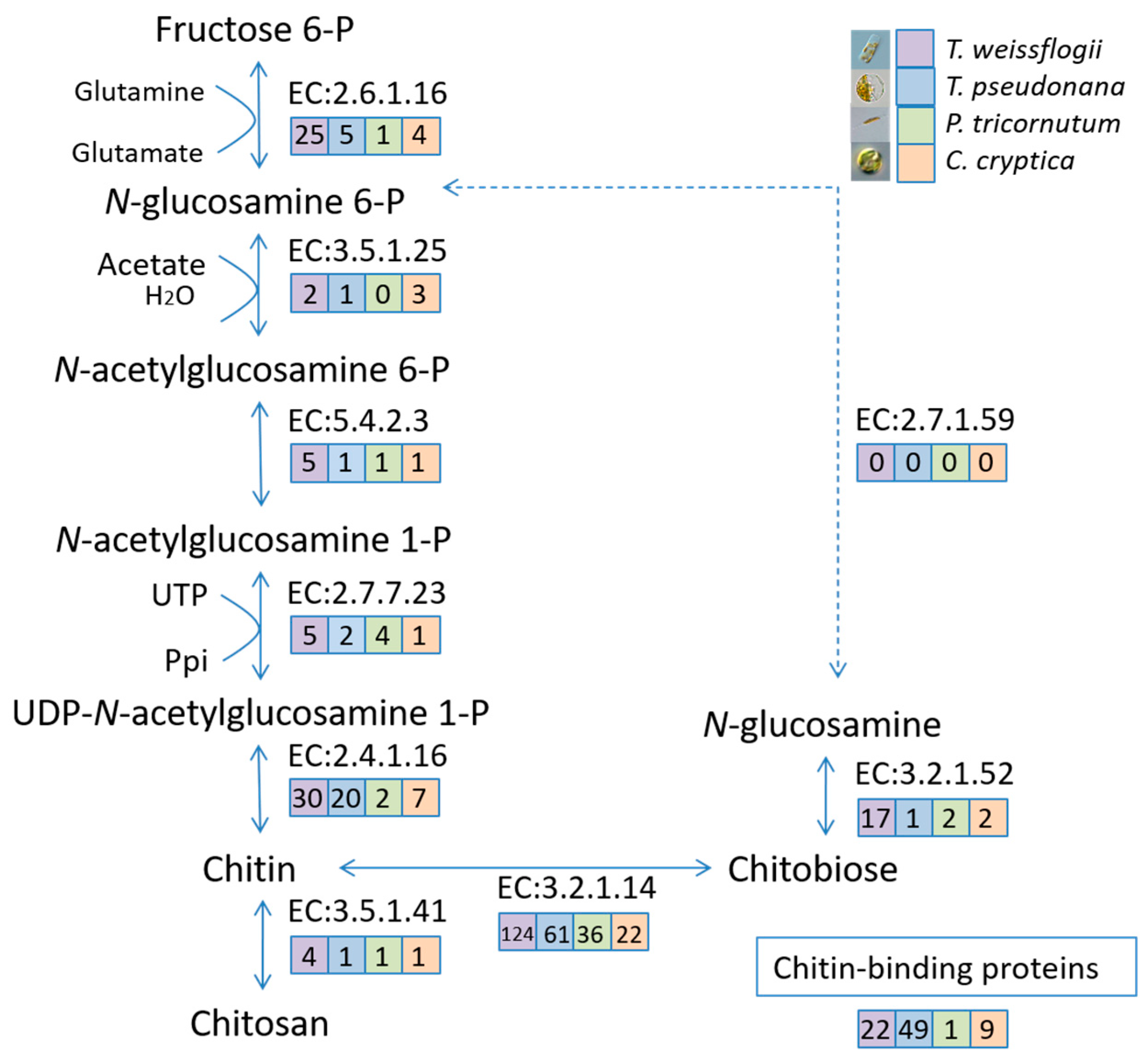
2.7. Chitin-Related Gene Mining
3. Materials and Methods
3.1. Glycosidic Linkage Analysis
3.2. Staining of Chitin and Chitosan
3.3. T. weissflogii Collection and RNA Extraction
3.4. Library Construction and Sequencing
3.5. Data Processing
3.6. Functional Annotation
3.7. Gene Type Analyses
3.8. Detection of Alternative Splicing and Simple Sequence Repeats
3.9. Mining of Chitin Metabolism Genes in Diatoms
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wagner, G.P. Evolution and multi-functionality of the chitin system. In Molecular Ecology and Evolution: Approaches and Applications; Schierwater, B., Streit, B., Wagner, G.P., DeSalle, R., Eds.; Birkhäuser: Basel, Switzerland, 1994; Volume 69, pp. 559–577. [Google Scholar]
- Muzzarelli, R.A.A.; Boudrant, J.; Meyer, D.; Manno, N.; DeMarchis, M.; Paoletti, M.G. Current views on fungal chitin/chitosan, human chitinases, food preservation, glucans, pectins and inulin: A tribute to Henri Braconnot, precursor of the carbohydrate polymers science, on the chitin bicentennial. Carbohydr. Polym. 2012, 87, 995–1012. [Google Scholar] [CrossRef]
- Tang, W.J.; Fernandez, J.; Sohn, J.J.; Amemiya, C.T. Chitin is endogenously produced in vertebrates. Curr. Biol. 2015, 25, 897–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigemasa, Y.; Minami, S. Applications of chitin and chitosan for biomaterials. Biotechnol. Genet. Eng. Rev. 1996, 13, 383–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuong, H.N.; Minh, N.C.; Van Hoa, N.; Trung, T.S. Preparation and characterization of high purity beta-chitin from squid pens (Loligo chenisis). Int. J. Biol. Macromol. 2016, 93, 442–447. [Google Scholar] [CrossRef]
- Shao, Z.; Thomas, Y.; Hembach, L.; Xing, X.; Duan, D.; Moerschbacher, B.M.; Bulone, V.; Tirichine, L.; Bowler, C. Comparative characterization of putative chitin deacetylases from Phaeodactylum tricornutum and Thalassiosira pseudonana highlights the potential for distinct chitin-based metabolic processes in diatoms. New Phytol. 2019, 221, 1890–1905. [Google Scholar] [CrossRef] [PubMed]
- Revol, J.F.; Chanzy, H. High-Resolution Electron Microscopy of β-Chitin Microfibrils. Biopolymers 1986, 25, 1599–1601. [Google Scholar] [CrossRef]
- Brunner, E.; Richthammer, P.; Ehrlich, H.; Paasch, S.; Simon, P.; Ueberlein, S.; van Pee, K.H. Chitin-based organic networks: An integral part of cell wall biosilica in the diatom Thalassiosira pseudonana. Angew. Chem. Int. Ed. Engl. 2009, 48, 9724–9727. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Kimura, S.; Wada, M. Electron diffraction and high-resolution imaging on highly-crystalline beta-chitin microfibril. J. Struct. Biol. 2011, 176, 83–90. [Google Scholar] [CrossRef]
