Strains, plasmids and proteins: Mtb ClpC1-WT and ClpC1-F444S were overproduced from pET24a expression vector in E. coli BL21 cells at 30 °C. The proteins were purified using Ni-IDA (Macherey-Nagel) using 50 mM Na-phosphate pH 8.0, 300 mM NaCl, 5 mM β-mercaptoethanol as lysis and washing buffer and the same buffer supplemented with 250 mM imidazole as elution buffer. Elution fractions were pooled and further purified by size exclusion chromatography (Superdex S200, GE Healthcare) in buffer A (50 mM HEPES pH 7.5, 150 mM KCl, 20 mM MgCl2, 2 mM DTT, 5% (v/v) glycerol). Sa ClpP was purified from E. coli MC4100 ΔclpB cells after overproduction from pDS56 expression vector. Sa ClpP was purified using the same protocol as Mtb ClpC1. Protein concentrations refer to monomers and were determined with the Bio-Rad Bradford assay using BSA as standard.
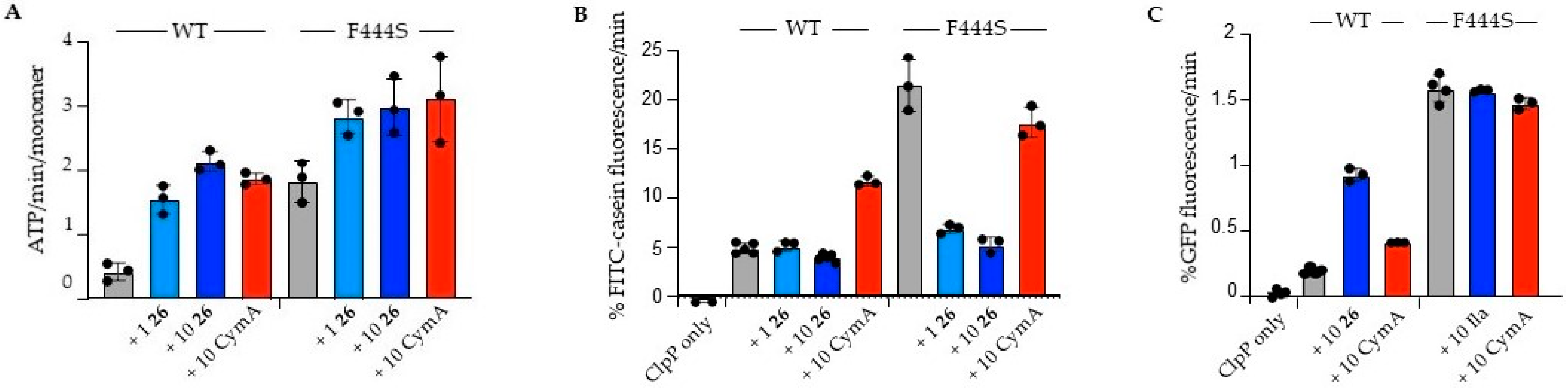
ATPase Assay: The ATPase activity of ClpC1 was determined using coupled reactions of pyruvate kinase (PK) and lactate dehydrogenase (LDH) in the presence of phosphoenolpyruvate (PEP) and NADH in buffer A. The reactions included 1 M ClpC1, 1.5 µM Sa ClpP and 1–10 µM 26 or cyclomarin A as indicated. Reactions were started with the injection of 2 mM ATP and changes in NADH absorbance was followed at 340 nm in a BMG Biotech CLARIOstar plate reader. ATPase rates were calculated from the linear decrease in A340 in at least three independent experiments and standard deviations were calculated. All reactions included 1% (v/v) DMSO to equal buffer conditions in presence of 26 or cyclomarin A. The presence of DMSO did not affect ClpC1 activities.
Degradation Assays: All degradation assays were performed in buffer A in presence of an ATP regenerating system (20 ng/μL pyruvate kinase, 3 mM PEP, 2 mM ATP). All reactions included 1% (v/v) DMSO to equal buffer conditions in presence of 26 or cyclomarin A. 0.1 μM FITC-casein was incubated with 1 µM ClpC1, 1.5 µM Sa ClpP and 1–10 µM 26 or cyclomarin A as indicated. The increase in FITC-casein fluorescence upon its degradation was monitored in BMG Biotech CLARIOstar plate reader by using 483 and 520/530 nm as excitation and emission wavelengths, respectively. For data processing the initial fluorescence intensities were set to 100. FITC-casein degradation rates were determined from the initial slopes of the fluorescence signal increase in at least three independent experiments and standard deviations were calculated. Degradation of 0.5 μM GFP-SsrA was performed in presence of 3 μM ClpC1, 4.5 μM Sa ClpP and 20 μM 26 or cyclomarin A as indicated in a Perkin Elmer LS55 Spectrofluorometer. Degradation was initiated by addition of an ATP regenerating system. GFP fluorescence was monitored by using 400 and 510 nm as excitation and emission wavelengths, respectively. Initial GFP fluorescence was set to 100. Degradation rates were determined from the initial slopes of fluorescence signal decrease in at least three independent experiments and standard deviations were calculated.
Anisotropy measurements: Binding of ClpC1 to FITC-casein (100 nM) was monitored by fluorescence anisotropy measurements using a BMG Biotech CLARIOstar plate reader. ClpC1 was incubated in buffer A for 5 min at 30 °C in presence of 2 mM ATPγS and 10–20 μM 26 or cyclomarin A as indicated. All reactions included 1% (v/v) DMSO to equal buffer conditions in presence of 26 or cyclomarin A. Polarization of FITC-casein was determined in black 384 well plates (excitation: 482 nm; emission: 530 nm, Target mP: 35). A sample containing FITC-casein only served as reference. Kd values were determined using nonlinear regression curve fitting (Prism 9.3.1, Graphpad Software, San Diego, CA, USA).
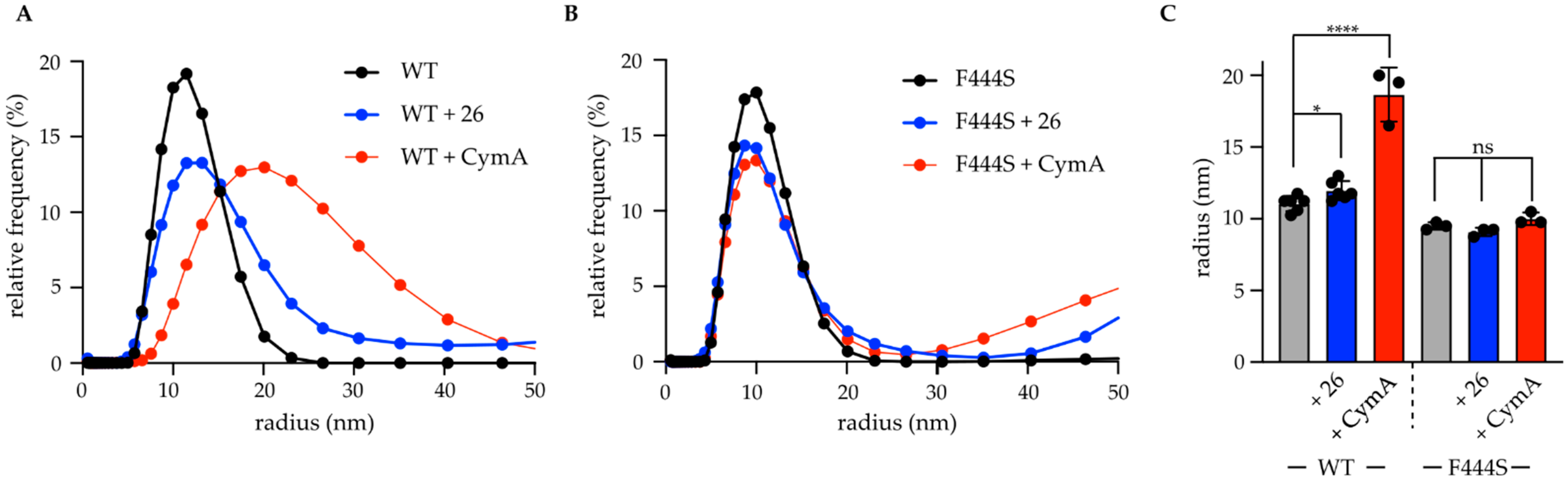
Dynamic light scattering measurements: 3 μM ClpC1-WT/F444S were incubated in buffer A with 2 mM ATP and 10 μM 26 or cyclomarin A as indicated. DLS measurements were performed in a Prometheus Pana system (Nanotemper) and particle sizes were determined at 25 °C. Statistical analysis was conducted using Prism 9.3.1.
Cytotoxicity evaluation: HepG2 cells (hepatocellular carcinoma cell line) were cultured under conditions recommended by the depositor. Briefly, cells were seeded at 6 × 103 cells per well of 96-well plates in 180 μL complete medium (DMEM, Sigma-Aldrich, St. Louis, MO, USA). Each compound was tested in a serial dilution as well as the internal solvent control. After 5 d incubation, 20 μL of 5 mg/mL MTT (thiazolyl blue tetrazolium bromide) in PBS was added per well and cells were further incubated for 2 h at 37 °C. The medium was then discarded and cells were washed with 100 μl PBS before adding 100 μL 2-propanol/10 N HCl (250:1) in order to dissolve formazan granules. The absorbance at 570 nm was measured using a microplate reader (Tecan Infinite M200Pro; Tecan, Männedorf, Switzerland), and cell viability was expressed as percentage relative to the respective methanol control. IC50 values were determined by sigmoidal curve fitting.
4.1. Synthesis of the Amino Acid Building Blocks
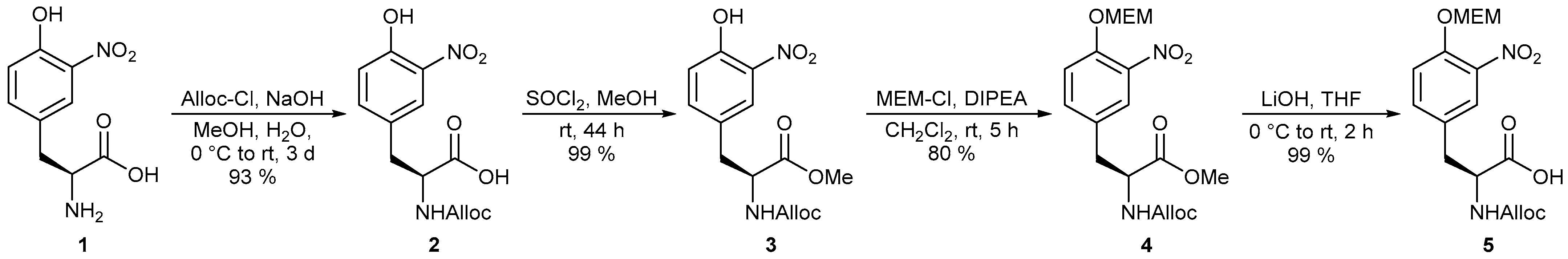
4.1.1. Synthesis of (S)-2-{[(allyloxy)carbonyl]amino}-3-(4-hydroxy-3-nitrophenyl)-propa noic Acid (2)
Alloc-Cl (ρ = 1.134 g/mL, 0.26 mL, 2.4 mmol) was slowly added to a solution of 3-nitrotyrosine (500 mg, 2.21 mmol) (1) in 1.2 mL (4.8 mmol) 4 M aq. NaOH solution at 0 °C. After the reaction mixture had been stirred for 10 min at this temperature, the ice bath was removed and 1.2 mL (4.8 mmol) 4 M aq. NaOH solution and 0.2 mL water were added. Alloc-Cl (94 μL, 0.88 mmol) was added again to the reaction mixture after 1 h at room temperature and 2.4 mL MeOH after 2 h. After three days the solution was diluted with 10 mL sat. aq. NaHCO3 solution. The aqueous phase was washed with diethyl ether, cooled to 0 °C and acidified to pH 3 with conc. HCl solution. After the aqueous phase had been extracted three times with EtOAc, the combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. For further purification, the crude product was lyophilized to yield 639 mg (2.06 mmol, 93%) of Alloc-protected tyrosine (2) as a yellow solid. Rf = 0.41 (DCM/MeOH 9:1); [α]D20 = +84.5 (c = 1.0, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 3.08 (1 H, dd, J = 14.2, 6.5 Hz, H-3), 3.25 (1 H, dd, J = 14.1, 5.3 Hz, H-3’), 4.52–4.60 (2 H, m, H-11), 4.67 (1 H, dt, J = 6.7, 6.4 Hz, H-2), 5.20–5.30 (1 H, m, NH), 5.23 (1 H, d, J = 10.4 Hz, H-13), 5.29 (1 H, d, J = 18.5 Hz, H-13’), 5.89 (1 H, ddt, J = 17.0, 10.6, 5.5 Hz, H-12), 7.11 (1 H, d, J = 8.6 Hz, H-8), 7.43 (1 H, dd, J = 8.7, 1.7 Hz, H-9), 7.93 (1 H, bs, H-5), 10.49 (1 H, s, OH); 13C-NMR (CDCl3, 100 MHz) δ 36.8 (t, C-3), 54.3 (d, C-2), 66.2 (t, C-11), 118.3 (t, C-13), 120.3 (d, C-8), 125.4 (d, C-5), 128.1 (s, C-4), 132.2 (d, C-12), 133.4 (s, C-6), 138.7 (d, C-9), 154.3 (s, C-7, C-10), 174.8 (s, C-1); HRMS (CI) m/z calculated for C13H15N2O7 [M + H]+ 311.0874, found 311.0881.
