
Semi-Synthesis, Cytotoxic Evaluation, and Structure—Activity Relationships of Brefeldin A Derivatives with Antileukemia Activity


 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
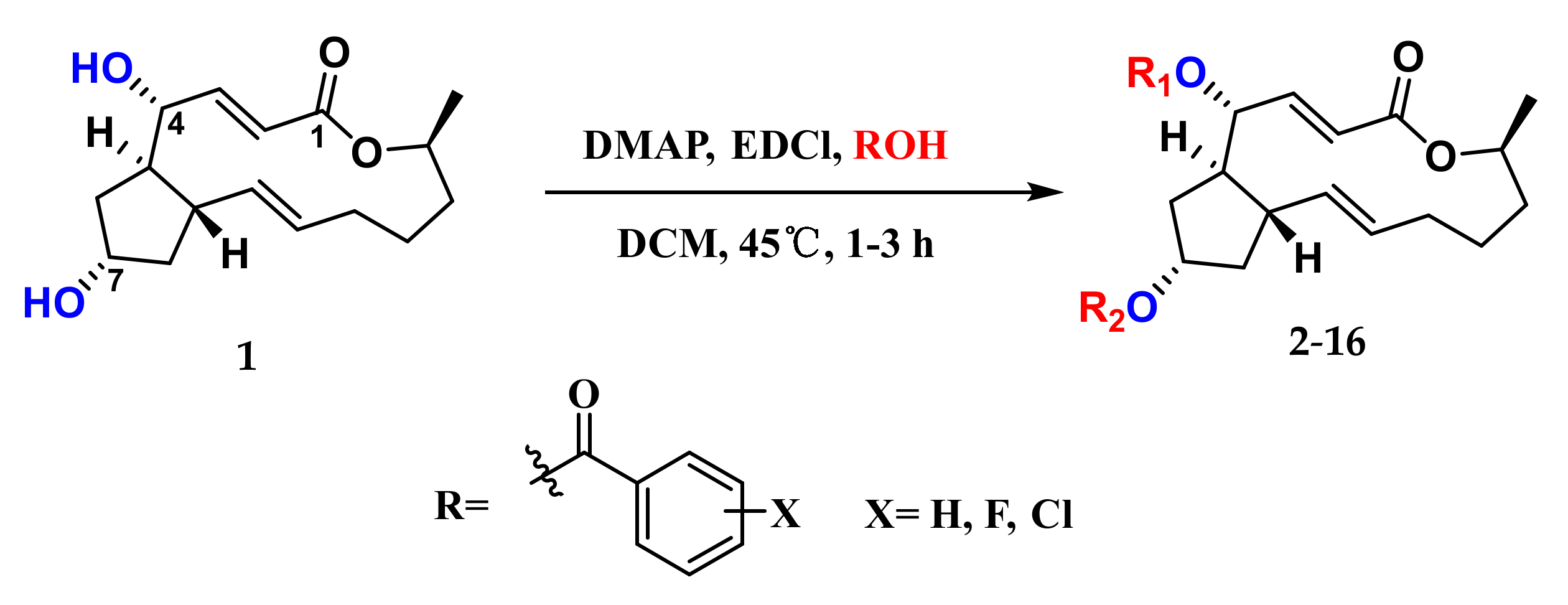
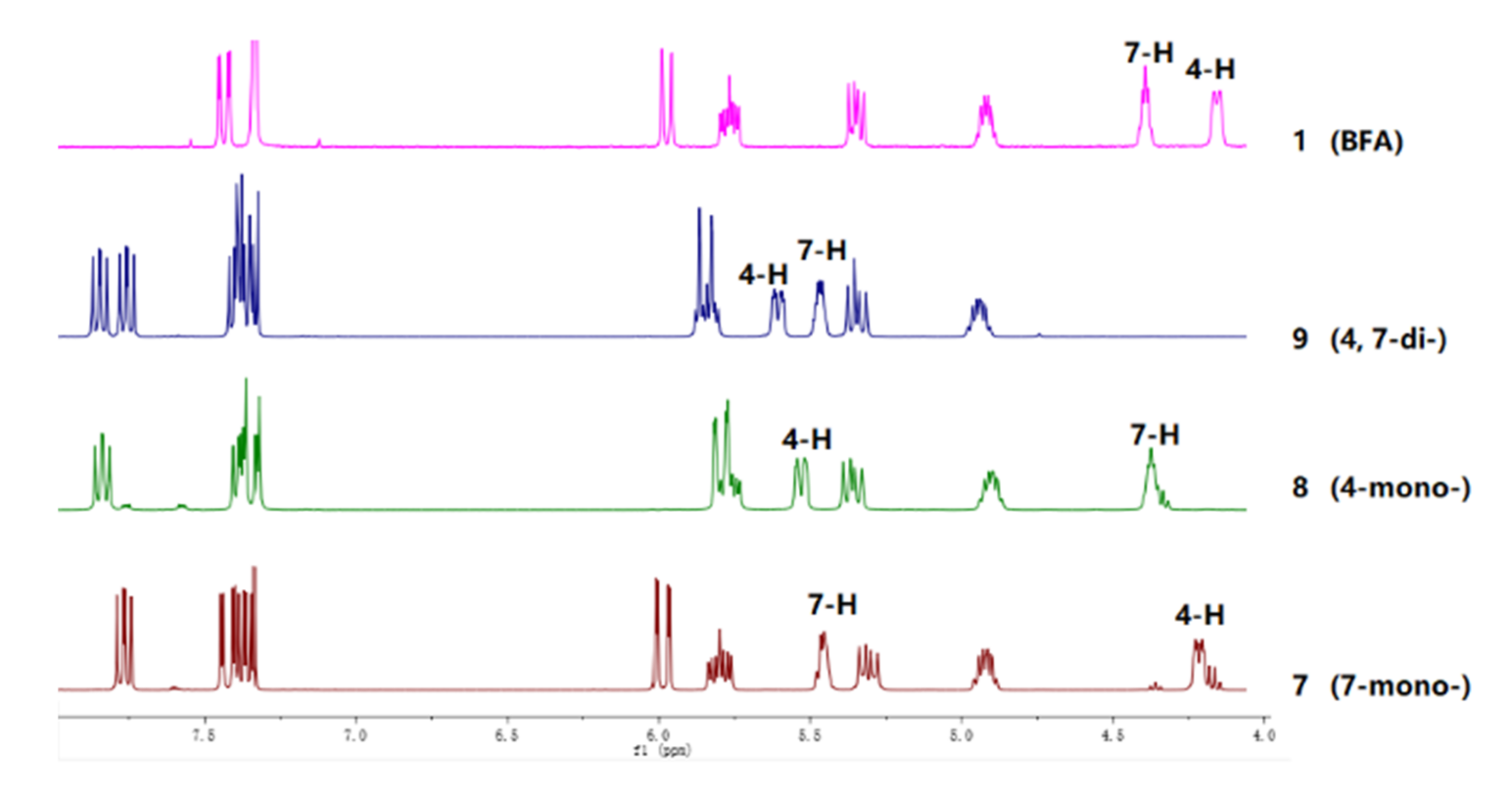
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Cytotoxic Activity
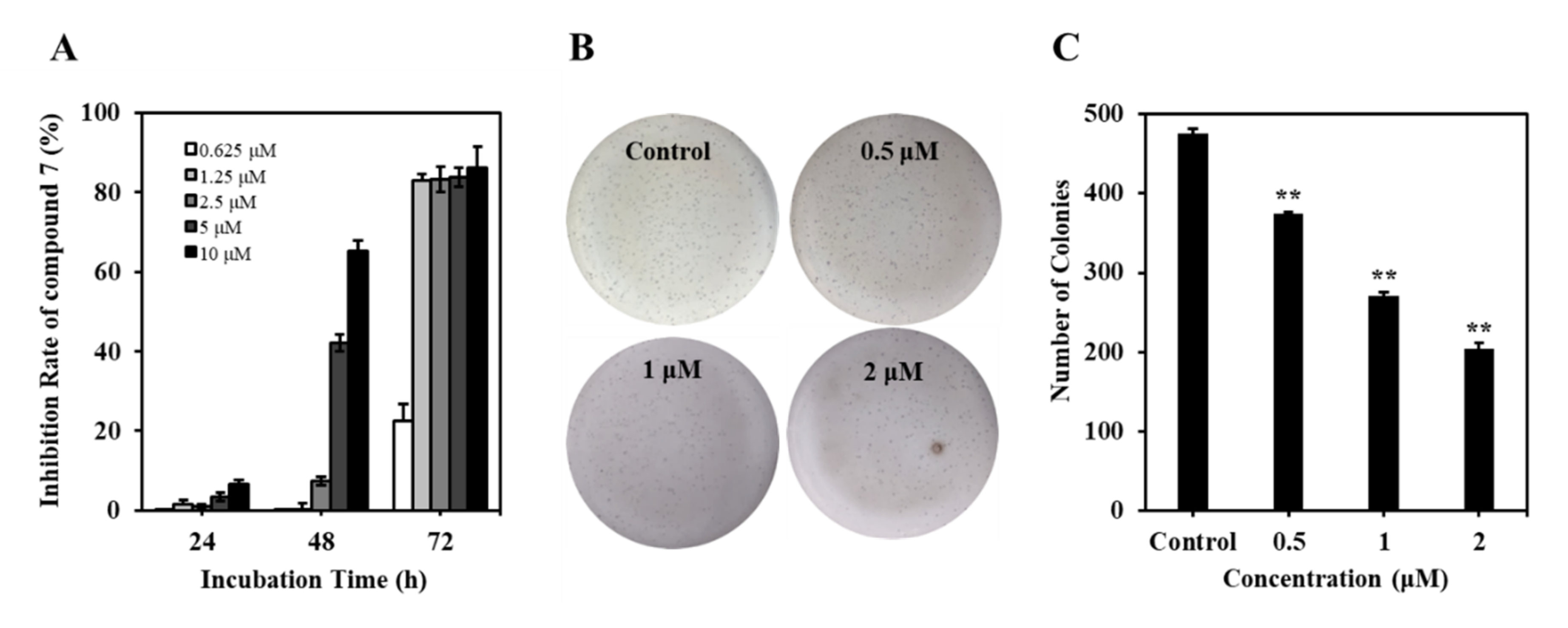
2.2.2. Compound 7 Inhibited the Proliferation of K562 Cells
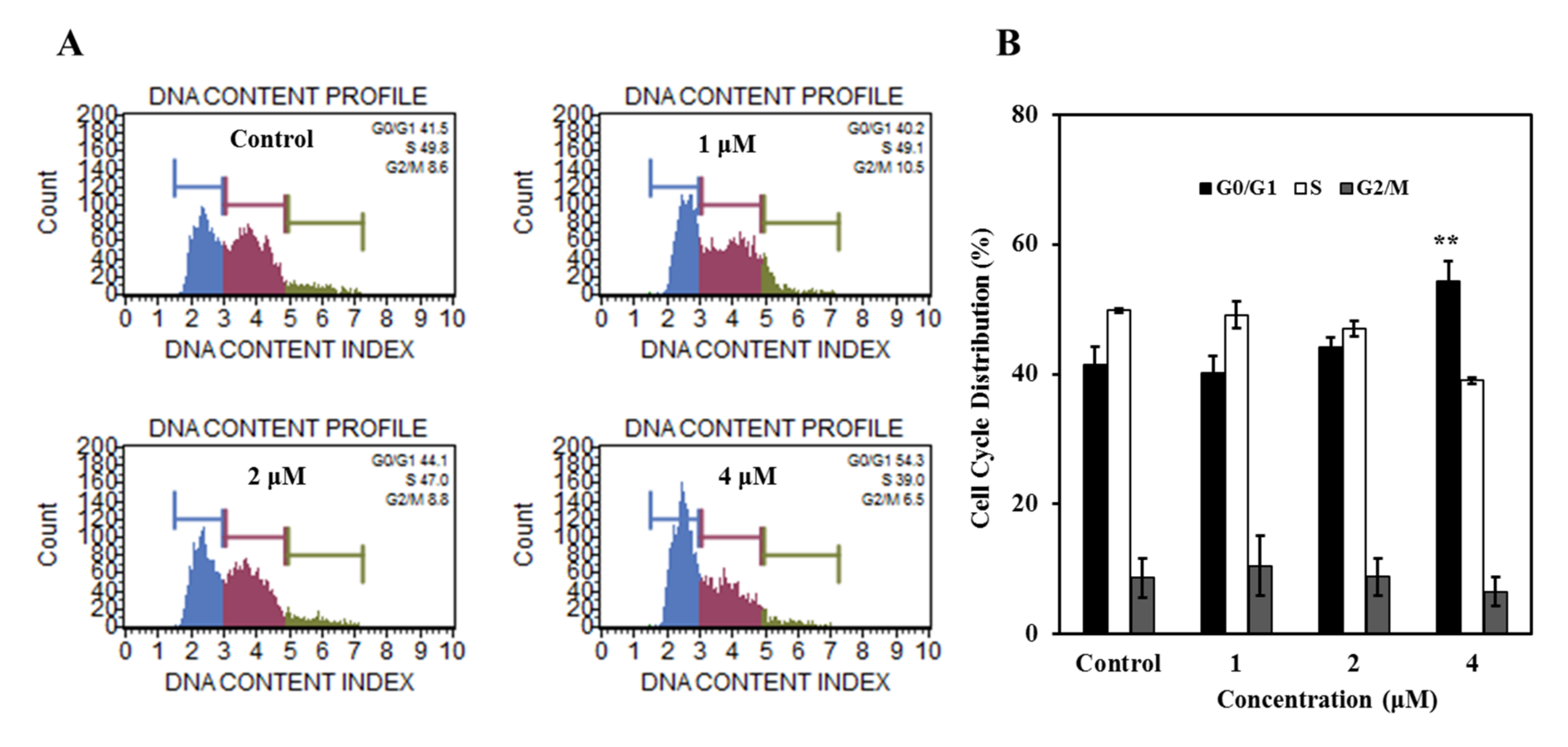
2.2.3. Compound 7 Induced G0/G1 Phase Cell Cycle Arrest in K562 Cells
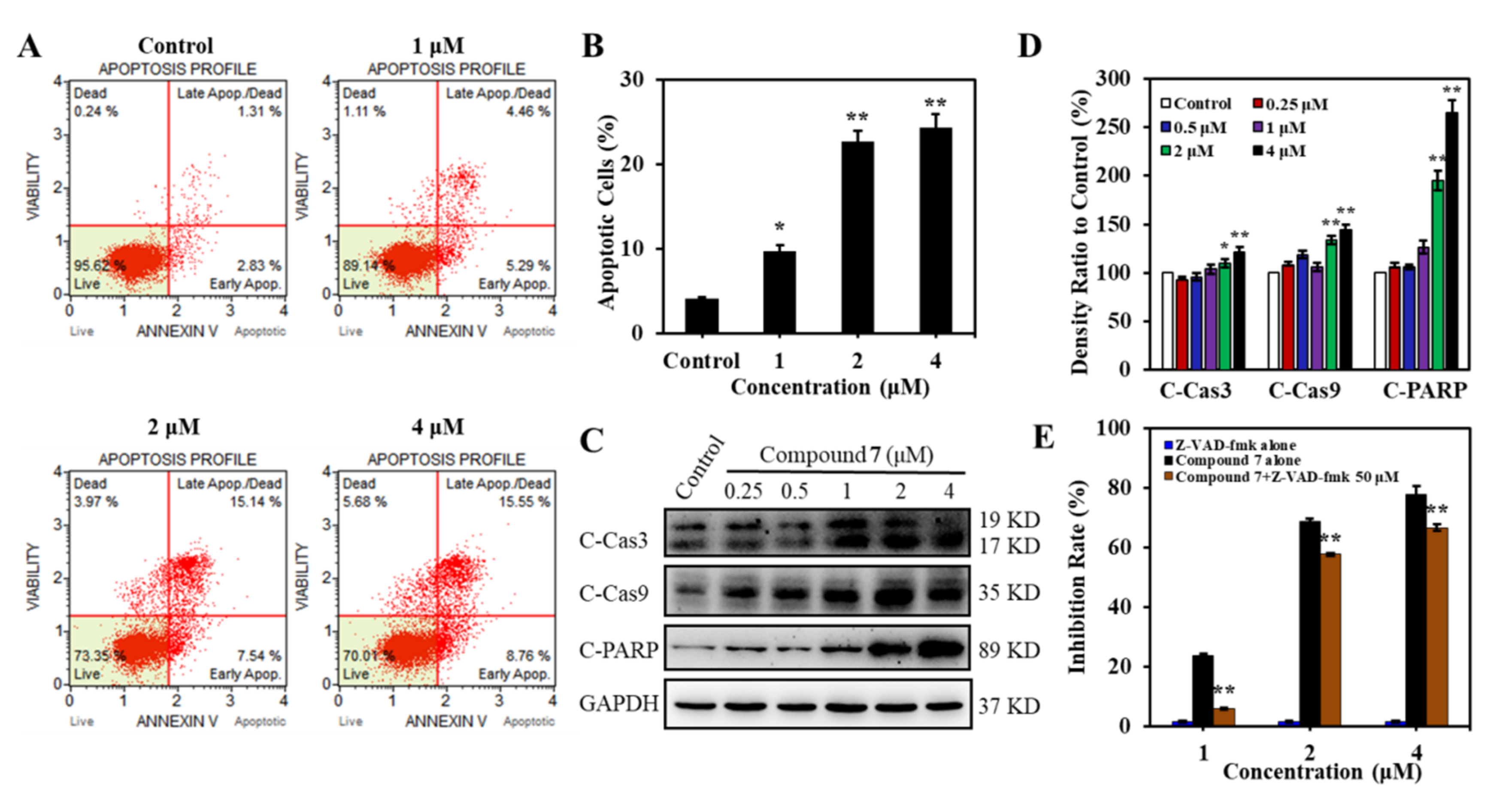
2.2.4. Compound 7 Induced Caspase-Dependent Apoptosis in K562 Cells
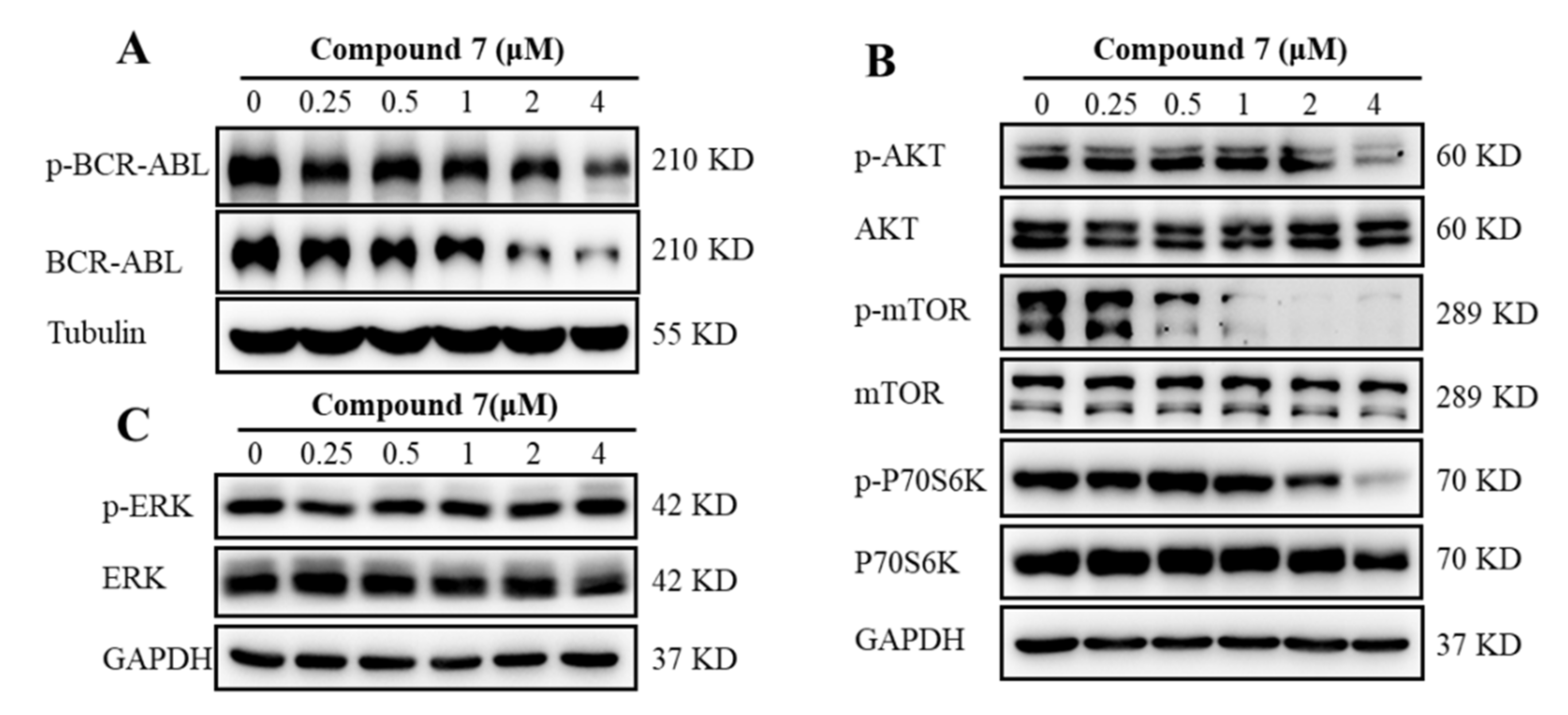
2.2.5. Compound 7 Inactivated BCR-ABL and Affected Its Downstream Signaling Pathways in K562 Cells
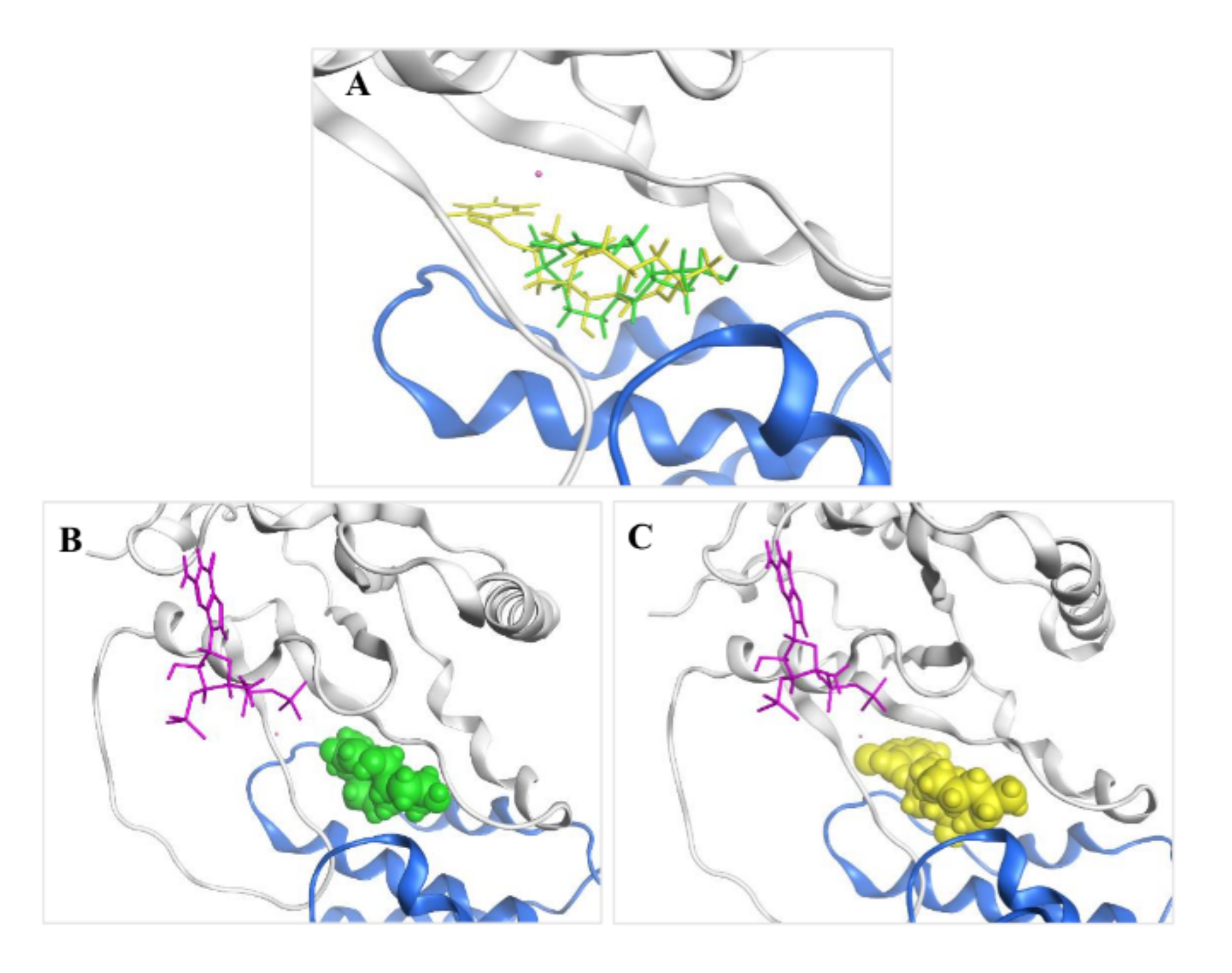
2.3. Molecular Modeling and Ligand Docking of Compound 7 into the Binding Site of 1
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. General Synthetic Methods for Compounds 2–16
3.5. Biological Assays
3.5.1. Reagents
3.5.2. Cell Lines and Cell Culture
3.5.3. Cell Proliferation Assay
3.5.4. Soft Agar Colony Formation Assay
3.5.5. Cell Cycle Analysis
3.5.6. Cell Apoptosis Analysis
3.5.7. Western Blotting Assay
3.5.8. Statistical Analysis
3.6. Solubility Measurement
3.7. Molecular Modeling and Protein–Ligand Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quintás-Cardama, A.; Cortes, J. Molecular biology of BCR-ABL1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugo, T.; Pendergast, A.; Muller, A.; Witte, O. Tyrosine kinase