- McLachlan, J.; Craigie, J.S. Chitan fibres in Cyclotella cryptica and growth of C. cryptica and Thalassiosira fluviatilis. In Some Contemporary Studies in Marine Science; Barnes, H., Ed.; George Allen and Unwin Ltd.: London, UK, 1966; pp. 511–517. [Google Scholar]
- Smucker, R.A. Chitin primary production. Biochem. Syst. Ecol. 1991, 19, 357–369. [Google Scholar] [CrossRef]
- McLachlan, J.; McInnes, A.G.; Falk, M. Studies on chitan (chitinpoly-n-acetylglucosamine) fibers of diatom Thalassiosira fluviatilis Hustedt. 1. Production and isolation of chitan fibers. Can. J. Bot. 1965, 43, 707. [Google Scholar] [CrossRef]
- Durkin, C.; Mock, T.; Armbrust, E. Chitin in Diatoms and Its Association with the Cell Wall. Eukaryot. Cell 2009, 8, 1038–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traller, J.C.; Cokus, S.J.; Lopez, D.A.; Gaidarenko, O.; Smith, S.R.; McCrow, J.P.; Gallaher, S.D.; Podell, S.; Thompson, M.; Cook, O.; et al. Genome and methylome of the oleaginous diatom Cyclotella cryptica reveal genetic flexibility toward a high lipid phenotype. Biotechnol. Biofuels 2016, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, C.; Behrenfeld, M.; Randerson, J.; Falkowski, P. Primary production of the biosphere: Integrating terrestrial and oceanic components. Science 1998, 281, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.M.; Tréguer, P.; Brzezinski, M.A.; Leynaert, A.; Quéguiner, B. Production and dissolution of biogenic silica in the ocean: Revised global estimates, comparison with regional data and relationship to biogenic sedimentation. Glob. Biogeochem. Cycles 1995, 9, 359–372. [Google Scholar] [CrossRef]
- Armbrust, E.; Berges, J.; Bowler, C.; Green, B.; Martinez, D.; Putnam, N.; Zhou, S.; Allen, A.; Apt, K.; Bechner, M.; et al. The Genome of the Diatom Thalassiosira Pseudonana: Ecology, Evolution, and Metabolism. Science 2004, 306, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef]
- Paajanen, P.; Strauss, J.; van Oosterhout, C.; McMullan, M.; Clark, M.D.; Mock, T. Building a locally diploid genome and transcriptome of the diatom Fragilariopsis cylindrus. Sci. Data 2017, 4, 170149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.; Feng, J.; Zhang, B.; Huang, Y.; Cheng, J.; Zhang, C. Transcriptome and Gene Expression Analysis of an Oleaginous Diatom Under Different Salinity Conditions. BioEnergy Res. 2013, 7, 192–205. [Google Scholar] [CrossRef]
- Nanjappa, D.; Sanges, R.; Ferrante, M.I.; Zingone, A. Diatom flagellar genes and their expression during sexual reproduction in Leptocylindrus danicus. BMC Genom. 2017, 18, 813. [Google Scholar] [CrossRef] [Green Version]