4.1.2. Synthesis of Methyl (S)-2-{[(allyloxy)carbonyl]amino}-3-(4-hydroxy-3-nitrophenyl)-propanoate (3)
To a solution of tyrosine derivative 2 (200 mg, 650 μmol) in 1.3 mL MeOH was slowly added thionyl chloride (57 μL, 0.77 mmol) at room temperature. Subsequently, stirring was continued overnight at this temperature. After 44 h (TLC), the solvent was removed under reduced pressure and the crude product was purified by flash chromatography (silica gel, DCM/MeOH 99:1). The desired methyl ester (3) (209 mg, 640 μmol, quant., mp: 70 °C) could be isolated as a yellow solid. Rf = 0.37 (DCM/MeOH 99:1); [α]D20 = +66.5 (c = 1.0, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 3.04 (1 H, dd, J = 14.1, 6.0 Hz, H-3), 3.18 (1 H, dd, J = 14.1, 5.4 Hz, H-3), 3.76 (3 H, s, H-14), 4.50–4.60 (2 H, m, H-11), 4.63 (1 H, dt, J = 7.0, 6.6 Hz, H-2), 5.22 (1 H, dd, J = 10.4, 1.2 Hz, H-13), 5.24–5.35 (1 H, m, NH), 5.29 (1 H, d, J = 16.0 Hz, H-13), 5.89 (1 H, ddt, J = 17.1, 10.6, 5.4 Hz, H-12), 7.09 (1 H, d, J = 8.6 Hz, H-8), 7.37 (1 H, dd, J = 8.6, 2.2 Hz, H-9), 7.87 (1 H, d, J = 2.0 Hz, H-5), 10.49 (1 H, s, OH); 13C-NMR (CDCl3, 100 MHz) δ 37.2 (t, C-3), 52.6 (q, C-14), 54.6 (d, C-2), 66.0 (t, C-11), 118.1 (t, C-13), 120.2 (d, C-8), 125.2 (d, C-5), 128.4 (s, C-4), 132.4 (d, C-12), 133.3 (s, C-6), 138.6 (d, C-9), 154.2 (s, C-7), 155.4 (s, C-10), 171.4 (s, C-1); HRMS (CI) m/z calculated for C14H17N2O7 [M + H]+ 325.1030, found 325.1041.
4.1.3. Synthesis of Methyl (S)-2-{[(allyloxy)carbonyl]amino}-3-{4-[(2-methoxyethoxy)met hoxy]-3-nitrophenyl}propanoate (4)
To a solution of tyrosine (3) (300 mg, 930 μmol) in 2.4 mL DCM was added successively DIPEA (339 µL, 1.94 mmol) and MEM-Cl (ρ = 1.09 g/mL, 115 µL, 1.02 mmol) at room temperature. After complete conversion (5 h, TLC), the reaction mixture was diluted with DCM. The organic phase was washed with water and brine, dried over Na2SO4 and concentrated under reduced pressure. Final purification by flash chromatography (silica gel, PE/EtOAc 1:1) afforded the MEM-protected tyrosine (4) (306 mg, 740 μmol, 80%) as a yellow oil. Rf = 0.25 (PE/EtOAc 1:1); [α]D20 = +35.3 (c = 1.0, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 3.06 (1 H, dd, J = 14.0, 6.0 Hz, H-3), 3.17 (1 H, dd, J = 14.4, 5.2 Hz, H-3’), 3.36 (3 H, s, H-13), 3.53–3.59 (2 H, m, H-12), 3.76 (3 H, s, H-18), 3.84–3.90 (2 H, m, H-11), 4.57 (2 H, d, J = 5.5 Hz, H-15), 4.63 (1 H, dt, J = 7.3, 6.6 Hz, H-2), 5.22 (1 H, dd, J = 10.4, 1.2 Hz, H-17), 5.24–5.31 (1 H, m, NH), 5.29 (1 H, d, J = 17.0 Hz, H-17’), 5.36 (2 H, s, H-10), 5.90 (1 H, ddt, J = 17.0, 10.6, 5.5 Hz, H-16), 7.25–7.33 (2 H, m, H-8, H-9), 7.57 (1 H, bs, H-5); 13C-NMR (CDCl3, 100 MHz) δ 37.0 (t, C-3), 52.6 (q, C-18), 54.6 (d, C-2), 59.0 (q, C-13), 66.0 (t, C-15), 68.4 (t, C-11), 71.4 (t, C-12), 94.3 (t, C-10), 117.5 (t, C-17), 118.1 (d, C-8), 125.9 (d, C-5), 129.7 (s, C-4), 132.4 (d, C-16), 134.8 (d, C-9), 140.4 (s, C-6), 149.5 (s, C-7), 155.4 (s, C-14), 171.4 (s, C-1); HRMS (CI) m/z calculated for C18H25N2O9 [M+H]+ 413.1555, found 413.1571.
4.1.4. Synthesis of (S)-2-{[(allyloxy)carbonyl]amino}-3-{4-[(2-methoxyethoxy)methoxy]-3-nitro-phenyl}propanoic Acid (5)
To a solution of methyl ester (4) (4.82 g, 11.7 mmol) in 146 mL THF was added dropwise a 1 M aq. LiOH solution (14.0 mL, 14.0 mmol) at 0 °C. During the reaction, the reaction mixture was slowly warmed to room temperature (2 h). The solvent was removed in vacuo. The residue was dissolved in water and the aqueous phase was acidified with 1 M aq. KHSO4 solution to pH 3–4. Subsequently, the aqueous phase was extracted three times with DCM. After the combined organic phases had been dried over Na2SO4, the solvent was removed under reduced pressure to obtain 5 (4.66 g, 11.7 mmol, quant.) as a yellow resin. Rf = 0.28 (DCM/MeOH 9:1); [α]D20 = +59.6 (c = 1.0, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ 3.09 (1 H, dd, J = 14.2, 6.0 Hz, H-3), 3.23 (1 H, dd, J = 14.3, 5.1 Hz, H-3’), 3.36 (3 H, s, H-13), 3.55–3.61 (2 H, m, H-12), 3.84–3.91 (2 H, m, H-11), 4.57 (2 H, d, J = 4.9 Hz, H-15), 4.66 (1 H, dt, J = 7.0, 6.4 Hz, H-2), 5.23 (1 H, dd, J = 10.4, 1.1 Hz, H-17), 5.25–5.39 (1 H, m, NH), 5.30 (1 H, d, J = 16.9 Hz, H-17’), 5.36 (2 H, s, H-10), 5.89 (1 H, ddt, J = 17.2, 10.6, 5.5 Hz, H-16), 6.15 (1 H, bs, COOH), 7.29 (1 H, d, J = 8.7 Hz, H-8), 7.34 (1 H, dd, J = 8.7, 2.1 Hz, H-9), 7.63 (1 H, bs, H-5); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 2.99–3.14 (1 H, m, H-3), 3.15–3.27 (1 H, m, H-3’), 4.48–4.60 (2 H, m, H-15), 7.64–7.70 (1 H, m, H-5); 13C-NMR (CDCl3, 100 MHz) δ 36.6 (t, C-3), 54.3 (d, C-2), 58.9 (q, C-13), 66.1 (t, C-15), 68.3 (t, C-11), 71.4 (t, C-12), 94.3 (t, C-10), 117.6 (t, C-17), 118.2 (d, C-8), 126.0 (d, C-5), 129.7 (s, C-4), 132.3 (d, C-16), 134.9 (d, C-9), 140.4 (s, C-6), 149.5 (s, C-7), 155.7 (s, C-14), 174.1 (s, C-1); HRMS (CI) m/z calculated for C17H23N2O9 [M + H]+ 399.1398, found 399.1396.
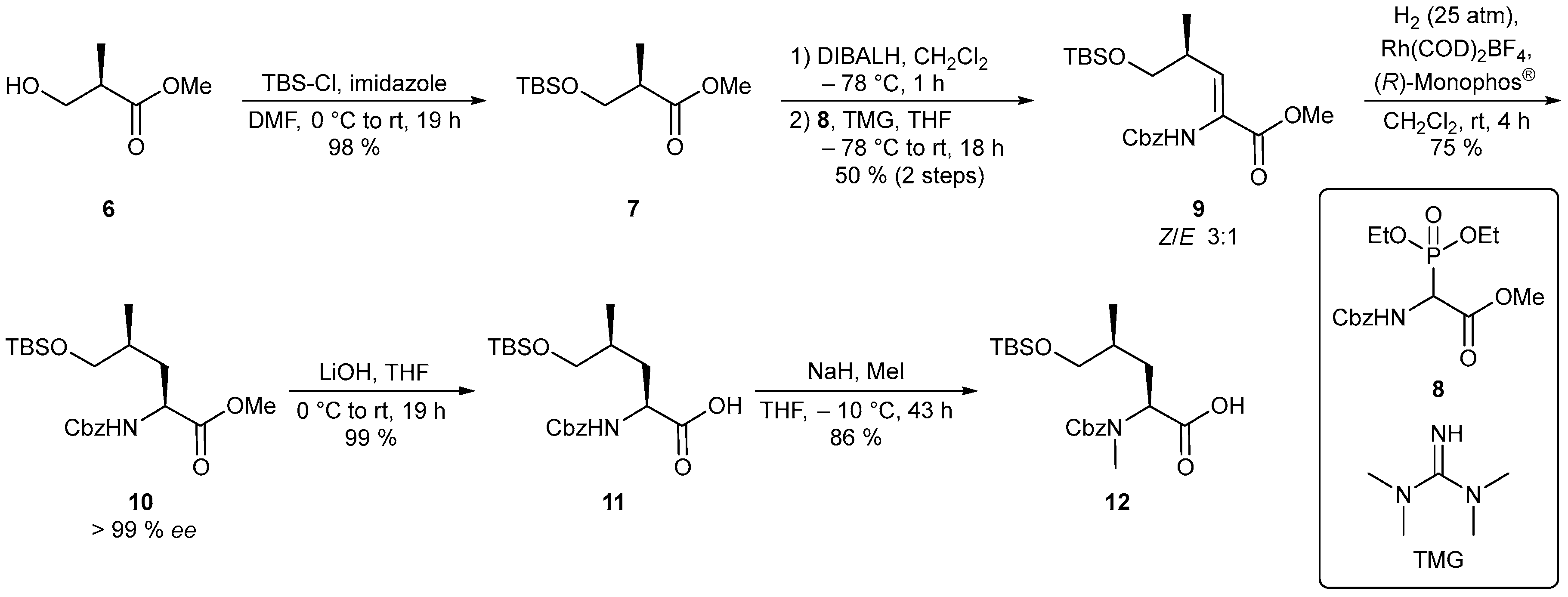
4.1.5. Synthesis of Methyl (R)-3-[(tert-butyldimethylsilyl)oxy]-2-methylpropanoate (7) [82]
To a solution of (R)-Roche ester (6) (5.00 g, 42.3 mmol) in anhydrous DMF was added imidazole (3.31 g, 48.7 mmol) at room temperature. Subsequent addition of TBS-Cl (7.02 g, 46.6 mmol) was carried out at 0 °C. The reaction mixture was slowly warmed to room temperature overnight. After 19 h (TLC), the solution was dissolved in diethyl ether. The organic phase was washed successively with water and brine and dried over Na2SO4. After the solvent was removed under reduced pressure, the crude product was purified by flash chromatography (silica gel, PE/Et2O 95:5). The TBS-protected Roche ester (7) (9.63 g, 41.4 mmol, 98%) was obtained as a colorless liquid. Rf = 0.29 (PE/Et2O 95:5); [α]D20 = −21.5 (c = 1.0, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 0.03 (3 H, s, H-4), 0.04 (3 H, s, H-4’), 0.87 (9 H, s, H-6), 1.14 (3 H, d, J = 7.0 Hz, H-7), 2.65 (1 H, ddq, J = 6.8, 6.8, 6.8 Hz, H-2), 3.65 (1 H, dd, J = 9.7, 6.0 Hz, H-3), 3.67 (3 H, s, H-8), 3.77 (1 H, dd, J = 9.7, 6.9 Hz, H-3’); 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-4, C-4’), 13.5 (q, C-7), 18.2 (s, C-5), 25.8 (q, C-6), 42.5 (d, C-2), 51.5 (q, C-8), 65.2 (t, C-3), 175.5 (s, C-1).
4.1.6. Synthesis of Methyl (S,Z)-2-{[(benzyloxy)carbonyl]amino}-5-[(tert-butyldimethylsilyl)oxy]-4-methylpent-2-enoate (9)
To a solution of TBS-protected Roche ester (7) (9.80 g, 42.2 mmol) in 422 mL anhydrous DCM, a DIBALH solution (ρ = 0.701 g/mL, 46.4 mL, 46.4 mmol, 1 M in hexane) was slowly added at –78 °C within 60 min. Stirring was continued at this temperature for 40 min. After complete conversion (1 h, TLC), 21 mL of MeOH and 120 mL of a 10% Na/K tartrate solution were added to the reaction mixture at –78 °C. The cooling bath was removed and the reaction mixture was allowed to warm to room temperature. After the DCM layer became clear, the phases were separated and the aqueous phase was extracted three times with DCM. The combined organic phases were washed with 1 M aq. KHSO4 solution as well as with sat. aq. NaHCO3 solution and dried over Na2SO4. The solvent was removed in vacuo and the crude aldehyde was dissolved in 320 mL dry THF. The Cbz-protected phosphonate (8) (16.7 g, 46.4 mmol) was dissolved in 100 mL anhydrous THF and cooled to –78 °C. Tetramethylguanidine (ρ = 0.918 g/mL, 5.6 mL, 44 mmol) was added. After the solution was stirred for 20 min at this temperature, the aldehyde solution was slowly added. Subsequently the reaction mixture was warmed to room temperature overnight. The solution was diluted with diethyl ether and water. The aqueous phase was extracted three times with diethyl ether and the combined organic phases were dried over Na2SO4. The solvent was removed in vacuo. After purification by flash chromatography (silica gel, PE/EtOAc 8:2), 8.57 g (21.0 mmol, 50 %, 75 % Z according to 1H-NMR) of dehydroamino acid (9) was obtained as a colorless liquid. Rf = 0.28 (PE/EtOAc 8:2); [α]D20 = +63.0 (c = 1.0, CHCl3); Mixture of configuration isomers: (Z)-9: 1H-NMR (CDCl3, 400 MHz) δ 0.03 (3 H, s, H-6), 0.05 (3 H, s, H-6’), 0.88 (9 H, s, H-8), 1.01 (3 H, d, J = 6.7 Hz, H-9), 2.74–2.88 (1 H, m, H-4), 3.42 (1 H, dd, J = 9.2, 9.2 Hz, H-5), 3.64 (1 H, dd, J = 9.5, 4.7 Hz, H-5’), 3.69–3.81 (3 H, m, H-10), 5.12 (1 H, d, J = 12.4, H-12), 5.17 (1 H, d, J = 12.4 Hz, H-12), 6.20 (1 H, d, J = 9.4 Hz, H-3), 7.01 (1 H, bs, NH), 7.28–7.40 (5 H, m, H-14, H-15, H-16); (E)-9 (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 0.07 (3 H, s, H-6), 0.10 (3 H, s, H-6’), 0.84 (3 H, d, J = 7.0 Hz, H-9), 0.90 (9 H, s, H-8), 1.88–2.00 (1 H, m, H-4), 3.55 (1 H, dd, J = 9.8, 8.0 Hz, H-5); (Z)-9: 13C-NMR (CDCl3, 100 Hz) δ -5.6 (q, C-6), -5.5 (q, C-6’), 15.8 (q, C-9), 18.3 (s, C-7), 25.8 (q, C-8), 35.2 (d, C-4), 52.2 (q, C-10), 67.1 (t, C-12), 68.5 (t, C-5), 128.0 (d, C-14), 128.1 (d, C-16), 128.4 (d, C-15), 136.1 (s, C-13), 137.0 (s, C-2; d, C-3), 154.2 (s, C-11), 165.0 (s, C-1); (E)-9 (selected signals): 13C-NMR (CDCl3, 100 Hz) δ -3.6 (q, C-6, C-6‘), 13.1 (q, C-9), 25.6 (q, C-8), 37.0 (d, C-4), 52.5 (q, C-10), 68.3 (t, C-12), 68.8 (t, C-5); HRMS (CI) m/z calculated for C21H34NO5Si [M + H]+ 408.2201, found 408.2204.