activity and transformation potency of BCR-ABL oncogene products. Science 1990, 247, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.; Rothman, P. JAK-STAT signaling activated by ABL oncogenes. Oncogene 2000, 19, 2523–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J. Imatinib and beyond-exploring the full potential of targeted therapy for CML. Nat. Rev. Clin. Oncol. 2009, 6, 535–543. [Google Scholar] [CrossRef]
- Isfort, S.; Amsberg, G.K.-V.; Schafhausen, P.; Koschmieder, S.; Brümmendorf, T.H. Bosutinib: A novel second-generation tyrosine kinase inhibitor. Small Mol. Oncol. 2014, 201, 81–97. [Google Scholar]
- Blay, J.; von Mehren, M. Nilotinib: A novel, selective tyrosine kinase inhibitor. Semin. Oncol. 2011, 38, S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Tran, C.; Lee, F.; Chen, P.; Norris, D.; Sawyers, C. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Liang, L.; Sun, Y.; Si, R.; Zhang, Q.; Wang, J.; Fu, J.; Zhang, J.; Zhang, J. Discovery of novel BCR-ABL inhibitors with flexible linker. Part 1: Confirmation optimization of phenyl-1H-indazol-3-amine as hinge binding moiety. Eur. J. Med. Chem. 2019, 178, 232–242. [Google Scholar] [CrossRef]
- Hou, X.M.; Liang, T.M.; Guo, Z.Y.; Wang, C.Y.; Shao, C.L. Discovery, absolute assignments, and total synthesis of asperversiamides A-C and their potent activity against Mycobacterium marinum. Chem. Commun. 2019, 55, 1104–1107. [Google Scholar] [CrossRef]
- Hai, Y.; Wei, M.Y.; Wang, C.Y.; Gu, Y.C.; Shao, C.L. The intriguing chemistry and biology of sulfur-containing natural products from marine microorganisms (1987–2020). Mar. Life Sci. Technol. 2021, 3, 488–518. [Google Scholar] [CrossRef]