- Galachyants, Y.P.; Zakharova, Y.R.; Volokitina, N.A.; Morozov, A.A.; Likhoshway, Y.V.; Grachev, M.A. De novo transcriptome assembly and analysis of the freshwater araphid diatom Fragilaria radians, Lake Baikal. Sci. Data 2019, 6, 183. [Google Scholar] [CrossRef] [Green Version]
- Keeling, P.J.; Burki, F.; Wilcox, H.M.; Allam, B.; Allen, E.E.; Amaral-Zettler, L.A.; Armbrust, E.V.; Archibald, J.M.; Bharti, A.K.; Bell, C.J.; et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 2014, 12, e1001889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Di Dato, V.; Di Costanzo, F.; Barbarinaldi, R.; Perna, A.; Ianora, A.; Romano, G. Unveiling the presence of biosynthetic pathways for bioactive compounds in the Thalassiosira rotula transcriptome. Sci. Rep. 2019, 9, 9893. [Google Scholar] [CrossRef] [PubMed]
- Di Dato, V.; Musacchia, F.; Petrosino, G.; Patil, S.; Montresor, M.; Sanges, R.; Ferrante, M.I. Transcriptome sequencing of three Pseudo-nitzschia species reveals comparable gene sets and the presence of Nitric Oxide Synthase genes in diatoms. Sci. Rep. 2015, 5, 12329. [Google Scholar] [CrossRef] [PubMed]
- Buhmann, M.T.; Poulsen, N.; Klemm, J.; Kennedy, M.R.; Sherrill, C.D.; Kroger, N. A tyrosine-rich cell surface protein in the diatom Amphora coffeaeformis identified through transcriptome analysis and genetic transformation. PLoS ONE 2014, 9, e110369. [Google Scholar] [CrossRef]
- Tanaka, T.; Maeda, Y.; Veluchamy, A.; Tanaka, M.; Abida, H.; Marechal, E.; Bowler, C.; Muto, M.; Sunaga, Y.; Tanaka, M.; et al. Oil accumulation by the oleaginous diatom Fistulifera solaris as revealed by the genome and transcriptome. Plant Cell 2015, 27, 162–176. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Mo, S.H.; Lin, X.Y.; Niu, J.; Yin, J.; Wei, D. The PacBio Full-Length Transcriptome of the Tea Aphid as a Reference Resource. Front. Genet. 2020, 11, 558394. [Google Scholar] [CrossRef]
- Chen, C.; Shi, X.; Zhou, T.; Li, W.; Li, S.; Bai, G. Full-length transcriptome analysis and identification of genes involved in asarinin and aristolochic acid biosynthesis in medicinal plant Asarum sieboldii. Genome 2020. [Google Scholar] [CrossRef]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef]
- Babu, M.M. Structure, evolution and dynamics of transcriptional regulatory networks. Biochem. Soc. Trans. 2010, 38, 1155–1178. [Google Scholar] [CrossRef]
- Rayko, E.; Maumus, F.; Maheswari, U.; Jabbari, K.; Bowler, C. Transcription factor families inferred from genome sequences of photosynthetic stramenopiles. New Phytol. 2010, 188, 52–66. [Google Scholar] [CrossRef]