4.1.7. Synthesis of Methyl (2S,4S)-2-{[(benzyloxy)carbonyl]amino}-5-[(tert-butyldimethylsilyl)oxy]-4-methylpentanoate (10)
Rh(COD)2BF4 (171 mg, 420 μmol) and (R)-Monophos® (302 mg, 840 μmol) were dissolved under argon-atmosphere in 76 mL dry DCM. Dehydroamino acid (9) (8.58 g, 21.0 mmol) dissolved in 19 mL dry DCM was added. The reaction mixture was stirred in an autoclave under 25 bar H2-atmosphere for 4 h at room temperature. The solvent was removed under reduced pressure and the residue was purified by flash chromatography (silica gel, PE/EtOAc 8:2) to afford 10 (6.55 g, 16.0 mmol, 76%, >99% ee) as a colorless oil. Rf = 0.36 (PE/EtOAc 8:2); [α]D20 = –12.5 (c = 1.0, CHCl3); To determine the enantiomeric excess of amino acid 10, the Cbz protecting group was replaced by an acetate group to allow gas chromatographic measurement. For this purpose, hydroxyleucine (10) (30 mg, 73 μmol) was dissolved in 0.7 mL dry THF and 10 wt-% Pd-C (3 mg, 10% on activated charcoal) was added to the reaction mixture. Stirring was carried out at 1 bar H2 atmosphere for 26 h at room temperature. The mixture was filtered through Celite and the solvent was removed in vacuo. Subsequently, the residue was dissolved in 1.5 mL pyridine followed by the addition of acetic anhydride (ρ = 1.08 g/mL, 10.4 μL, 110 μmol) at room temperature. After 21 h (TLC), the reaction mixture was diluted with EtOAc and the organic phase was washed twice with 1 M aq. HCl solution, dried over Na2SO4 and concentrated under reduced pressure. The acetate-protected hydroxyleucine (14 mg, 44 μmol, 60%) was obtained as a yellow resin, Rf = 0.31 (PE/EtOAc 8:2). GC-FID: Column: Agilent CP-Chirasil-Dex CB; Carrier gas: N2; T0 [1 min] = 110 °C, 2.0 °C/min to 180 °C, injector: 250 °C, detector: 275 °C; N-acetyl-(2R,4S): tr = 32.42 min, N-acetyl-(2S,4S): tr = 32.76 min; 1H-NMR (CDCl3, 500 MHz) δ 0.03 (3 H, s, H-6), 0.03 (3 H, s, H-6’), 0.88 (9 H, s, H-8), 0.94 (3 H, d, J = 6.6 Hz, H-9), 1.53–1.62 (1 H, m, H-3), 1.70–1.80 (2 H, m, H-3’, H-4), 3.36 (1 H, dd, J = 9.8, 6.3 Hz, H-5), 3.48 (1 H, dd, J = 9.8, 5.0 Hz, H-5’), 3.73 (3 H, s, H-10), 4.34–4.41 (1 H, m, H-2), 5.10 (2 H, s, H-12), 5.32 (1 H, d, J = 8.2 Hz, NH), 7.29–7.38 (5 H, m, H-14, H-15, H-16); 13C-NMR (CDCl3, 125 MHz) δ -5.5 (q, C-6), -5.5 (q, C-6’), 16.4 (q, C-9), 18.3 (s, C-7), 25.9 (q, C-8), 32.7 (d, C-4), 36.1 (t, C-3), 52.3 (q, C-10), 52.4 (d, C-2), 66.9 (t, C-12), 67.9 (t, C-5), 128.1 (d, C-14), 128.1 (d, C-16), 128.5 (d, C-15), 136.3 (s, C-13), 156.1 (s, C-11), 173.6 (s, C-1); HRMS (CI) m/z calculated for C21H36NO5Si [M + H]+ 410.2357, found 410.2374.
4.1.8. Synthesis of (2S,4S)-2-{[(benzyloxy)carbonyl]amino}-5-[(tert-butyldimethyl-silyl)oxy]-4-methylpentanoic Acid (11)
To a solution of methyl ester 10 (2.35 g, 5.74 mmol) in 57 mL THF was added a 1 M aq. LiOH solution (6.9 mL, 6.9 mmol) at 0 °C. With stirring, the reaction mixture was slowly warmed to room temperature (19 h). The solvent was removed under reduced pressure. The residue dissolved in water and acidified with 1 M aq. KHSO4 solution to pH 3. The aqueous phase was extracted three times with DCM. After the combined organic phases were dried over Na2SO4, the solvent was removed in vacuo again. Without further purification, 2.27 g (5.74 mmol, quant.) of 11 was obtained as a colorless oil. Rf = 0.12 (PE/EtOAc 8:2); [α]D20 = +8.8 (c = 1.0, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ 0.01–0.12 (6 H, m, H-6, H-6‘), 0.89 (9 H, s, H-8), 0.94 (3 H, d, J = 6.4 Hz, H-9), 1.59–1.73 (1 H, m, H-3), 1.73–1.92 (2 H, m, H-3’, H-4), 3.43 (1 H, dd, J = 9.5, 7.2 Hz, H-5), 3.53–3.59 (1 H, m, H-5’), 4.39–4.49 (1 H, m, H-2), 5.11 (2 H, s, H-11), 5.49 (1 H, d, J = 7.7 Hz, NH), 6.93 (1 H, bs, COOH), 7.28–7.42 (5 H, m, H-13, H-14, H-15); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 4.35 (1 H, bs, H-2), 5.64 (1 H, bs, NH); 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-6), -5.5 (q, C-6’), 16.8 (q, C-9), 18.3 (s, C-7), 25.9 (q, C-8), 32.6 (d, C-4), 36.9 (t, C-3), 52.4 (d, C-2), 67.0 (t, C-11), 68.5 (t, C-5), 128.0 (d, C-13), 128.1 (d, C-15), 128.5 (d, C-14), 136.2 (s, C-12), 156.2 (s, C-10), 176.2 (s, C-1); HRMS (CI) m/z calculated for C20H34NO5Si [M + H]+ 396.2201, found 396.2210.
4.1.9. Synthesis of (2S,4S)-2-{[(benzyloxy)carbonyl](methyl)amino}-5-[(tert-butyldimethylsilyl)oxy]-4-methylpentanoic Acid (12)
Cbz-protected amino acid 11 (5.58 g, 14.1 mmol) was dissolved in 141 mL anhydrous THF. After cooling down to –10 °C, NaH (2.26 g, 56.5 mmol, 60% in mineral oil) was added to the reaction mixture in portions, followed by methyl iodide (ρ = 2.28 g/mL, 7.1 mL, 113 mmol). After 43 h (LCMS) at this temperature, the reaction mixture was diluted with EtOAc and poured onto water. The organic phase was extracted three times with water. The combined aqueous phases were acidified to pH 3 using 1 M aq. KHSO4 solution and extracted three times with DCM. Subsequently, the combined organic phases were washed with 5% Na2SO3 solution and brine and dried over Na2SO4. The solvent was removed under reduced pressure to obtain the desired N-methylated amino acid 12 (4.97 g, 12.1 mmol, 86%) as a yellow oil. [α]D20 = –4.2 (c = 1.0, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ 0.04 (6 H, s, H-6), 0.88 (9 H, s, H-8), 0.92 (3 H, d, J = 6.2 Hz, H-9), 1.50–1.60 (1 H, m, H-4), 1.58–1.68 (1 H, m, H-3) 2.01–2.13 (1 H, m, H-3’), 2.89 (3 H, s, H-10), 3.36 (1 H, dd, J = 9.7, 7.3 Hz, H-5), 3.50 (1 H, dd, J = 9.8, 4.9 Hz, H-5’), 5.01 (1 H, dd, J = 11.9, 4.0 Hz, H-2), 5.08–5.23 (2 H, m, H-12), 7.27–7.40 (5 H, m, H-14, H-15, H-16); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 0.02 (6 H, s, H-6), 0.82 (3 H, d, J = 5.8 Hz, H-9), 0.88 (9 H, s, H-8), 2.90 (3 H, s, H-10), 3.32 (1 H, d, J = 9.7, 6.6 Hz, H-5), 3.47 (1 H, dd, J = 11.4, 4.4 Hz, H-5’), 4.85 (1 H, dd, J = 11.5, 3.4 Hz, H-2); Major rotamer: 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-6), 15.6 (q, C-9), 18.3 (s, C-7), 25.9 (q, C-8), 30.1 (q, C-10), 31.9 (t, C-3), 32.6 (d, C-4), 56.1 (d, C-2), 67.6 (t, C-12), 68.1 (t, C-5), 127.7 (d, C-14), 128.0 (d, C-16), 128.5 (d, C-15), 136.5 (s, C-13), 157.3 (s, C-11), 177.0 (s, C-1); Minor rotamer (selected signals): 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-6), 15.4 (q, C-9), 18.2 (s, C-7), 30.3 (q, C-10), 32.3 (t, C-3), 32.4 (d, C-4), 55.9 (d, C-2), 67.7 (t, C-12), 68.1 (t, C-5), 127.9 (d, C-14), 128.1 (d, C-16), 128.5 (d, C-15), 136.5 (s, C-13), 156.4 (s, C-11), 177.1 (s, C-1); HRMS (CI) m/z calculated for C21H36NO5Si [M + H]+ 410.2357, found 410.2367.
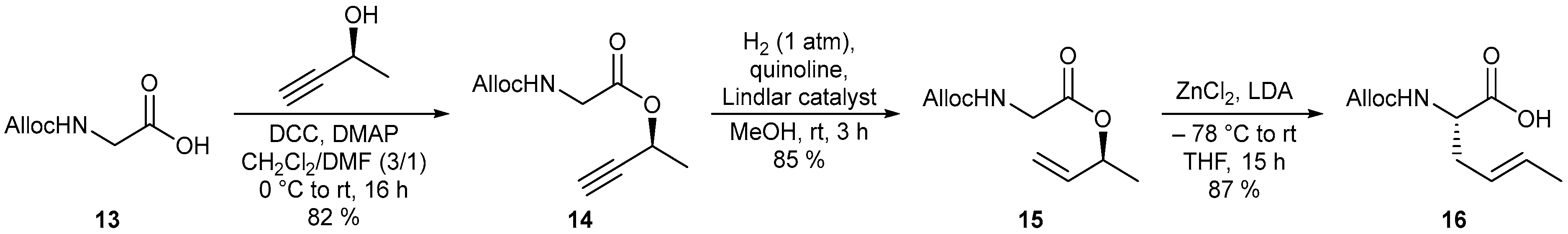
4.1.10. Synthesis of (S)-But-3-yn-2-yl [(allyloxy)carbonyl]glycinate (14)
To a solution of propargyl alcohol (1.61 g, 23.0 mmol) in 23 mL anhydrous DCM was added Alloc glycine (13) (4.03 g, 25.3 mmol) and DMAP (141 mg, 1.15 mmol) at room temperature. The reaction mixture was cooled to 0 °C, followed by the addition of DCC (5.23 g, 25.3 mmol) and 8 mL dry DMF. Overnight it was warmed to room temperature (16 h). The reaction mixture was filtered through Celite and the solvent was subsequently removed under reduced pressure. The residue was diluted with EtOAc and the organic phase was washed three times with water and once with brine. After drying over Na2SO4, the crude product was concentrated under reduced pressure. Flash chromatographic purification (silica gel, PE/EtOAc 8:2) afforded the desired propargyl ester 14 (3.99 g, 18.9 mmol, 82%) as a colorless oil. Rf = 0.15 (PE/EtOAc 8:2); [α]D20 = –95.0 (c = 1.0, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 1.53 (3 H, d, J = 6.7 Hz, H-7), 2.48 (1 H, d, J = 2.1 Hz, H-10), 3.97 (1 H, dd, J = 18.1, 5.5 Hz, H-2), 4.03 (1 H, dd, J = 18.3, 5.7 Hz, H-2’), 4.59 (2 H, d, J = 5.6 Hz, H-4), 5.19–5.27 (1 H, m, NH), 5.22 (1 H, dd, J = 10.4, 1.2 Hz, H-6), 5.31 (1 H, dd, J = 17.2, 1.5 Hz, H-6’), 5.49 (1 H, dq, J = 6.7, 6.7 Hz, H-8), 5.91 (1 H, ddt, J = 17.2, 10.4, 5.5 Hz, H-5); 13C-NMR (CDCl3, 100 MHz) δ 21.1 (q, C-7), 42.7 (t, C-2), 61.3 (d, C-8), 66.0 (t, C-4), 73.5 (d, C-10), 81.4 (s, C-9), 117.9 (t, C-6), 132.5 (d, C-5), 156.1 (s, C-3), 169.0 (s, C-1); HRMS (CI) m/z calculated for C10H14NO4 [M + H]+ 212.0917, found 212.0924.