- Singleton, V.L.; Bohonos, N.; Ullstrup, A.J. Decumbin, a new compound from a species of Penicillium. Nature 1958, 181, 1072–1073. [Google Scholar] [CrossRef]
- Trisuwan, K.; Rukachaisirikul, V.; Sukpondma, Y.; Phongpaichit, S.; Preedanon, S.; Sakayaroj, J. Lactone derivatives from the marine-derived fungus Penicillium sp. PSU-F44. Chem. Pharm. Bull. 2009, 57, 1100–1102. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.F.; Qin, L.L.; Ding, W.J.; Liu, Y.; Ma, Z.J. New analogues of brefeldin A from sediment-derived fungus Penicillium sp. DT-F29. Nat. Prod. Res. 2016, 30, 2311–2315. [Google Scholar] [CrossRef]
- Cheng, X.; Yu, L.; Wang, Q.; Ding, W.; Chen, Z.; Ma, Z. New brefeldins and penialidins from marine fungus Penicillium janthinellum DT-F29. Nat. Prod. Res. 2018, 32, 282–286. [Google Scholar] [CrossRef]
- Zhang, J.M.; Jiang, Y.Y.; Huang, Q.F.; Lu, X.X.; Wang, G.H.; Shao, C.L.; Liu, M. Brefeldin A delivery nanomicelles in hepatocellular carcinoma therapy: Characterization, cytotoxic evaluation in vitro, and antitumor efficiency in vivo. Pharmacol. Res. 2021, 172, 105800–105810. [Google Scholar] [CrossRef]
- Mossessova, E.; Corpina, R.A.; Goldberg, J. Crystal structure of ARF1·Sec7 complexed with brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 2003, 12, 1403–1411. [Google Scholar] [CrossRef]
- Renault, L.; Guibert, B.; Cherfils, J. Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 2003, 426, 525–530. [Google Scholar] [CrossRef]
