
Six New Antimicrobial Metabolites from the Deep-Sea Sediment-Derived Fungus Aspergillus fumigatus SD-406

 , and
, and 
Abstract
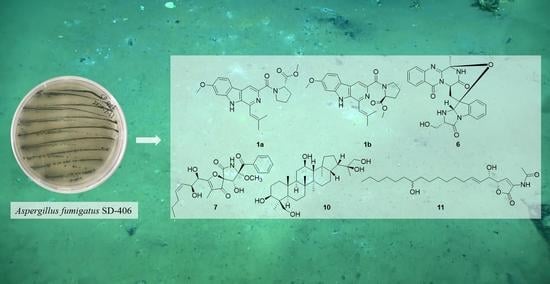
:
1. Introduction
2. Results and Discussion
2.1. Structure Elucidation
2.2. Antimicrobial Activity
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction and Isolation
3.4. Acidic Hydrolysis of Compound 1
3.5. X-ray Crystallographic Analysis of Compound 10
3.6. ECD Calculation of Compound 6 and OR Calculation of Compound 10
3.7. Computational NMR Chemical Shift and DP4+ Analyses
3.8. Bioassay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yurchenko, A.N.; Girich, E.V.; Yurchenko, E.A. Metabolites of marine sediment-derived Fungi: Actual trends of biological activity studies. Mar. Drugs 2021, 19, 88. [Google Scholar] [CrossRef]
- Chi, L.-P.; Li, X.-M.; Wan, Y.-P.; Li, X.; Wang, B.-G. Ophiobolin sesterterpenoids and farnesylated phthalide derivatives from the deep sea cold-seep-derived fungus Aspergillus insuetus SD-512. J. Nat. Prod. 2020, 83, 3652–3660. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Li, X.-M.; Li, X.; Yang, S.-Q.; Shi, X.-S.; Li, H.-L.; Wang, B.-G. Antibacterial alkaloids and polyketide derivatives from the deep sea-derived fungus Penicillium cyclopium SD-413. Mar. Drugs 2020, 18, 553. [Google Scholar] [CrossRef]
- Chi, L.-P.; Li, X.-M.; Li, L.; Li, X.; Wang, B.-G. Cytotoxic thiodiketopiperazine derivatives from the deep sea-derived fungus Epicoccum nigrum SD-388. Mar. Drugs 2020, 18, 160. [Google Scholar] [CrossRef] [Green Version]
- Niu, S.; Si, L.; Liu, D.; Zhou, A.; Zhang, Z.; Shao, Z.; Wang, S.; Zhang, L.; Zhou, D.; Lin, W. Spiromastilactones: A new class of influenza virus inhibitors from deep-sea fungus. Eur. J. Med. Chem. 2016, 108, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.-B.; Kakeya, H.; Osada, H. Novel mammalian cell cycle inhibitors, tryprostatins A, B and other diketopiperazines produced by Aspergillus fumigatus. J. Antibiot. 1996, 49, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Abraham, W.R.; Arfmann, H.A. 12,13-Dihydroxy-fumitremorgin C from Aspergillus fumigatus. Phytochemistry 1990, 29, 1025–1026. [Google Scholar] [CrossRef]
- Cui, C.-B.; Kakeya, H.; Osada, H. Novel mammalian cell cycle inhibitors, cyclotroprostatins A–D, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron 1997, 53, 59–72. [Google Scholar] [CrossRef]
- Numata, A.; Takahashi, C.; Matsushita, T.; Miyamoto, T.; Kawai, K.; Usami, Y.; Matsumura, E.; Inoue, M.; Ohishi, H.; Shingu, T. Fumiquinazolines, novel metabolites of a fungus isolated from a saltfish. Tetrahedron Lett. 1992, 33, 1621–1624. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, W.-L.; Fang, Y.-C.; Zhu, T.-J.; Gu, Q.-Q.; Zhu, W.-M. Cytotoxic alkaloids and antibiotic nordammarane triterpenoids from the marine-derived fungus Aspergillus sydowi. J. Nat. Prod. 2008, 71, 985–989. [Google Scholar] [CrossRef]