4.1.11. Synthesis of (S)-But-3-en-2-yl [(allyloxy)carbonyl]glycinate (15)
To a solution of propargyl ester 14 (3.94 g, 18.6 mmol) in 48 mL MeOH were successively added quinoline (ρ = 1.09 g/mL, 0.66 mL, 5.6 mmol) and Lindlar catalyst (272 mg) at room temperature. Subsequently, stirring was performed under 1 atm H2 atmosphere for 3 h. The reaction mixture was filtered through Celite, the solvent was removed under reduced pressure and the crude product was purified by flash chromatography (silica gel, PE/EtOAc 9:1 → 85:15). Allyl ester 15 (3.37 g, 15.8 mmol, 85%) was isolated as a colorless oil. Rf = 0.22 (PE/EtOAc 8:2); [α]D20 = –25.3 (c = 1.0, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ 1.34 (3 H, d, J = 6.5 Hz, H-7), 3.93–4.00 (2 H, m, H-2), 4.59 (2 H, d, J = 5.5 Hz, H-4), 5.17 (1 H, d, J = 10.5 Hz, H-10), 5.19–5.25 (1 H, m, NH), 5.22 (1 H, dd, J = 10.5, 1.2 Hz, H-6), 5.26 (1 H, d, J = 17.2 Hz, H-10’), 5.31 (1 H, dd, J = 17.2, 1.2 Hz, H-6’), 5.41 (1 H, dq, J = 6.4, 6.4 Hz, H-8), 5.83 (1 H, ddd, J = 17.3, 10.7, 6.3 Hz, H-9), 5.92 (1 H, ddt, J = 17.2, 10.4, 5.3 Hz, H-5); 13C-NMR (CDCl3, 100 MHz) δ 19.9 (q, C-7), 42.9 (t, C-2), 65.9 (t, C-4), 72.5 (d, C-8), 116.5 (t, C-10), 117.8 (t, C-6), 132.6 (d, C-5), 137.0 (d, C-9), 156.1 (s, C-3), 169.3 (s, C-1); HRMS (CI) m/z calculated for C10H16NO4 [M + H]+ 214.1074, found 214.1083.
4.1.12. Synthesis of (S,E)-2-{[(allyloxy)carbonyl]amino}hex-4-enoic Acid (16)
Preparation of LDA solution: Under N2 atmosphere, n-BuLi (ρ = 0.680 g/mL, 22.6 mL, 36.2 mmol, 1.6 M in hexane) was added to a solution of diisopropylamine (ρ = 0.717 g/mL, 5.28 mL, 37.4 mmol) in 40 mL dry THF at –15 °C. Afterwards, the reaction mixture was stirred at this temperature for 15 min. Preparation of substrate solution: ZnCl2 (1.97 g, 14.5 mmol) was first dried in high vacuum at approx. 250 °C and then dissolved in 40 mL anhydrous THF. At room temperature the allyl ester 15 (2.57 g, 12.1 mmol) was added, which was also dissolved in 40 mL dry THF. Reaction procedure: Both reaction solutions were cooled to –78 °C. Subsequently, the LDA solution was slowly added to the substrate solution via cannula. Under stirring, the reaction was allowed to warm up to room temperature overnight. After complete conversion (15 h, TLC), the reaction mixture was hydrolyzed by 120 mL of 1 M aq. KHSO4 solution and then diluted with EtOAc. The aqueous phase was extracted three times with EtOAc and the combined organic phases were washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (silica gel, DCM/MeOH 97:3 → 95:5) to afford crotyl-glycine 16 (2.23 g, 10.5 mmol, 87%) as a yellow resin. Rf = 0.36 (DCM/MeOH 95:5); [α]D20 = +14.3 (c = 1.0, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ 1.68 (3 H, d, J = 6.2 Hz, H-7), 2.42–2.60 (2 H, m, H-10), 4.40 (1 H, dt, J = 7.0, 5.7 Hz, H-2), 4.59 (2 H, d, J = 5.3 Hz, H-4), 5.18–5.27 (1 H, m, NH), 5.23 (1 H, dd, J = 10.4, 1.0 Hz, H-6), 5.27–5.39 (1 H, m, H-9), 5.31 (1 H, d, J = 16.8 Hz, H-6’), 5.60 (1 H, dq, J = 15.0, 6.5 Hz, H-8), 5.92 (1 H, ddt, J = 17.1, 10.5, 5.4 Hz, H-5), 5.97 (1 H, bs, COOH); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 4.29 (1 H, bs, H-2); 13C-NMR (CDCl3, 100 MHz) δ 18.0 (q, C-7), 35.1 (t, C-10), 53.3 (d, C-2), 66.0 (t, C-4), 118.0 (t, C-6), 124.1 (d, C-9), 130.7 (d, C-8), 132.5 (d, C-5), 155.9 (s, C-3), 176.3 (s, C-1); HRMS (CI) m/z calculated for C10H16NO4 [M + H]+ 214.1074, found 214.1081.
4.2. Synthesis of the Linear Heptapeptide
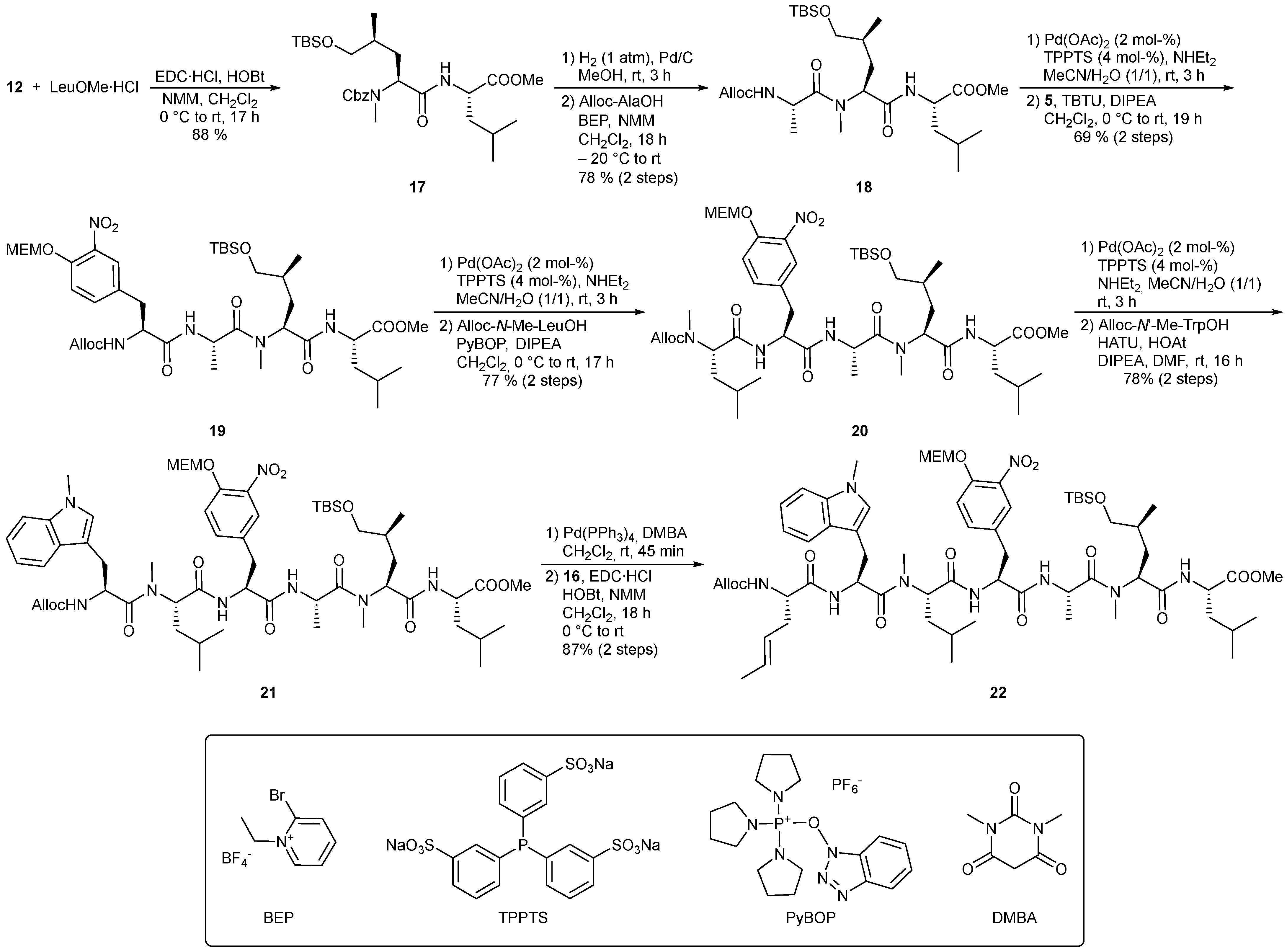
4.2.1. Synthesis of Methyl {[2S,4S]-2-({[benzyloxy]carbonyl}(methyl)amino)-5-[(tert-butyldimethyl-silyl)oxy]-4-methylpentanoyl}-L-leucinate (17)
To a solution of H-Leu-OMe·HCl (143 mg, 790 μmol) in 7.8 mL anhydrous DCM was added hydroxyleucine 12 (355 mg, 880 μmol) at room temperature. The reaction mixture was cooled to 0 °C, followed by successive addition of NMM (ρ = 0.91 g/mL, 295 mL, 2.68 mmol), HOBt (133 mg, 880 μmol) and EDC·HCl (166 mg, 880 μmol). Overnight, the solution was allowed to warm to room temperature (17 h). The reaction mixture was diluted with EtOAc. The organic phase was washed with 1 M aq. KHSO4 solution, sat. aq. NaHCO3 solution and brine, dried over Na2SO4 and concentrated under reduced pressure. After purification by flash chromatography (silica gel, cyclohexane → cyclohexane/EtOAc 8:2), 372 mg (690 μmol, 88%) of the dipeptide 17 was obtained as a colorless resin. Rf = 0.35 (PE/EtOAc 7:3); [α]D20 = –53.4 (c = 1.0, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ -0.01–0.06 (6 H, m, H-8), 0.73–0.98 (9 H, m, H-11, H-21), 0.88 (9 H, s, H-10), 1.44–1.53 (2 H, m, H-5, H-6), 1.49–1.58 (1 H, m, H-20), 1.49–1.66 (2 H, m, H-19), 1.92–2.10 (1 H, m, H-5’), 2.84 (3 H, s, H-12), 3.28–3.41 (1 H, m, H-7), 3.41–3.50 (1 H, m, H-7’), 3.71 (3 H, s, H-22), 4.51–4.59 (1 H, m, H-2), 4.79–4.88 (1 H, m, H-4), 5.09–5.26 (2 H, m, H-14), 6.38 (1 H, d, J = 7.5 Hz, NH), 7.29–7.40 (5 H, m, H-16, H-17, H-18); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 0.89 (9 H, s, H-10), 4.70–4.79 (1 H, m, H-4), 6.08 (1 H, d, J = 7.1 Hz, NH); 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-8), 16.1 (q, C-11), 18.3 (s, C-9), 21.8 (q, C-21), 22.8 (q, C-21’), 24.9 (d, C-20), 25.9 (q, C-10), 29.4 (q, C-12), 30.8 (t, C-5), 32.4 (d, C-6), 41.2 (t, C-19), 50.6 (d, C-2), 52.3 (q, C-22), 56.3 (d, C-4), 67.6 (t, C-14), 68.3 (t, C-7), 127.8 (d, C-16), 128.1 (d, C-18), 128.5 (d, C-17), 136.4 (s, C-15), 157.4 (s, C-13), 170.8 (s, C-3), 173.1 (s, C-1); HRMS (CI) m/z calculated for C28H49N2O6Si [M + H]+ 537.3354, found 537.3355.