- Fujiwara, T.; Oda, K.; Yokota, S.; Takatsuki, A.; Ikehara, Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J. Biol. Chem. 1988, 263, 18545–18552. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Yuan, L.C.; Bonifacino, J.S.; Klausner, R.D. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: Evidence for membrane cycling from Golgi to ER. Cell 1989, 56, 801–813. [Google Scholar] [CrossRef]
- Driouich, A.; Zhang, G.F.; Staehelin, L.A. Effect of brefeldin A on the structure of the Golgi apparatus and on the synthesis and secretion of proteins and polysaccharides in sycamore maple (Acer pseudoplatanus) suspension-cultured cells. Plant Physiol. 1993, 101, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Rouhana, J.; Padilla, A.; Estaran, S.; Bakari, S.; Delbecq, S.; Boublik, Y.; Chopineau, J.; Pugniere, M.; Chavanieu, A. Kinetics of interaction between ADP-ribosylation factor-1 (Arf1) and the Sec7 domain of Arno guanine nucleotide exchange factor, modulation by allosteric factors, and the uncompetitive inhibitor brefeldin A. J. Biol. Chem. 2013, 288, 4659–4672. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Dominguez, N.; Parnell, C.; Teng, Y. Drugging the small GTPase pathways in cancer treatment: Promises and challenges. Cells 2019, 8, 255. [Google Scholar] [CrossRef] [Green Version]
- Anadu, N.O.; Davisson, V.J.; Cushman, M. Synthesis and anticancer activity of Brefeldin A ester derivatives. J. Med. Chem. 2006, 49, 3897–3905. [Google Scholar] [CrossRef]
- Kikuchi, S.; Shinpo, K.; Tsuji, S.; Yabe, I.; Niino, M.; Tashiro, K. Brefeldin A-induced neurotoxicity in cultured spinal cord neurons. J. Neurosci. Res. 2003, 71, 591–599. [Google Scholar] [CrossRef]