- Wang, F.-Z.; Li, D.-H.; Zhu, T.-J.; Zhang, M.; Gu, Q.-Q. Pseurotin A1 and A2, two new 1-oxa-7-azaspiro[4.4]non-2-ene-4,6-diones from the holothurian-derived fungus Aspergillus fumigatus WFZ-25. Can. J. Chem. 2011, 89, 72–76. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Matsushita, T.; Doi, M.; Minoura, K.; Shingu, T.; Kumeda, Y.; Numata, A. Fumiquinazolines A–G, novel metabolites of a fungus separated from a Pseudolabrus marine fish. J. Chem. Soc. Perkin Trans. 1 1995, 18, 2345–2353. [Google Scholar] [CrossRef]
- Bifulco, P.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ohshima, M.; Yuasa, K.; Kikuchi, T.; Tanaka, R. Assignment of the CD Cotton effect to the chiral center in pseurotins, and the stereochemical Revision of pseurotin A2. Mar. Drugs 2016, 14, 74. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Kitada, H.; Kajimoto, T.; Numata, A.; Tanaka, R. The relationship between the CD Cotton effect and the absolute configuration of FD-838 and its seven stereoisomers. J. Org. Chem. 2010, 75, 4146–4153. [Google Scholar] [CrossRef]
- Xu, X.; Han, J.; Wang, Y.; Lin, R.; Yang, H.; Li, J.; Wei, S.; Polyak, S.W.; Song, F. Two new spiro-heterocyclic γ-lactams from a marine-derived Aspergillus fumigatus strain CUGBMF170049. Mar. Drugs 2019, 17, 289. [Google Scholar] [CrossRef] [Green Version]
- Bloch, P.; Tamm, C. Isolation and structure of pseurotin A, a microbial metabolite of Pseudeurotium ovalis STOLK with an unusual heterospirocyclic system. Helv. Chim. Acta 1981, 64, 304–315. [Google Scholar] [CrossRef]
- Masuda, K.; Yamashita, H.; Shiojima, K.; Itoh, T.; Ageta, H. Fern constituents: Triterpenoids isolated from rhizomes of pyrrosia lingua. I. Chem. Pharm. Bull. 1997, 45, 590–594. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, H.-T.; Wang, D.; Yang, C.-R.; Zhang, Y.-J. Sphingofungins G and H: New five-membered lactones from Aspergillus penicilliodes Speg. Nat. Prod. Res. 2019, 33, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Berova, N.; Breinholt, J.; Jensen, G.W.; Kjaer, A.; Lo, L.-C.; Nakanishi, K.; Nielsen, R.I.; Olsen, C.E.; Pedersen, C.; Stidsen, C.E. Malonofungin: An antifungal aminomalonic acid from Phaeoramularia fusimaculans. Acta Chem. Scand. 1994, 48, 240–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crystallographic Data of Compound 10 Have Been Deposited in the Cambridge Crystallographic Data Centre as CCDC 2124932. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 30 November 2021).
- Sheldrick, G.M. SADABS, Software for Empirical Absorption Correction; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXTL, Structure Determination Software Programs; Bruker Analytical X-ray System Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97 and SHELXS-97, Program for X-ray Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Meng, L.-H.; Li, X.-M.; Zhang, F.-Z.; Wang, Y.-N.; Wang, B.-G. Talascortenes A–G, highly oxygenated diterpenoid acids from the sea-anemone-derived endozoic fungus Talaromyces scorteus AS-242. J. Nat. Prod. 2020, 83, 2528–2536. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1a (Major) | 1b (Minor) | ||
|---|---|---|---|---|