4.2.2. Synthesis of Methyl {[2S,4S]-2-({S}-2-{[(allyloxy)carbonyl]amino}-N-methylpropan amido)-5-[(tert-butyldimethylsilyl)oxy]-4-methylpentanoyl}-L-leucinate (18)
To a solution of dipeptide 17 (807 mg, 1.50 mmol) in 15 mL MeOH was added 10 wt-% Pd-C (80.7 mg, 10% on activated charcoal). Until complete conversion (3 h, TLC), the reaction mixture was stirred under 1 atm H2 atmosphere at room temperature. The reaction solution was filtered through Celite and the solvent was removed under reduced pressure. The residue was dissolved in 30 mL dry DCM. After addition of Alloc-AlaOH (286 mg, 1.65 mmol) the reaction mixture was cooled to −20 °C, followed by addition of NMM (ρ = 0.91 g/mL, 0.36 mL, 3.3 mmol) and BEP (453 mg, 1.65 mmol). Overnight, it was slowly warmed to room temperature. After 18 h (TLC), the reaction mixture was diluted with DCM. The organic phase was washed with water, sat. aq. NaHCO3 solution as well as with brine, dried over Na2SO4 and concentrated under reduced pressure. Tripeptide 18 (654 mg, 1.17 mmol, 78%) was obtained as a colorless resin after purification by flash chromatography (silica gel, PE/EtOAc 7:3). Rf = 0.35 (PE/EtOAc 6:4); [α]D20 = −90.3 (c = 1.0, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ 0.03 (6 H, s, H-16), 0.85 (3 H, d, J = 6.2 Hz, H-19), 0.86–0.96 (6 H, m, H-22), 0.87 (9 H, s, H-18), 1.31 (3 H, d, J = 6.7 Hz, H-7), 1.43–1.53 (1 H, m, H-14), 1.48–1.59 (3 H, m, H-13, H-20, H-21), 1.57–1.70 (1 H, m, H-20’), 2.00–2.09 (1 H, m, H-13’), 2.96 (3 H, s, H-12), 3.31-3.40 (1 H, m, H-15), 3.47 (1 H, dd, J = 9.7, 4.8 Hz, H-15’), 3.71 (3 H, s, H-23), 4.50–4.59 (1 H, m, H-2), 4.56 (2 H, d, J = 5.3 Hz, H-9), 4.65 (1 H, dq, J = 7.1, 7.1 Hz, H-6), 5.14–5.22 (1 H, m, H-4), 5.21 (1 H, dd, J = 10.4, 1.2 Hz, H-11), 5.30 (1 H, dd, J = 17.2, 1.5 Hz, H-11’), 5.71 (1 H, d, J = 7.8 Hz, NHb), 5.91 (1 H, ddt, J = 17.4, 10.5, 5.3 Hz, H-10), 6.36 (1 H, d, J = 8.1 Hz, NHa); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 0.87 (9 H, s, H-18), 1.35 (3 H, d, J = 6.9 Hz, H-7), 2.77 (3 H, s, H-12), 3.52 (1 H, dd, J = 9.9, 4.9 Hz, H-15’), 3.71 (3 H, s, H-23), 4.43–4.50 (3 H, m, H-2, H-9), 4.68–4.74 (1 H, m, H-6), 4.74 (1 H, dd, J = 8.1, 6.8 Hz, H-4) 5.24–5.31 (1 H, m, H-11’), 5.36 (1 H, d, J = 7.0 Hz, NHb), 5.80–5.90 (1 H, m, H-10), 7.88 (1 H, d, J = 7.6 Hz, NHa); Major rotamer: 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-16), 16.2 (q, C-19), 18.3 (s, C-17), 18.5 (q, C-7), 21.6 (q, C-22), 22.8 (q, C-22’), 24.8 (d, C-21), 25.9 (q, C-18), 30.1 (q, C-12), 30.6 (t, C-13), 32.3 (d, C-14), 41.3 (t, C-20), 47.2 (d, C-6), 50.5 (d, C-2), 52.3 (q, C-23), 54.1 (d, C-4), 65.6 (t, C-9), 68.2 (t, C-15), 117.7 (t, C-11), 132.7 (d, C-10), 155.3 (s, C-8), 170.2 (s, C-3), 173.0 (s, C-1), 173.5 (s, C-5); Minor rotamer (selected signals): 13C-NMR (CDCl3, 100 MHz) δ 16.9 (q, C-19), 18.0 (q, C-7), 21.2 (q, C-22), 23.0 (q, C-22’), 24.7 (d, C-21), 28.9 (q, C-12), 32.1 (d, C-14), 32.3 (t, C-13), 39.9 (t, C-20), 45.8 (d, C-6), 51.3 (d, C-2), 52.1 (q, C-23), 58.1 (d, C-4), 66.1 (t, C-9), 68.1 (t, C-15), 118.1 (t, C-11), 132.2 (d, C-10), 156.5 (s, C-8), 169.7 (s, C-3), 173.2 (s, C-1), 173.4 (s, C-5); HRMS (CI) m/z calculated for C27H52N3O7Si [M + H]+ 558.3569, found 558.3586.
4.2.3. Synthesis of Methyl {[2S,4S]-2-({S}-2-[(S)-2-{[(allyloxy)carbonyl]amino}-3-{4-[(2-methoxy-ethoxy)methoxy]-3-nitrophenyl}propanamido]-N-methylpropanamido)-5-[(tert-butyldimethylsilyl)oxy]-4-methylpentanoyl}-L-leucinate (19)
Tripeptide 18 (600 mg, 1.08 mmol) was reacted according to the general procedure for allyl deprotection with diethylamine (ρ = 0.707 g/mL, 0.56 mL, 5.4 mmol), TPPTS (24 mg, 43 μmol) and Pd(OAc)2 (1.1 mL, 22 μmol, 0.02 M in MeCN) in 10.8 mL of MeCN and water for 3 h. The solvent was removed in vacuo and the residue was dissolved in 10.8 mL anhydrous DCM. Afterwards nitrotyrosine 5 (472 mg, 1.18 mmol) was added at room temperature. The reaction mixture was cooled to 0 °C, DIPEA (ρ = 0.742 g/mL, 400 μL, 2.26 mmol) was directly added and after 10 min TBTU (380 mg, 1.18 mmol). Overnight, the reaction solution was slowly warmed to room temperature. After complete conversion (19 h, LCMS), the solvent was removed in vacuo and the crude product was dissolved in EtOAc. The organic phase was successively washed with 1 M aq. KHSO4 solution, sat. aq. NaHCO3 solution and brine and dried over Na2SO4. The solvent was removed under reduced pressure. Purification by reverse phase flash chromatography (C18 silica gel, H2O/MeCN 9:1 → 5:95) afforded tetrapeptide 19 (637 mg, 750 μmol, 69%) as a yellow resin. [α]D20 = −51.3 (c = 0.75, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ 0.04 (6 H, s, H-29), 0.84 (3 H, d, J = 6.1 Hz, H-32), 0.85–0.96 (6 H, m, H-35), 0.88 (9 H, s, H-31), 1.30 (3 H, d, J = 6.7 Hz, H-24), 1.42–1.57 (4 H, m, H-26, H-27, H-33, H-34), 1.57–1.65 (1 H, m, H-33’), 2.01–2.12 (1 H, m, H-26’), 2.96–3.06 (1 H, m, H-13), 3.00 (3 H, s, H-25), 3.10 (1 H, dd, J = 14.1, 5.7 Hz, H-13’), 3.32–3.40 (1 H, m, H-28), 3.36 (3 H, s, H-23), 3.46–3.54 (1 H, m, H-28’), 3.54–3.58 (2 H, m, H-22), 3.72 (3 H, s, H-36), 3.84–3.89 (2 H, m, H-21), 4.38–4.59 (4 H, m, H-2, H-8, H-10), 4.76–4.91 (1 H, m, H-6), 5.17 (1 H, dd, J = 10.3, 5.5 Hz, H-4), 5.20 (1 H, dd, J = 10.4, 1.2 Hz, H-12), 5.26 (1 H, d, J = 17.2 Hz, H-12’), 5.31–5.36 (1 H, m, NHc), 5.35 (2 H, s, H20), 5.86 (1 H, ddt, J = 17.2, 10.7, 5.5 Hz, H-11), 6.46 (1 H, d, J = 8.1 Hz, NHa), 7.04–7.13 (1 H, m, NHb), 7.25–7.30 (1 H, m, H-18), 7.33 (1 H, dd, J = 8.6, 2.2 Hz, H-19), 7.60 (1 H, d, J = 1.2 Hz, H-15); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 0.03 (6 H, s, H-29), 0.87 (9 H, s, H-31), 1.36 (3 H, d, J = 6.7 Hz, H-24), 3.71 (3 H, s, H-36), 6.65 (1 H, d, J = 5.9 Hz, NHb), 7.34–7.39 (1 H, m, H-19), 7.67 (1 H, d, J = 2.1 Hz, H-15), 7.92 (1 H, d, J = 7.5 Hz, NHa); 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-29), 16.2 (q, C-32), 18.1 (q, C-24), 18.3 (s, C-30), 21.7 (q, C-35), 22.8 (q, C-35’), 24.8 (d, C-34), 25.9 (q, C-31), 30.3 (q, C-25), 31.1 (t, C-26), 32.4 (d, C-27), 37.4 (t, C-13), 41.3 (t, C-33), 46.1 (d, C-6), 50.5 (d, C-2), 52.3 (q, C-36), 54.3 (d, C-4), 59.0 (d, C-8, C-23), 66.0 (t, C-10), 68.2 (t, C-28), 68.4 (t, C-21), 71.4 (t, C-22), 94.3 (t, C-20), 117.5 (d, C-18), 118.1 (t, C-12), 126.0 (d, C-15), 130.1 (s, C-14), 132.3 (d, C-11), 134.8 (d, C-19), 140.4 (s, C-16), 149.4 (s, C-17), 155.6 (s, C-9), 169.3 (s, C-7), 170.3 (s, C-3), 172.9 (s, C-5), 173.0 (s, C-1); HRMS (ESI) m/z calculated for C40H68N5O13Si [M + H]+ 854.4577, found 854.4577.
4.2.4. Synthesis of Methyl {[2S,4S]-2-({S}-2-[(S)-2-{[S]-2-({[allyloxy]carbonyl}(methyl)amino)-4-methylpentanamido}-3-{4-[(2-methoxyethoxy)methoxy]-3-nitrophenyl}-propan-amido]-N-methylpropanamido)-5-[(tert-butyldimethylsilyl)oxy]-4-methyl-pentanoyl}-L-leucinate (20)
Tetrapeptide 19 (494 mg, 580 μmol) was reacted according to the general procedure for allyl deprotection with diethylamine (ρ = 0.707 g/mL, 0.30 mL, 2.9 mmol), TPPTS (13.2 mg, 23.0 μmol) and Pd(OAc)2 (580 μL, 12 μmol, 0.02 M in MeCN) in 5.8 mL of MeCN and water for 3 h. After removal of the solvent under reduced pressure, the residue was dissolved in 5.8 mL anhydrous DCM. To the reaction mixture was added Alloc-N-Me-LeuOH (177 mg, 690 μmol) and it was cooled to 0 °C. DIPEA (ρ = 0.742 g/mL, 313 µL, 1.79 mmol) was directly added and after 10 min PyBOP (361 mg, 690 μmol). Overnight, the reaction mixture was slowly warmed to room temperature. After 17 h (LCMS), the solvent was removed in vacuo and the crude product was dissolved in EtOAc. The organic phase was washed with 1 M aq. KHSO4 solution, sat. aq. NaHCO3 solution and brine, dried over Na2SO4 and concentrated. Pentapeptide 20 (434 mg, 440 μmol, 77%) was obtained as a yellow foam after purification by reverse phase flash chromatography (C18 silica gel, H2O/MeCN 9:1 → 5:95). [α]D20 = −70.1 (c = 1.0, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ 0.04 (6 H, s, H-35), 0.84 (3 H, d, J = 6.0 Hz, H-38), 0.85-0.95 (12 H, m, H-18, H-41), 0.87 (9 H, s, H-37), 1.29 (3 H, d, J = 6.7 Hz, H-30), 1.40–1.56 (2 H, m, H-17, H-40), 1.46–1.56 (3 H, m, H-32, H-33, H39), 1.56–1.71 (3 H, m, H-16, H-39’), 2.01–2.12 (1 H, m, H-32’), 2.66 (3 H, s, H-15), 2.95 (1 H, dd, J = 14.3, 7.5 Hz, H-19), 2.98 (3 H, s, H-31), 3.09–3.19 (1 H, m, H-19’), 3.32–3.40 (1 H, m, H-34), 3.36 (3 H, s, H-29), 3.49 (1 H, dd, J = 10.1, 4.4 Hz, H-34’), 3.52–3.57 (2 H, m, H-28), 3.72 (3 H, s, H-42), 3.82–3.88 (2 H, m, H-27), 4.44–4.57 (1 H, m, H-2), 4.50–4.65 (3 H, m, H-10, H-12), 4.65–4.73 (1 H, m, H-8), 4.81 (1 H, dq, J = 7.0, 7.0 Hz, H-6), 5.15 (1 H, dd, J = 10.0, 5.3 Hz, H-4), 5.22 (1 H, d, J = 10.5 Hz, H-14), 5.30 (1 H, d, J = 17.9 Hz, H-14’), 5.34 (2 H, s, H-26), 5.83–6.01 (1 H, m, H-13), 6.39 (1 H, d, J = 7.7 Hz, NHa), 6.64 (1 H, d, J = 7.2 Hz, NHc), 7.00–7.08 (1 H, m, NHb), 7.21–7.30 (1 H, m, H-24), 7.28–7.37 (1 H, m, H-25), 7.56 (1 H, s, H-21); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 0.02 (6 H, s, H-35), 0.87 (9 H, s, H-37), 0.89–0.92 (3 H, m, H-38), 1.35 (3 H, d, J = 6.9 Hz, H-30), 3.71 (3 H, s, H-42), 7.62 (1 H, bs, H-21); Major rotamer: 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q; C-35), 16.2 (q, C-38), 18.0 (q, C-30), 18.3 (s, C-36), 21.7 (q, C-18, C-41), 22.8 (q, C-18’, C-41’), 24.8 (d, C-17, C-40), 25.9 (q, C-37), 29.6 (q; C-15), 30.2 (q, C-31), 30.8 (t, C-32), 32.3 (d, C-33), 36.3 (t; C-16), 36.8 (t, C-19), 41.3 (t, C-39), 46.1 (d, C-6), 50.5 (d, C-2), 52.3 (q, C-42), 53.4 (d, C-8), 54.2 (d, C-4), 56.9 (d, C-10), 59.0 (q, C-29), 66.6 (t, C-12), 68.2 (t, C-34), 68.4 (t, C-27), 71.3 (t, C-28), 94.3 (t, C-26), 117.5 (d, C-24), 117.6 (t, C-14), 125.9 (d, C-21), 130.3 (s, C-20), 132.6 (d, C-13), 134.6 (d, C-25), 140.4 (s, C-22), 149.3 (s, C-23), 157.1 (s, C-11), 169.0 (s, C-7, C-9), 170.2 (s, C-3), 172.8 (s, C-5), 173.0 (s, C-1); Minor rotamer (selected signals): 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q; C-35), 16.9 (q, C-38), 17.6 (q, C-30); HRMS (ESI) m/z calculated for C47H81N6O14Si [M + H]+ 981.5575, found 981.5574.