- He, B.; Wang, Y.; Zheng, Y.; Chen, W.; Zhu, Q. Synthesis and cytotoxic evaluation of acylated Brefeldin A derivatives as potential anticancer agents. Chem. Biol. Drug Des. 2013, 82, 307–316. [Google Scholar] [CrossRef]
- Zhu, J.W.; Nagasawa, H.; Nagura, F.; Mohamad, S.B.; Uto, Y.; Ohkura, K.; Hori, H. Elucidation of strict structural requirements of Brefeldin A as an inducer of differentiation and apoptosis. Bioorg. Med. Chem. 2000, 8, 455–463. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, X.; Yang, Y.-Q.; Hu, Q.; Huang, J.-H. Enantioselective total synthesis of (+)-brefeldin A and 7-epi-brefeldin A. J. Org. Chem. 2004, 69, 3857–3865. [Google Scholar] [CrossRef]
- Proksa, B.; Uhrin, D.; Adamcova, J.; Fuska, J. Oxidation of brefeldin A. Pharmazie 1992, 47, 582–584. [Google Scholar] [PubMed]
- Fox, B.M.; Vroman, J.A.; Fanwick, P.E.; Cushman, M. Preparation and evaluation of sulfide derivatives of the antibiotic brefeldin A as potential prodrug candidates with enhanced aqueous solubilities. J. Med. Chem. 2001, 44, 3915–3924. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.W.; Hori, H.; Nojiri, H.; Tsukuda, T.; Taira, Z. Synthesis and activity of brefeldin A analogs as inducers of cancer cell differentiation and apoptosis. Bioorg. Med. Chem. Lett. 1997, 7, 139–144. [Google Scholar] [CrossRef]
- Seehafer, K.; Rominger, F.; Helmchen, G.; Langhans, M.; Robinson, D.G.; Oezata, B.; Bruegger, B.; Strating, J.R.P.M.; van Kuppeveld, F.J.M.; Klein, C.D. Synthesis and biological properties of novel brefeldin A analogues. J. Med. Chem. 2013, 56, 5872–5884. [Google Scholar] [CrossRef]