| δC, Type a | δH (J in Hz) b | δC, Type a | δH (J in Hz) b | |
| 1-NH | 11.75, s | 11.55, s | ||
| 2 | 134.6, C | 134.4, C | ||
| 3 | 138.5, C | 138.2, C | ||
| 5 | 172.6, C | 172.9, C | ||
| 6 | 59.6, CH | 4.55, dd, (8.5, 4.6) | 60.7, CH | 5.21, dd, (8.6, 3.5) |
| 7 | 28.3, CH2 | β 1.87, m; α 2.25, m | 31.3, CH2 | β 1.97, m; α 2.28, m |
| 8 | 25.2, CH2 | 1.89, m | 21.6, CH2 | 1.83, m |
| 9 | 49.6, CH2 | β 4.01, m; α 3.89, m | 47.8, CH2 | 3.69, m |
| 11 | 166.7, C | 166.2, C | ||
| 12 | 141.9, C | 141.8, C | ||
| 13 | 113.3, CH | 8.33, s | 113.4, CH | 8.43, s |
| 14 | 128.1, C | 128.3, C | ||
| 15 | 114.9, C | 114.9, C | ||
| 16 | 122.7, CH | 8.17, d, (8.7) | 122.7, CH | 8.17, d, (8.6) |
| 17 | 109.6, CH | 6.88, dd, (8.7, 2.2) | 109.6, CH | 6.87, dd, (8.6, 2.2) |
| 18 | 160.4, C | 160.4, C | ||
| 19 | 94.8, CH | 7.04, d, (2.2) | 94.8, CH | 7.04, d, (2.2) |
| 20 | 142.3, C | 142.5, C | ||
| 21 | 119.5, CH | 6.79, s | 120.0, CH | 6.59, s |
| 22 | 141.1, C | 140.4, C | ||
| 23 | 20.2, CH3 | 2.19, s | 20.0, CH3 | 1.91, s |
| 24 | 27.1, CH3 | 2.06, s | 26.5, CH3 | 2.03, s |
| 25 | 55.3, CH3 | 3.88, s | 55.3, CH3 | 3.87, s |
| 26 | 51.6, CH3 | 3.67, s | 51.4, CH3 | 3.46, s |
| No. | 6 | No. | 7 | ||
|---|---|---|---|---|---|
| δC, Type a | δH (J in Hz) b | δC, Type a | δH (J in Hz) b | ||
| 1 | 169.9, C | 2 | 186.9, C | ||
| 2-NH | 9.97, s | 3 | 111.5, C | ||
| 3 | 83.7, C | 4 | 196.7, C | ||
| 4 | 150.3, C | 5 | 91.1, C | ||
| 6 | 146.4, C | 6 | 166.5, C | ||
| 7 | 128.2, CH | 7.82, dd, (8.3, 1.2) | 7-NH | 9.94, s | |
| 8 | 134.7, CH | 7.91, td, (8.3, 1.6) | 8 | 92.4, C | |
| 9 | 128.0, CH | 7.65, td, (8.3, 1.2) | 8-OCH3 | 51.6, CH3 | 3.25, s |
| 10 | 126.2, CH | 8.21, dd, (8.3, 1.6) | 9 | 74.9, CH | 4.40, d, (9.4) |
| 11 | 120.9, C | 9-OH | 6.22, s, (9.4) | ||
| 12 | 160.0, C | 10 | 71.9, CH | 4,34, t, (5.3) | |
| 14 | 51.3, CH | 5.34, dd, (6.7, 1.2) | 11 | 68.3, CH | 4.45, m |
| 15 | 30.7, CH2 | β 3.31, m ; α 1.90, m | 12 | 129.8, CH | 5.42, dd, (8.7, 11.0) |
| 16 | 23.9, CH3 | 1.90, s | 13 | 131.9, CH | 5.43, dd, (6.9, 11.0) |
| 17 | 86.4, C | 14 | 29.2, CH2 | 1.99, m | |
| 18 | 87.2, CH | 5.18, d, (8.6) | 15 | 22.2, CH2 | 1.30, m |
| 19-NH | 2.32, t, (8.6) | 16 | 13.5, CH3 | 0.83, t, (7.4) | |
| 20 | 64.3, CH | 3.64, m | 17 | 5.6, CH3 | 1.64, s |
| 21 | 169.6, C | 18 | 196.3, C | ||
| 23 | 135.7, C | 19 | 133.4, C | ||
| 24 | 114.7, CH | 7.32, dd, (7.3, 1.2) | 20 | 130.2, CH | 8.25, d, (7.8) |
| 25 | 129.6, CH | 7.35, td, (7.3, 1.2) | 21 | 128.3, CH | 7.53, t, (7.8) |
| 26 | 125.8, CH | 7.24, td, (7.3, 1.2) | 22 | 133.7, CH | 7.67, t, (7.8) |
| 27 | 124.8, CH | 7.31, dd. (7.3, 1.2) | 23 | 128.3, CH | 7.53, t, (7.8) |
| 28 | 138.8, C | 24 | 130.2, CH | 8.25, d, (7.8) | |
| 29 | 61.8, CH2 | β 3.21, m; α 3.41, m | |||
| 29-OH | 4.16, t, (5.5) | ||||
| No. | 10 | No. | 11 | ||
|---|---|---|---|---|---|
| δC, Type a | δH (J in Hz) b | δC, Type a | δH (J in Hz) b | ||
| 1 | 38.1, CH2 | α 0.89, m; β 1.61, m | NH | 10.03, s | |
| 2 | 27.3, CH2 | α 1.58, m; β 1.58, m | 1 | 168.9, C | |