4.2.5. Synthesis of Methyl {[2S,4S]-2-({S}-2-[(S)-2-{[S]-2-({S}-2-[({allyloxy}carbonyl)amino]-N-methyl-3-[1-methyl-1H-indol-3-yl]propanamido)-4-methylpentanamido}-3-(4-{[2-methoxyethoxy]methoxy}-3-nitrophenyl)propanamido]-N-methylpropanamido)-5-[(tert-butyldimethylsilyl)oxy]-4-methylpentanoyl}-L-leucinate (21)
According to the general procedure for allyl deprotection, pentapeptide 20 (30 mg, 31 μmol) was reacted with diethylamine (ρ = 0.707 g/mL, 16.0 μL, 0.15 mmol), TPPTS (0.7 mg, 1.2 μmol) and Pd(OAc)2 (31 μL, 0.61 μmol, 0.02 M in MeCN) in 0.3 mL of MeCN and water for 3 h. After the solvent was removed in vacuo, the residue was dissolved in 0.3 mL dry DMF. Alloc-N’-Me-TrpOH (18.5 mg, 61.0 μmol), HATU (23.2 mg, 61.0 μmol), HOAt (4.2 mg, 31.0 μmol) and DIPEA (ρ = 0.742 g/mL, 27 µL, 0.15 mmol) were added successively to the reaction mixture at room temperature. After complete conversion (16 h, LCMS), the reaction solution was diluted with EtOAc. The organic phase was washed once with 1 M aq. KHSO4, three times with water and once with sat. aq. NaHCO3 solution and brine, dried over Na2SO4 and concentrated under reduced pressure. Hexapeptide 21 (28 mg, 24 μmol, 78%) could be isolated as a yellow foam after purification by reverse phase flash chromatography (C18 silica gel, H2O/MeCN 9:1 → 5:95). [α]D20 = −28.3 (c = 1.0, CHCl3); Major rotamer: 1H-NMR (CDCl3, 400 MHz) δ 0.03 (6 H, s, H-47), 0.70–0.93 (12 H, m, H-30, H-53), 0.79–0.86 (3 H, m, H-50), 0.88 (9 H, s, H-49), 1.31 (3 H, d, J = 6.7 Hz, H-42), 1.42–1.71 (8 H, m, H-28, H-29, H-44, H-45, H-51, H-52), 2.00–2.12 (1 H, m, H-44’), 2.41 (3 H, s, H-27), 2.65–3.32 (4 H, m, H-17, H-31), 2.98 (3 H, s, H43), 3.31-3.40 (1 H, m, H-46), 3.33 (3 H, s, H-41), 3.42–3.55 (1 H, m, H-46’), 3.46–3.50 (2 H, m, H-40), 3.67–3.75 (6 H, m, H-26, H-54), 3.76–3.81 (2 H, m, H-39), 4.38–4.75 (5 H, m, H-2, H-8, H-10, H-14), 4.75–5.00 (2 H, m, H-6, H-12), 5.10–5.22 (1 H, m, H-4), 5.12–5.31 (2 H, m, H-16), 5.22 (2 H, s, H-38), 5.78–5.95 (2 H, m, H-15, NHd), 6.27 (1 H, d, J = 8.1 Hz, NHc), 6.40 (1 H, d, J = 7.7 Hz, NHa), 6.88 (1 H, s, H-19), 7.10 (1 H, dd, J = 6.9 Hz, H-23), 7.16–7.29 (3 H, m, H-36, H-37, NHb), 7.18–7.26 (1 H, m, H-22), 7.23–7.31 (1 H, m, H-21), 7.30–7.35 (1 H, m, H-33), 7.63–7.69 (1 H, m, H-24); Minor rotamer (selected signals): 1H-NMR (CDCl3, 400 MHz) δ 0.02 (6 H, s, H-47), 0.45 (6 H, d, J = 6.6 Hz, H-30, H-53), 0.54 (6 H, d, J = 6.6 Hz, H-30’, H-53’), 0.86 (9 H, s, H-49), 0.97 (3 H, d, J = 6.6 Hz, H-50), 1.41 (3 H, d, J = 6.5 Hz, H-42), 2.36 (3 H, s, H-27), 2.93 (3 H, s, H43), 3.37 (3 H, s, H-41), 3.50–3.59 (2 H, m, H-40), 3.81–3.93 (2 H, m, H-39), 5.28–5.39 (2 H, m, H-38), 6.85 (1 H, s, H-19), 7.54 (1 H, d, J = 8.1 Hz, H-24), 7.58 (1 H, d, J = 2.1 Hz, H-33); Major rotamer: 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-47), 16.1 (q, C-50), 18.1 (q, C-42), 18.3 (s, C-48), 21.7 (q, C-30, C-53), 22.8 (q, C-30’, C-53’), 24.8 (d, C-29, C-52), 25.9 (q, C-49), 28.7 (t, C-17), 28.8 (q, C-27), 30.1 (q, C-43), 30.6 (t, C-44), 32.3 (d, C-45), 32.7 (q, C-26), 36.1 (t, C-31), 36.6 (t, C-28), 41.3 (t, C-51), 45.9 (d, C-6), 50.5 (d, C-2), 51.6 (d, C-12), 52.3 (q, C-54), 54.0 (d, C-8), 54.3 (d, C-4), 58.4 (d, C-10), 59.0 (q, C-41), 66.3 (t, C-14), 68.2 (t, C-46), 68.3 (t, C-39), 71.3 (t, C-40), 94.2 (t, C-38), 108.2 (s, C-18), 109.4 (d, C-21), 117.5 (d, C-36), 117.8 (t, C-16), 118.3 (d, C-24), 119.4 (d, C-23), 122.0 (d, C-22), 125.8 (d, C-33), 127.6 (d, C-19), 127.9 (s, C-25), 130.9 (s, C-32), 132.7 (d, C-15), 134.6 (d, C-37), 136.9 (s, C-20), 140.4 (s, C-34), 149.1 (s, C-35), 156.9 (s, C-13), 169.0 (s, C-9), 169.3 (s, C-7), 170.1 (s, C-3), 173.0 (s, C-5), 173.0 (s, C-1), 173.3 (s, C-11); Minor rotamer (selected signals): 13C-NMR (CDCl3, 100 MHz) δ -5.5 (q, C-47), 16.7 (q, C-50), 17.5 (q, C-42), 21.7 (q, C-30, C-53), 23.0 (q, C-30’, C-53’), 24.6 (d, C-29, C-52), 36.4 (t, C-28), 41.2 (t, C-51), 45.5 (d, C-6), 50.5 (d, C-2), 58.9 (q, C-41), 71.4 (t, C-40), 94.5 (t, C-38); HRMS (ESI) m/z calculated for C59H93N8O15Si [M + H]+ 1181.6524, found 1181.6522.
4.2.6. Synthesis of Methyl {[2S,4S]-2-({S}-2-[(S)-2-{[S]-2-({S}-2-[(S,E)-2-{[(allyloxy)carbonyl]amino}-hex-4-enamido]-N-methyl-3-{1-methyl-1H-indol-3-yl}propanamido)-4-methyl-pentanamido}-3-[4-({2-methoxyethoxy}methoxy)-3-nitrophenyl]propanamido]-N-methylpropanamido)-5-[(tert-butyldimethylsilyl)oxy]-4-methylpentanoyl}-L-leucinate (22)
To a solution of hexapeptide 21 (226 mg, 190 μmol) in 1.9 mL dry DCM was added 1,3-dimethylbarbituric acid (90 mg, 0.57 mmol) and Pd(Ph3P)4 (6.6 mg, 5.7 µmol) at room temperature. After 45 min the reaction mixture was diluted with EtOAc and the organic phase was washed three times with sat. aq. NaHCO3 solution. Subsequently, a back extraction from the aqueous phase was carried out with EtOAc. The combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved again in 1.9 mL dry DCM and Alloc-crotylglycine 16 (44.9 mg, 210 μmol) was added. The reaction mixture was cooled to 0 °C, treated with NMM (ρ = 0.91 g/mL, 51 µL, 0.46 mmol), HOBt (32.2 mg, 210 μmol) and EDC·HCl (40.4 mg, 210 μmol) and warmed to room temperature overnight. After 18 h (LCMS), the solution was diluted with EtOAc. The organic phase was washed with 1 M aq. KHSO4 solution, sat. aq. NaHCO3 solution and brine, dried over Na2SO4 and concentrated. Heptapeptide 22 (215 mg, 170 μmol, 87%) was obtained as a yellow foam after purification by reverse phase flash chromatography (C18 silica gel, H2O/MeCN 9:1 → 5:95). [α]D20 = −76.8 (c = 0.75, CHCl3); Major rotamer: 1H-NMR (CDCl3, 500 MHz) δ 0.04 (6 H, s, H-53), 0.73–0.82 (6 H, m, H-36), 0.82–0.87 (3 H, m, H-56), 0.82–1.01 (6 H, m, H-59), 0.89 (9 H, s, H-55), 1.08–1.22 (1 H, m, H-35), 1.39 (3 H, d, J = 6.7 Hz, H-48), 1.43–1.62 (6 H, m, H-34, H-50, H-51, H-57, H-58), 1.61 (3 H, d, J = 5.3 Hz, H-22), 1.66–1.76 (1 H, m, H-34’), 2.11–2.19 (1 H, m, H-50’), 2.25–2.38 (1 H, m, H-19), 2.38–2.48 (1 H, m, H-19’), 2.81 (3 H, s, H-33), 2.91-2.99 (1 H, m, H-37), 3.04–3.11 (1 H, m, H-37’), 3.10–3.20 (2 H, m, H-23), 3.12 (3 H, s, H-49), 3.32–3.40 (1 H, m, H-52), 3.34 (3 H, s, H-47), 3.43–3.53 (1 H, m, H-52’), 3.48–3.58 (2 H, m, H-46), 3.68–3.75 (6 H, m, H-32, H-60), 3.79–3.89 (2 H, m, H-45), 4.31–4.42 (1 H, m, H-14), 4.49–4.57 (2 H, m, H-2, H-10), 4.50–4.59 (2 H, m, H-16), 4.54–4.64 (1 H, m, H-8), 4.92–5.02 (1 H, m, H-6), 5.17–5.28 (3 H, m, H-4, H-12, H-18), 5.25–5.38 (4 H, m, H-18’, H-20, H-44), 5.52 (1 H, dq, J = 8.4, 6.8 Hz, H-21), 5.66 (1 H, bs, NHe), 5.90 (1 H, ddt, J = 16.9, 10.5, 5.3 Hz, H-17), 6.27 (1 H, d, J = 8.4 Hz, NHc), 6.83 (1 H, s, H-25), 6.95 (1 H, bs, NHa), 7.11 (1 H, dd, J = 7.0, 7.0 Hz, H-29), 7.17–7.24 (1 H, m, H-28), 7.20–7.34 (3 H, m, H-27, H-42, H-43), 7.45 (1 H, bs, H-39), 7.49 (1 H, bs, NHb), 7.68 (1 H, d, J = 7.9 Hz, H-30), 7.68–7.72 (1 H, m, NHd); Minor rotamer (selected signals): 1H-NMR (CDCl3, 500 MHz) δ 0.01–0.08 (6 H, m, H-53), 0.74–0.81 (3 H, m, H-56), 0.87 (9 H, s, H-55), 1.27 (3 H, d, J = 6.9 Hz, H-48), 1.64 (3 H, d, J = 6.0 Hz, H-22), 2.02–2.10 (1 H, m, H-50’), 2.76 (3 H, s, H-33), 2.92–2.96 (3 H, m, H-49), 2.96–3.02 (2 H, m, H-23), 3.36 (3 H, s, H-47), 4.81 (1 H, dq, J = 6.9, 6.9 Hz, H-6), 5.10–5.19 (2 H, m, H-4, H-12), 6.86 (1 H, s, H-25); Major rotamer: 13C-NMR (CDCl3, 125 MHz) δ -5.5 (q, C-53), 15.9 (q, C-56), 17.8 (q, C-48), 18.0 (q, C-22), 18.3 (s, C-54), 21.6 (q, C-59), 21.8 (q, C-36), 22.9 (q, C-59’), 23.0 (q, C-36’), 24.7 (d, C-35), 24.9 (d, C-58), 25.9 (q, C-55), 28.4 (t, C-23), 29.2 (q, C-33), 30.6 (q, C-49), 31.6 (t, C-50), 32.5 (d, C-51), 32.7 (q, C-32), 35.9 (t, C-37), 36.7 (t, C-19), 36.8 (t, C-34), 40.9 (t, C-57), 45.8 (d, C-6), 49.8 (d, C-12), 50.4 (d, C-2), 52.2 (q, C-60), 53.9 (d, C-8), 54.2 (d, C-4), 54.4 (d, C-14), 58.7 (d, C-10), 59.0 (q, C-47), 65.8 (t, C-16), 68.2 (t, C-52), 68.4 (t, C-45), 71.3 (t, C-46), 94.3 (t, C-44), 108.9 (s, C-24), 109.3 (d, C-27), 117.6 (d, C-42), 117.8 (t, C-18), 118.9 (d, C-30), 119.3 (d, C-29), 121.9 (d, C-28), 124.9 (d, C-20), 125.9 (d, C-39), 127.7 (d, C-25), 127.9 (s, C-31), 129.7 (d, C-21), 130.8 (s, C-38), 132.6 (d, C-17), 134.7 (d, C-43), 136.9 (s, C-26), 140.4 (s, C-40), 149.2 (s, C-41), 155.9 (s, C-15), 169.1 (s, C-7), 170.4 (s, C-9), 170.9 (s, C-3), 171.7 (s, C-13), 172.7 (s, C-11), 173.1 (s, C-1), 173.7 (s, C-5); Minor rotamer (selected signals): 13C-NMR (CDCl3, 125 MHz) δ 16.0 (q, C-56), 26.0 (q, C-55), 28.9 (q, C-33), 30.0 (q, C-49), 30.6 (t, C-50), 59.0 (q, C-47), 68.4 (t, C-45), 71.4 (t, C-46), 94.4 (t, C-44), 109.4 (d, C-27), 140.5 (s, C-40), 149.0 (s, C-41); HRMS (ESI) m/z calculated for C65H102N9O16Si [M + H]+ 1292.7208, found 1292.7222.