- Haruma, T. Antitumor Agent Containing Acyl Thiourea Compound and Antitumor Effect Potentiator for Use with Immunocheckpoint Regulator. WO Patent WO2019049956A1, 14 March 2019. [Google Scholar]
- Van Pee, K.-H.; Ludwig-Mueller, J. Halogenated indole derivatives. Curr. Top. Phytochem. 2002, 5, 1–21. [Google Scholar]
- Zhang, J.M.; Wang, C.F.; Wei, M.Y.; Dong, H.; Gu, Y.C.; Mo, X.M.; Shao, C.L.; Liu, M. Brefeldin A induces apoptosis, inhibits BCR-ABL activation, and triggers BCR-ABL degradation in chronic myeloid leukemia K562 cells. Anticancer Agents Med. Chem. 2021, in press. [Google Scholar] [CrossRef]
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94, 107–121. [Google Scholar] [CrossRef]
- Martin, G.S. Cell signaling and cancer. Cancer Cell 2003, 4, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Wang, F.; Zhao, Z.; Zhao, X.; Qiu, J.; Nie, C.; Wei, Y. BIM-mediated AKT phosphorylation is a key modulator of arsenic trioxide-induced apoptosis in cisplatin-sensitive and -resistant ovarian cancer cells. PLoS ONE 2011, 6, e20586. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Yu, S.; Wang, L.; He, M.; Cao, X.; Li, Y.; Xiao, H. Exendin-4 inhibits growth and augments apoptosis of ovarian cancer cells. Mol. Cell Endocrinol. 2016, 436, 240–249. [Google Scholar] [CrossRef]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell. Mol. Life Sci. 2015, 72, 2337–2347. [Google Scholar] [CrossRef]