- Cruz de Carvalho, M.H.; Sun, H.X.; Bowler, C.; Chua, N.H. Noncoding and coding transcriptome responses of a marine diatom to phosphate fluctuations. New Phytol. 2016, 210, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarthou, G.; Timmermans, K.R.; Blain, S.; Tréguer, P. Growth physiology and fate of diatoms in the ocean: A review. J. Sea Res. 2005, 53, 25–42. [Google Scholar] [CrossRef]
- Allen, A.E.; Vardi, A.; Bowler, C. An ecological and evolutionary context for integrated nitrogen metabolism and related signaling pathways in marine diatoms. Curr. Opin. Plant Biol. 2006, 9, 264–273. [Google Scholar] [CrossRef]
- Black, D.L. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, A.; Maheswari, U.; Dorrell, R.G.; Vieira, F.R.J.; Maumus, F.; Kustka, A.; McCarthy, J.; Allen, A.E.; Kersey, P.; Bowler, C.; et al. Integrative analysis of large scale transcriptome data draws a comprehensive landscape of Phaeodactylum tricornutum genome and evolutionary origin of diatoms. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharopova, N.; McMullen, M.D.; Schultz, L.; Schroeder, S.; Sanchez-Villeda, H.; Gardiner, J.; Bergstrom, D.; Houchins, K.; MeliaHancock, S.; Musket, T.; et al. Development and mapping of SSR markers for maize. Plant Mol. Biol. 2002, 48, 463–481. [Google Scholar] [CrossRef]
- Karre, S.; Kumar, A.; Dhokane, D.; Kushalappa, A.C. Metabolo-transcriptome profiling of barley reveals induction of chitin elicitor receptor kinase gene (HvCERK1) conferring resistance against Fusarium graminearum. Plant Mol. Biol. 2017, 93, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Giubergia, S.; Phippen, C.; Nielsen, K.F.; Gram, L. Growth on Chitin Impacts the Transcriptome and Metabolite Profiles of Antibiotic-Producing Vibrio coralliilyticus S2052 and Photobacterium galatheae S2753. mSystems 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.M.; Li, Y.J.; Zhang, X.R.; Chu, J.; Ma, J.H.; Liu, Z.X.; Wang, J.; Sheng, S.; Wu, F.A. Identification and Functional Study of Chitin Metabolism and Detoxification-Related Genes in Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) Based on Transcriptome Analysis. Int. J. Mol. Sci. 2020, 21, 1904. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Shao, Z.; Lu, C.; Duan, D. Genome-wide identification of chitinase genes in Thalassiosira pseudonana and analysis of their expression under abiotic stresses. BMC Plant Biol. 2021, 21, 87. [Google Scholar] [CrossRef] [PubMed]