4.3. Synthesis of Cyclic Ilamycin Derivatives
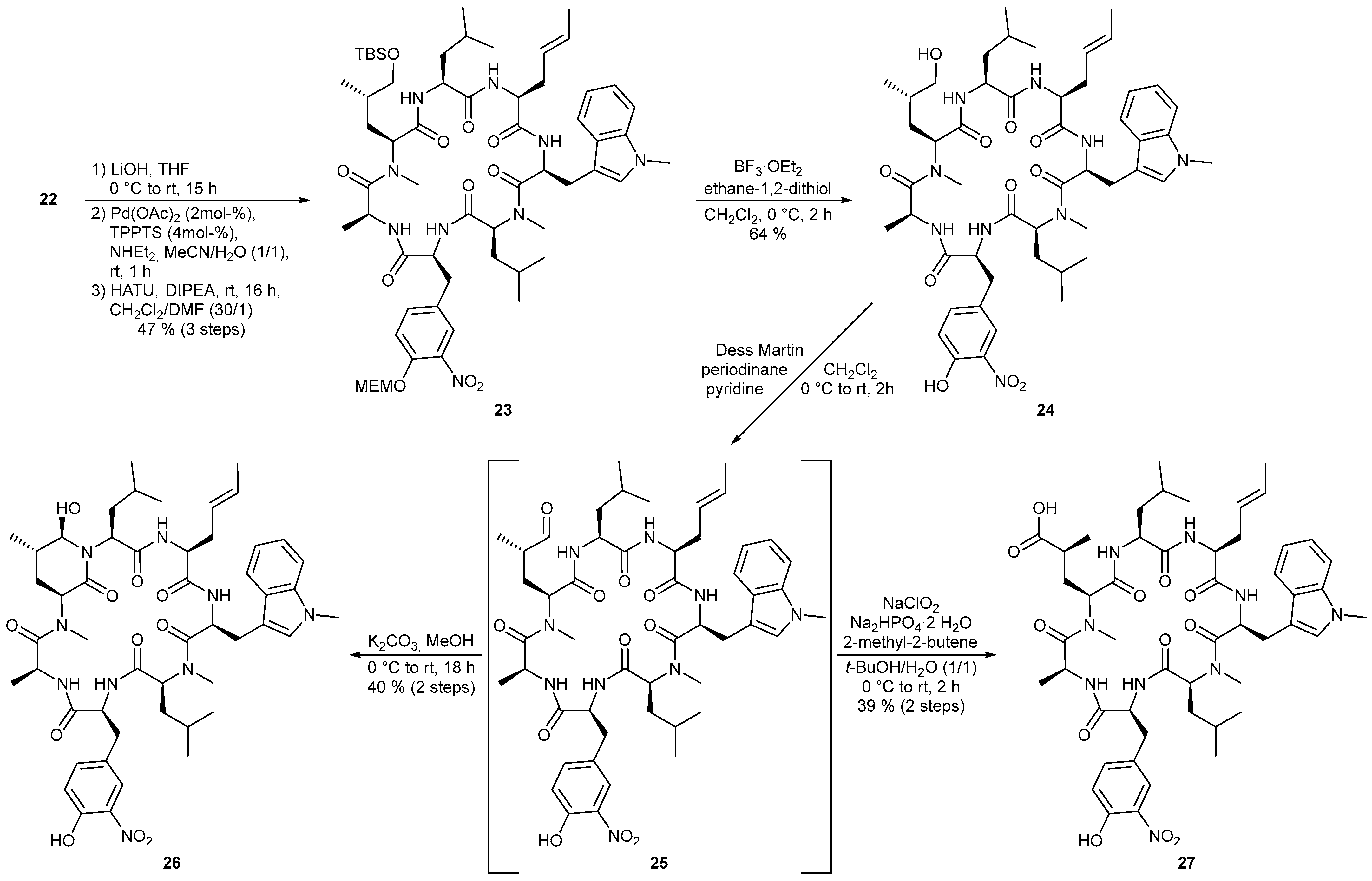
4.3.1. Synthesis of (3S,6S,9S,12S,15S,18S,21S)-15-[(E)-But-2-en-1-yl]-21-[(S)-3-{(tert-butyldimethyl-silyl)oxy}-2-methylpropyl]-9,18-diisobutyl-6-[4-({2-methoxyethoxy}methoxy)-3-nitrobenzyl]-1,3,10-trimethyl-12-[(1-methyl-1H-indol-3-yl)methyl]-1,4,7,10,13,16,19-heptaazacyclohenicosane-2,5,8,11,14,17,20-heptaone (23)
To a solution of linear heptapeptide 22 (181 mg, 140 μmol) in 1.4 mL THF was added a 1 M aq. LiOH solution (210 µL, 210 μmol) at 0 °C. Overnight the reaction mixture was slowly warmed to room temperature. After 15 h (LCMS), the solvent was removed under reduced pressure. According to the general procedure for allyl deprotection the residue was reacted with diethylamine (ρ = 0.707 g/mL, 73 µL, 0.70 mmol), TPPTS (3.2 mg, 5.6 µmol) and Pd(OAc)2 (140 µL, 2.8 µmol, 0.02 M in MeCN) in 1.4 mL of MeCN and water for 1 h. The reaction mixture was concentrated and the crude product was dried under high vacuum. HATU (186 mg, 490 μmol) and DIPEA (ρ = 0.717 g/mL, 110 μL, 630 μmol) were dissolved in 135 mL dry DCM in a three-neck flask at room temperature. Via syringe pump the heptapeptide, which was dissolved in 4.5 mL anhydrous DMF, was added dropwise over 4 h to the reaction solution. Stirring was performed overnight at room temperature. After complete conversion (16 h, LCMS), the solvent was removed in vacuo and the residue was dissolved in EtOAc. The organic phase was washed once with 1 M aq. KHSO4 solution, three times with water and once with sat. aq. NaHCO3 solution and brine, dried over Na2SO4 and concentrated under reduced pressure. The crude product was first purified by reverse phase flash chromatography (C18 silica gel, H2O/MeCN 9:1 → 5:95). Further purification by preparative HPLC (Luna, H2O/MeCN 7:3 → MeCN) yielded 23 (77 mg, 65 μmol, 47%) as a yellowish foam. [α]D20 = –75.9 (c = 1.0, CHCl3); 1H-NMR ((CD3)2SO, 400 MHz) δ −0.41-(-0.30) (1 H, m, H-30), −0.02 (3 H, s, H 49), -0.02 (3 H, s, H-49’), 0.22 (3 H, d, J = 6.6 Hz, H-32), 0.44 (3 H, d, J = 6.6 Hz, H-32’), 0.77–0.86 (6 H, m, H-55), 0.81 (9 H, s, H-51), 0.81–0.92 (3 H, m, H-52), 0.94-1.07 (1 H, m, H-31), 1.13 (3 H, d, J = 6.7 Hz, H-44), 1.36–1.48 (1 H, m, H-47), 1.45–1.60 (4 H, m, H-30’, H-53, H-54), 1.46 (3 H, d, J = 6.2 Hz, H-18), 1.61–1.78 (2 H, m, H-46), 2.34–2.45 (1 H, m, H-15), 2.50 (3 H, s, H-29), 2.50–2.61 (1 H, m, H-15’), 2.53–2.65 (1 H, m, H-33), 2.59 (3 H, s, H-45), 2.96 (1 H, dd, J = 13.0, 9.6 Hz, H-33’), 3.04 (1 H, dd, J = 13.1, 4.1 Hz, H-19), 3.20 (3 H, s, H-43), 3.29 (1 H, dd, J = 13.5, 9.6 Hz, H-19’), 3.30–3.39 (1 H, m, H-48), 3.44 (2 H, t, J = 4.7 Hz, H-42), 3.48 (1 H, dd, J = 10.0, 4.8 Hz, H-48’), 3.69–3.76 (2 H, m, H-41), 3.71 (3 H, s, H-28), 4.22–4.35 (1 H, m, H-2), 4.39 (1 H, dt, J = 6.0, 5.0 Hz, H-14), 4.46–4.56 (2 H, m, H-8, H-10), 4.67–4.84 (2 H, m, H-6, H-12), 4.95 (1 H, t, J = 7.1 Hz, H-4), 5.03 (1 H, ddd, J = 15.3, 7.5, 7.5 Hz, H-16), 5.26–5.37 (1 H, m, H-17), 5.36 (2 H, s, H-40), 6.99 (1 H, dd, J = 7.5, 7.5 Hz, H-25), 7.05 (1 H, s, H-21), 7.13 (1 H, dd, J = 7.5, 7.5 Hz, H-24), 7.24–7.33 (2 H, m, H-38, H-39), 7.38 (1 H, d, J = 8.4 Hz, H-23), 7.44 (1 H, d, J = 1.6 Hz, H-35), 7.47 (1 H, d, J = 7.8 Hz, H-26), 7.48 (1 H, d, J = 6.2 Hz, NHe), 8.61 (1 H, d, J = 9.2 Hz, NHa), 8.74 (1 H, d, J = 8.0 Hz, NHc), 9.16 (1 H, d, J = 3.9 Hz, NHb), 9.30 (1 H, d, J = 6.2 Hz, NHd); 13C-NMR ((CD3)2SO, 100 MHz) δ -5.6 (q, C-49), -5.5 (q, C-49‘), 16.6 (q, C-44), 16.9 (q, C-52), 18.0 (q, C-18), 18.0 (s, C-50), 20.7 (q, C-32), 21.3 (q, C-55), 22.7 (q, C-55’), 22.9 (q, C-32’), 23.6 (d, C-31), 24.1 (d, C-54), 25.7 (q, C-51), 26.9 (t, C-19), 28.1 (q, C-45), 28.6 (q, C-29), 31.8 (d, C-47), 32.3 (q, C-28), 33.0 (t, C-46), 34.5 (t, C-15), 36.8 (t, C-30, C-33), 42.4 (t, C-53), 45.0 (d, C-6), 50.2 (d, C-12), 51.3 (d, C-14), 53.0 (d, C-2), 53.1 (d, C-8), 57.2 (d, C-4), 57.6 (d, C-10), 58.0 (q, C-43), 68.0 (t, C-41), 68.2 (t, C-48), 70.9 (t, C-42), 93.9 (t, C-40), 108.5 (s, C-20), 109.7 (d, C-23), 117.2 (d, C-38), 118.4 (d, C-26), 118.6 (d, C-25), 121.3 (d, C-24), 124.7 (d, C-16), 125.4 (d, C-35), 127.3 (s, C-27), 128.1 (d, C-21), 128.7 (d, C-17), 130.1 (s, C-34), 134.7 (d, C-39), 136.5 (s, C-22), 140.1 (s, C-36), 148.2 (s, C-37), 167.3 (s, C-9), 169.1 (s, C-3), 170.4 (s, C-7), 170.7 (s, C-1), 171.2 (s, C-13), 171.4 (s, C-11), 172.3 (s, C-5); HRMS (ESI) m/z calculated for C60H94N9O13Si [M + H]+ 1176.6735, found 1176.6732.