- Airiau, K.; Mahon, F.X.; Josselin, M.; Jeanneteau, M.; Belloc, F. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. Cell Death Dis. 2013, 4, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Okabe, S.; Tauchi, T.; Tanaka, Y.; Kitahara, T.; Kimura, S.; Maekawa, T.; Ohyashiki, K. Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with nilotinib against BCR-ABL-positive leukemia cells involves the ABL kinase domain mutation. Cancer Biol. Ther. 2014, 15, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, X.M.; Teuscher, F.; Li, D.L.; Diesel, A.; Ebel, R.; Proksch, P.; Wang, B.G. Chaetopyranin, a benzaldehyde derivative, and other related metabolites from Chaetomium globosum, an endophytic fungus derived from the marine red alga Polysiphonia urceolata. J. Nat. Prod. 2006, 69, 1622–1625. [Google Scholar] [CrossRef]
- Zhi, Y.; Wu, X.; Shen, W.; Wang, Y.; Zhou, X.; He, P.; Pan, J.; Chen, Z.; Li, W.; Zhou, Z. Synthesis and pharmacological evaluation of novel epidermal growth factor receptor inhibitors against prostate tumor cells. Oncol. Lett. 2018, 16, 6522–6530. [Google Scholar] [CrossRef]
- Lu, X.; Geng, J.; Zhang, J.; Miao, J.; Liu, M. Xanthohumol, a prenylated flavonoid from hops, induces caspase-dependent degradation of oncoprotein BCR-ABL in K56.62 cells. Antioxidants 2019, 8, 402. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compounds | IC50 (μM) a | Compounds | IC50 (μM) a | ||
| Structure | Substituents | K562 | Structure | Substituents | K562 |
| brefeldin A | 1 R1 = R2 = H | 0.24 |  | 10 R1 = H, R2 = R | 1.07 |
 | 2 R1 = H, R2 = R | 2.49 | 11 R1 = R, R2 = H | 1.33 | |