- Hardt, M.; Laine, R.A. Mutation of active site residues in the chitin-binding domain ChBDChiA1 from chitinase A1 of Bacillus circulans alters substrate specificity: Use of a green fluorescent protein binding assay. Arch Biochem. Biophys. 2004, 426, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Nampally, M.; Moerschbacher, B.M.; Kolkenbrock, S. Fusion of a novel genetically engineered chitosan affinity protein and green fluorescent protein for specific detection of chitosan in vitro and in situ. Appl. Environ. Microbiol. 2012, 78, 3114–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinf. Comput. Biol. 2006, 4, 649–664. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef] [PubMed]
- Alamancos, G.P.; Pages, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Linkage | Composition (Mol%) | ||
|---|---|---|---|---|
| Tw | Tp | Tr | ||
| 1 | 4-Glcp | 15.5 | 25.4 | 4.3 |
| 2 | 3-Glcp | 12.8 | 27.8 | 56.4 |
| 3 | 4-GlcNAcp | 6.9 | 0.6 | 1.4 |
| 4 | t-Galp | 6.7 | 2.2 | 3.1 |
| 5 | 2,3-Glcp | 5.5 | 1.5 | 2.0 |
| 6 | t-Manp | 5.4 | 1.7 | 1.7 |
| 7 | 2-Manp | 4.9 | 2.4 | 3.1 |
| 8 | 4-Xylp | 3.7 | 1.9 | 1.4 |
| 9 | t-Xylp | 3.3 | 2.9 | 0.8 |
| 10 | 6-Manp | 2.9 | ND | ND |
| Statistical Data | T. Weissflogii | |
|---|---|---|
| Raw reads | Subread number | 44,233,932 |
| Average length (bp) | 1583 | |
| N50 (bp) | 2003 | |
| CCSs | Number of reads Number of CCS bases CCS read length (mean) (bp) Number of passes (mean) | 1,021,310 2,172,901,990 2127 8 |
| Clustered reads | Number of polished high-quality isoforms | 110,527 |
| Number of polished low-quality isoforms | 338 | |
| Unigenes | Total number | 25,412 |
| Total length (bp) | 51,968,546 | |
| Maximum length (bp) | 11,939 | |
| Minimum length (bp) | 64 | |
| Average length (bp) | 2045.04 | |
| N50 length (bp) | 2417 | |
| GC content | 46.95% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.; Bowler, C.; Xing, X.; Bulone, V.; Shao, Z.; Duan, D. Full-Length Transcriptome of Thalassiosira weissflogii as a Reference Resource and Mining of Chitin-Related Genes. Mar. Drugs 2021, 19, 392. https://doi.org/10.3390/md19070392
Cheng H, Bowler C, Xing X, Bulone V, Shao Z, Duan D. Full-Length Transcriptome of Thalassiosira weissflogii as a Reference Resource and Mining of Chitin-Related Genes. Marine Drugs. 2021; 19(7):392. https://doi.org/10.3390/md19070392
Chicago/Turabian StyleCheng, Haomiao, Chris Bowler, Xiaohui Xing, Vincent Bulone, Zhanru Shao, and Delin Duan. 2021. "Full-Length Transcriptome of Thalassiosira weissflogii as a Reference Resource and Mining of Chitin-Related Genes" Marine Drugs 19, no. 7: 392. https://doi.org/10.3390/md19070392
APA StyleCheng, H., Bowler, C., Xing, X., Bulone, V., Shao, Z., & Duan, D. (2021). Full-Length Transcriptome of Thalassiosira weissflogii as a Reference Resource and Mining of Chitin-Related Genes. Marine Drugs, 19(7), 392. https://doi.org/10.3390/md19070392