| 3 | 78.6, CH | 3.16, m | 2 | 126.7, C | |
| 3-OH | 4.96, d, (4.8) | 3 | 127.4, CH | 7.32, d, (1.9) | |
| 4 | 42.1, C | 4 | 83.5, CH | 4.98, m | |
| 5 | 55.3, CH | 0.69, m | 5 | 71.5, CH | 4.15, d, (5.2) |
| 6 | 18.6, CH2 | α 1.56, m; β 1.45, m | 6 | 128.5, CH | 5.32, m |
| 7 | 33.0, CH2 | α 1.28, m; β 1.15, m | 7 | 132.9, CH | 5.65, m |
| 8 | 40.8, C | 8 | 31.3, CH2 | 1.98, m | |
| 9 | 48.5, CH | 1.20, m | 9 | 29.0, CH2 | 1.20–1.29, m |
| 10 | 36.2, C | 10 | 28.8, CH2 | 1.20–1.29, m | |
| 11 | 32.2, CH2 | α 1.67, m; β 1.21, m | 11 | 28.4, CH2 | 1.20–1.29, m |
| 12 | 67.9, CH | 3.67, m | 12 | 25.1, CH2 | 1.20–1.29, m |
| 12-OH | 3.82, overlap | 13 | 37.1, CH2 | 1.20–1.29, m | |
| 13 | 55.0, CH | 1.20, m | 14 | 69.5, CH | 3.34, m |
| 14 | 42.2, C | 15 | 37.1, CH2 | 1.20–1.29, m | |
| 15 | 34.7, CH2 | α 1.26, m; β 1.08, m | 16 | 25.1, CH2 | 1.20–1.29, m |
| 16 | 20.9, CH2 | α 1.53, m; β 1.94, m | 17 | 28.4, CH2 | 1.20–1.29, m |
| 17 | 54.1, CH | 1.29, m | 18 | 31.5, CH2 | 1.20–1.29, m |
| 18 | 42.9, C | 19 | 22.0, CH2 | 1.20–1.29, m | |
| 19 | 43.9, CH2 | α 1.93, m; β 1.03, m | 20 | 13.8, CH3 | 1.20–1.29, m |
| 20 | 25.6, CH2 | α 1.38, m; β 1.52, m | 21 | 169.5, C | |
| 21 | 43.2, CH | 2.13, m | 22 | 22.9, CH3 | 2.06, s |
| 22 | 74.4, C | ||||
| 22-OH | 3.59, s | ||||
| 23 | 62.8, CH2 | α 3.80, overlap; β 3.25, dd, (10.9, 7.7) | |||
| 23-OH | 4.06, dd, (7.7, 3.1) | ||||
| 24 | 22.8, CH3 | 1.05, s | |||
| 25 | 15.8, CH3 | 0.76, s | |||
| 26 | 16.3, CH3 | 0.88, s | |||
| 27 | 17.6, CH3 | 0.86, s | |||
| 28 | 16.0, CH3 | 0.81, s | |||
| 29 | 23.1, CH3 | 0.95, s | |||
| 30 | 69.2, CH2 | 3.17, m | |||
| 30-OH | 4.33, t, (5.7) | ||||
| Strain | Compound | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 4 | 6 | 7 | 10 | 11 | Positive Control | |
| Pseudomonas aeruginosab | - d | - | - | - | - | 8 | 4 |
| Vibrio alginolyticusb | 32 | - | - | - | 16 | 8 | 1 |
| Edwardsiella tardab | 64 | - | - | - | - | 8 | 1 |
| Fusarium oxysporumc | - | - | 64 | - | - | - | 2 |
| Fusarium graminearum Schw. c | 4 | 64 | - | 16 | 32 | - | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, L.-H.; Li, X.-M.; Chi, L.-P.; Li, X.; Wang, B.-G. Six New Antimicrobial Metabolites from the Deep-Sea Sediment-Derived Fungus Aspergillus fumigatus SD-406. Mar. Drugs 2022, 20, 4. https://doi.org/10.3390/md20010004
Yan L-H, Li X-M, Chi L-P, Li X, Wang B-G. Six New Antimicrobial Metabolites from the Deep-Sea Sediment-Derived Fungus Aspergillus fumigatus SD-406. Marine Drugs. 2022; 20(1):4. https://doi.org/10.3390/md20010004
Chicago/Turabian StyleYan, Li-Hong, Xiao-Ming Li, Lu-Ping Chi, Xin Li, and Bin-Gui Wang. 2022. "Six New Antimicrobial Metabolites from the Deep-Sea Sediment-Derived Fungus Aspergillus fumigatus SD-406" Marine Drugs 20, no. 1: 4. https://doi.org/10.3390/md20010004
APA StyleYan, L. -H., Li, X. -M., Chi, L. -P., Li, X., & Wang, B. -G. (2022). Six New Antimicrobial Metabolites from the Deep-Sea Sediment-Derived Fungus Aspergillus fumigatus SD-406. Marine Drugs, 20(1), 4. https://doi.org/10.3390/md20010004