| 3 R1 = R2 = R | 4.71 | 12 R1 = R2 = R | >10 | ||
| 4 R1 = R, R2 = H | 0.91 |  | 13 R1 = H, R2 = R | 1.11 | |
 | 5 R1 = H, R2 = R | 0.91 | 14 R1 = R, R2 = H | 1.76 | |
| 6 R1 = R, R2 = H | 1.22 | 15 R1 = R2 = R | >10 | ||
 | 7 R1 = H, R2 = R | 0.84 |  | 16 R1 = H, R2 = R | >10 |
| 8 R1 = R, R2 = H | 2.06 | Doxorubicin | 0.06 | ||
| 9 R1 = R2 = R | >10 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.-X.; Jiang, Y.-Y.; Wu, Y.-W.; Chen, G.-Y.; Shao, C.-L.; Gu, Y.-C.; Liu, M.; Wei, M.-Y. Semi-Synthesis, Cytotoxic Evaluation, and Structure—Activity Relationships of Brefeldin A Derivatives with Antileukemia Activity. Mar. Drugs 2022, 20, 26. https://doi.org/10.3390/md20010026
Lu X-X, Jiang Y-Y, Wu Y-W, Chen G-Y, Shao C-L, Gu Y-C, Liu M, Wei M-Y. Semi-Synthesis, Cytotoxic Evaluation, and Structure—Activity Relationships of Brefeldin A Derivatives with Antileukemia Activity. Marine Drugs. 2022; 20(1):26. https://doi.org/10.3390/md20010026
Chicago/Turabian StyleLu, Xu-Xiu, Yao-Yao Jiang, Yan-Wei Wu, Guang-Ying Chen, Chang-Lun Shao, Yu-Cheng Gu, Ming Liu, and Mei-Yan Wei. 2022. "Semi-Synthesis, Cytotoxic Evaluation, and Structure—Activity Relationships of Brefeldin A Derivatives with Antileukemia Activity" Marine Drugs 20, no. 1: 26. https://doi.org/10.3390/md20010026
APA StyleLu, X. -X., Jiang, Y. -Y., Wu, Y. -W., Chen, G. -Y., Shao, C. -L., Gu, Y. -C., Liu, M., & Wei, M. -Y. (2022). Semi-Synthesis, Cytotoxic Evaluation, and Structure—Activity Relationships of Brefeldin A Derivatives with Antileukemia Activity. Marine Drugs, 20(1), 26. https://doi.org/10.3390/md20010026