4.3.2. Synthesis of (3S,6S,9S,12S,15S,18S,21S)-15-[(E)-But-2-en-1-yl]-21-[(S)-3-hydroxy-2-methyl-propyl]-6-(4-hydroxy-3-nitrobenzyl)-9,18-diisobutyl-1,3,10-trimethyl-12-[(1-methyl-1H-indol-3-yl)methyl]-1,4,7,10,13,16,19-heptaazacyclohenicosane-2,5,8,11,14,17,20-heptaone (24)
To a solution of protected cyclic heptapeptide 23 (77 mg, 65 μmol) in 6.5 mL dry DCM was added 1,2-ethanedithiol (ρ = 1.12 g/mL, 165 mL, 1.96 mmol) as well as BF3·OEt2 (ρ = 1.12 g/mL, 166 µL, 1.31 mmol) at 0 °C. After complete conversion (2 h, LCMS) at this temperature, the reaction mixture was quenched with approx. 10 mL sat. aq. NaHCO3 solution. The aqueous phase was extracted three times with DCM and the combined organic phases were dried over Na2SO4 and concentrated under reduced pressure. The crude product was first purified by flash chromatography (silica gel, DCM/MeOH 95:5 → 9:1). Further purification was carried out by preparative HPLC (Luna, H2O/MeCN 9:1 → MeCN) to afford the deprotected cyclic heptapeptide 24 (41 mg, 42 μmol, 64%, mp. 168 °C) as a yellow solid. [α]D20 = −96.7 (c = 0.5, MeOH); Major rotamer: 1H-NMR ((CD3)2SO, 500 MHz) δ -0.22 (1 H, ddd, J = 12.9, 8.6, 4.3 Hz, H-30), 0.29 (3 H, d, J = 6.7 Hz, H-32), 0.46 (3 H, d, J = 6.6 Hz, H-32‘), 0.77–0.84 (3 H, m, H-48), 0.81–0.90 (6 H, m, H-45, H-48‘), 0.95–1.05 (1 H, m, H-31), 1.14 (3 H, d, J = 6.7 Hz, H-40), 1.24–1.33 (1 H, m, H-43), 1.45–1.59 (4 H, m, H-30‘, H-42, H-46, H-47), 1.47 (3 H, d, J = 6.3 Hz, H-18), 1.62 (1 H, dd, J = 9.3, 7.7 Hz, H-46‘), 1.72–1.82 (1 H, ddd, J = 14.3, 9.6, 4.4 Hz, H-42‘), 2.27–2.38 (1 H, m, H-15), 2.42–2.53 (1 H, m, H-15‘), 2.50 (3 H, s, H-29), 2.55–2.60 (1 H, m, H-33), 2.61 (3 H, s, H-41), 2.92 (1 H, dd, J = 13.2, 9.1 Hz, H-33‘), 3.04 (1 H, dd, J = 13.6, 5.2 Hz, H-19), 3.13–3.20 (1 H, m, H-44), 3.23–3.30 (2 H, m, H-19‘, H-44‘), 3.71 (3 H, s, H-28), 4.18–4.26 (1 H, m, H-2), 4.39 (1 H, dt, J = 5.4, 5.4 Hz, H-14), 4.46–4.53 (1 H, m, H-8), 4.54–4.61 (1 H, m, Hyleu-OH), 4.56 (1 H, t, J = 5.1 Hz, H-10), 4.70–4.77 (1 H, m, H-6), 4.77–4.86 (2 H, m, H-4, H-12), 5.03 (1 H, dt, J = 15.1, 7.3 Hz, H-16), 5.31 (1 H, dq, J = 8.7, 6.4 Hz, H-17), 6.95 (1 H, d, J = 8.5 Hz, H-38), 7.00 (1 H, dd, J = 7.4, 7.4 Hz, H-25), 7.07 (1 H, s, H-21), 7.13 (1 H, dd, J = 7.6, 7.6 Hz, H-24), 7.17 (1 H, dd, J = 8.7, 2.1 Hz, H-39), 7.38 (1 H, d, J = 8.2 Hz, H-23), 7.50 (1 H, d, J = 7.9 Hz, H-26), 7.51–7.55 (1 H, m, NHe), 7.54 (1 H, d, J = 2.1 Hz, H-35), 8.56-8.62 (2 H, m, NHa, NHc), 9.07 (1 H, d, J = 4.4 Hz, NHb), 9.23 (1 H, d, J = 6.7 Hz, NHd) 10.69 (1 H, s, Tyr-OH); Minor rotamer: 1H-NMR ((CD3)2SO, 500 MHz) δ 0.50 (3 H, d, J = 6.4 Hz, H-48), 0.56 (3 H, d, J = 6.4 Hz, H-48’), 1.18 (3 H, d, J = 6.9 Hz, H-40), 1.44 (3 H, d, J = 4.9 Hz, H-18), 2.59 (3 H, s, H-41), 3.72 (3 H, s, H-28); Major rotamer: 13C-NMR ((CD3)2SO, 125 MHz) δ 16.6 (q, C-40), 17.0 (q, C-45), 18.0 (q, C-18), 20.9 (q, C-32), 21.3 (q, C-48), 22.7 (q, C-48’), 22.9 (q, C-32’), 23.6 (d, C-31), 24.1 (d, C-47), 26.9 (t, C-19), 28.1 (q, C-41), 28.5 (q, C-29), 31.7 (d, C-43), 32.2 (q, C-28), 32.5 (t, C-42), 34.4 (t, C-15), 36.7 (t, C-33), 36.8 (t, C-30), 41.9 (t, C-46), 45.0 (d, C-6), 50.0 (d, C-12), 51.3 (d, C-14), 53.1 (d, C-2), 53.3 (d, C-8), 57.3 (d, C-4), 57.5 (d, C-10), 66.4 (t, C-44), 108.6 (s, C-20), 109.6 (d, C-23), 118.5 (d, C-26), 118.6 (d, C-25), 118.9 (d, C-38), 121.2 (d, C-24), 124.8 (d, C-16), 125.6 (d, C-35), 127.3 (s, C-34), 127.6 (s, C-27), 128.1 (d, C-21), 128.4 (d, C-17), 136.1 (d, C-39), 136.2 (s, C-36), 136.4 (s, C-22), 151.0 (s, C-37), 167.4 (s, C-9), 169.4 (s, C-3), 170.3 (s, C-7), 170.7 (s, C-1), 171.3 (s, C-13), 171.4 (s, C-11), 172.3 (s, C-5); Minor rotamer: 13C-NMR ((CD3)2SO, 125 MHz) δ 16.3 (q, C-40), 16.8 (q, C-45), 17.7 (q, C-18), 21.0 (q; C-48), 22.7 (q, C-48’); HRMS (ESI) m/z calculated for C50H72N9O11 [M + H]+ 974.5346, found 974.5344.
4.3.3. Synthesis of (2S,5S,8S,11S,14S,17S,20S,22S,23R)-5-[(E)-But-2-en-1-yl]-23-hydroxy-14-(4-hydroxy-3-nitrobenzyl)-2,11-diisobutyl-10,17,19,22-tetramethyl-8-[(1-methyl-1H-indol-3-yl)methyl]-1,4,7,10,13,16,19-heptaazabicyclo[18.3.1]tetracosane-3,6,9,12,15,18,24-heptaone (26)
To a solution of 24 (5.0 mg, 5.1 μmol) in 5.1 mL anhydrous DCM was added pyridine (ρ = 0.982 g/mL, 8.3 µL, 0.1 mmol) as well as Dess-Martin periodinane (10.9 mg, 26.0 μmol) at 0 °C. The ice bath was removed and the reaction mixture was stirred for 1 h at room temperature. After complete conversion (LCMS), the reaction solution was quenched with approx. 1.6 mL sat. aq. Na2SO3 solution. The aqueous phase was extracted three times with EtOAc and the combined organic phases were washed with sat. aq. CuSO4 solution and brine, dried over Na2SO4 and concentrated. After dissolving the residue in 2 mL MeOH, the reaction mixture was cooled again to 0 °C and K2CO3 (3.6 mg, 26 μmol) was added. Overnight the reaction solution was slowly warmed to room temperature (18 h). The reaction mixture was quenched with 1.5 mL sat. aq. NH4Cl solution and the aqueous phase was extracted three times with EtOAc. The combined organic phases were washed with 1 M aq. NH4Cl solution and brine and dried over Na2SO4. The solvent was removed in vacuo and the crude product was purified by reverse phase flash chromatography (C18 silica gel, H2O/MeCN 9:1 → MeCN). Further purification was carried out by preparative HPLC (Luna, H2O/MeCN 9:1 → MeCN). The desired ilamycin derivative 26 (2.0 mg, 2.1 μmol, 40%) was isolated as a yellow amorphous solid. [α]D20 = −98.7 (c = 0.25, CHCl3); Major rotamer: 1H-NMR (CD3OD, 500 MHz) δ -0.46 (1 H, ddd, J = 13.1, 8.7, 4.0 Hz, H-30), 0.24 (3 H, d, J = 6.6 Hz, H-32), 0.48 (3 H, d, J = 6.6 Hz, H-32’), 0.92 (3 H, d, J = 6.4 Hz, H-48), 0.90–1.00 (1 H, m, H-31), 1.02 (3 H, d, J = 6.7 Hz, H-48’), 1.09 (3 H, d, J = 6.7 Hz, H-45), 1.27 (3 H, d, J = 6.6 Hz, H-40), 1.38–1.48 (2 H, m, H-30’, H-47), 1.66 (3 H, dd, J = 6.1, 1.1 Hz, H-18), 1.83–1.89 (1 H, m, H-42), 1.88–1.96 (2 H, m, H-46), 1.94–2.00 (1 H, m, H-43), 2.23–2.29 (1 H, m, H-42’), 2.29 (3 H, s, H-29), 2.57–2.65 (1 H, m, H-15), 2.76–2.85 (1 H, m, H-15’), 2.91 (1 H, dd, J = 14.4, 10.3 Hz, H-33), 3.09 (1 H, dd, J = 14.2, 5.8 Hz, H-33’), 3.13–3.27 (2 H, m, H-19), 3.25 (3 H, s, H-41), 3.74 (3 H, s, H-28), 3.74–3.82 (1 H, m, H-4), 4.21 (1 H, dd, J = 10.3, 3.9 Hz, H-10), 4.57 (1 H, t, J = 7.4 Hz, H-14), 4.62 (1 H, dd, J = 10.2, 5.7 Hz, H-8), 4.77 (1 H, d, J = 1.5 Hz, H-44), 4.77–4.82 (1 H, m, H-6), 4.86–4.94 (1 H, m, H-12), 5.22 (1 H, dd, J = 11.3, 5.1 Hz, H-2), 5.52–5.60 (1 H, m, H-16), 5.60–5.68 (1 H, m, H-17), 7.00–7.07 (3 H, m, H-21, H-25, H-38), 7.13–7.19 (1 H, m, H-24), 7.21 (1 H, d, J = 8.2 Hz, H-23), 7.37 (1 H, dd, J = 9.0, 1.5 Hz, H-39), 7.44 (1 H, d, J = 7.9 Hz, H-26), 7.83 (1 H, d, J = 2.0 Hz, H-35); Minor rotamer: 1H-NMR (CD3OD, 500 MHz) δ 0.27 (3 H, d, J = 6.6 Hz, H-32), 0.45 (3 H, d, J = 6.1 Hz, H-32’), 1.61 (3 H, d, J = 6.4 Hz, H-18); 13C-NMR (CD3OD, 125 MHz) δ 17.7 (q, C-45), 18.0 (q, C-40), 18.5 (q, C-18), 21.4 (q, C-48), 21.8 (q, C-32), 23.4 (q, C-32’), 24.0 (q, C-48’), 25.9 (d, C-31), 26.0 (d, C-47), 27.0 (t, C-42), 29.5 (t, C-19), 29.5 (q, C-29), 33.0 (q, C-28), 34.4 (d, C-43), 35.2 (t, C-15), 36.0 (t, C-46), 37.4 (t, C-30), 38.0 (t, C-33), 38.6 (q, C-41), 47.8 (d, C-6), 52.3 (d, C-12), 54.4 (d, C-14), 55.6 (d, C-2), 57.4 (d, C-8), 59.7 (d, C-10), 63.3 (d, C-4), 79.6 (d, C-44), 109.7 (s, C-20), 110.6 (d, C-23), 119.9 (d, C-26), 120.3 (d, C-25), 121.6 (d, C-38), 123.0 (d, C-24), 126.6 (d, C-35), 127.8 (d, C-16), 129.0 (s, C-34), 129.3 (d, C-21), 129.4 (s, C-27), 129.5 (d, C-17), 135.9 (s, C-36), 138.5 (s, C-22), 139.0 (d, C-39), 156.3 (s, C-37), 170.2 (s, C-9), 172.0 (s, C-7), 172.2 (s, C-3), 172.7 (s, C-5), 173.3 (s, C-13), 173.3 (s, C-1), 174.4 (s, C-11); HRMS (ESI) m/z calculated for C50H70N9O11 [M + H]+ 972.5189, found 972.5189.
4.3.4. Synthesis of (S)-3-[(2S,5S,8S,11S,14S,17S,20S)-8-{(E)-But-2-en-1-yl}-17-[4-hydroxy-3-nitro-benzyl]-5,14-diisobutyl-1,13,20-trimethyl-11-{(1-methyl-1H-indol-3-yl)-methyl}-3,6,9,12,15,18,21-heptaoxo-1,4,7,10,13,16,19-heptaazacyclo-henicosan-2-yl]-2-methylpropanoic Acid (27)
To a solution of 24 (5.0 mg, 5.1 μmol) in 5.1 mL dry DCM was added pyridine (ρ = 0.982 g/mL, 8.3 µL, 0.1 mmol) as well as Dess-Martin periodinane (10.9 mg, 26.0 μmol) at 0 °C. The ice bath was removed and the reaction mixture was stirred for 1 h at room temperature. After complete conversion (LCMS), the reaction solution was quenched with approx. 1.6 mL sat. aq. Na2SO3 solution. The aqueous phase was extracted three times with EtOAc and the combined organic phases were washed with sat. aq. CuSO4 solution and brine, dried over Na2SO4 and concentrated. After the residue was dissolved in 0.3 mL of t-BuOH and water, NaH2PO4 dihydrate (16 mg, 0.1 mmol) and 2-methyl-2-butene (ρ = 0.66 g/mL, 27 µL, 0.26 mmol) were added to the reaction mixture at room temperature followed by the addition of sodium chlorite (5.8 mg, 51 μmol) at 0 °C. The ice bath was removed and stirring was continued for two hours at room temperature. The reaction mixture was diluted with approx. 2.6 mL EtOAc and 0.6 mL water and the phases were separated. The aqueous phase was extracted three times with EtOAc and the combined organic phases were washed with water and brine, dried over Na2SO4 and concentrated under reduced pressure. After purification by reverse phase flash chromatography (C18 silica gel, H2O/MeCN 9:1 → MeCN), the crude product was further purified via preparative HPLC (Luna, H2O/MeCN 9:1 → MeCN) to give the desired ilamycin derivative 27 (2.0 mg, 2.0 μmol, 39%) as a yellowish amorphous solid. [α]D20 = −79.0 (c = 0.25, CHCl3); Major rotamer: 1H-NMR (CD3OD, 500 MHz) δ -0.61 (1 H, dd, J = 12.8, 9.7, 3.1 Hz, H-30), 0.24 (3 H, d, J = 6.6 Hz, H-32), 0.44 (3 H, d, J = 6.7 Hz, H-32’), 0.95 (3 H, d, J = 5.8 Hz, H-48), 0.98 (3 H, d, J = 5.7 Hz, H-48’), 1.00–1.08 (1 H, m, H-31), 1.23 (3 H, d, J = 7.2 Hz, H-45), 1.27 (3 H, d, J = 6.9 Hz, H-40), 1.50–1.59 (1 H, m, H-30), 1.56 (3 H, d, J = 6.3 Hz, H-18), 1.68–1.78 (3 H, m, H-46, H-47), 1.82 (1 H, ddd, J = 13.0, 9.4, 4.5 Hz, H-42), 2.14–2.24 (1 H, m, H-42’), 2.42–2.49 (1 H, m, H-43), 2.46–2.54 (1 H, m, H-15), 2.61 (3 H, s, H-41), 2.69 (1 H, dd, J = 13.1, 4.7 Hz, H-33), 2.73 (3 H, s, H-29), 2.74–2.84 (1 H, m, H-15’), 3.03 (1 H, dd, J = 13.0, 9.9 Hz, H-33’), 3.15 (1 H, dd, J = 13.5, 3.7 Hz, H-19), 3.43 (1 H, dd, J = 13.1, 10.8 Hz, H-19’), 3.76 (3 H, s, H-28), 4.54–4.60 (3 H, m, H-8, H-10, H-14), 4.60–4.67 (1 H, m, H-2), 4.75–4.83 (1 H, m, H-6), 4.88–4.94 (1 H, m, H-12), 4.97 (1 H, dd, J = 8.5, 4.6 Hz, H-4), 5.17 (1 H, dt, J = 14.8, 7.0 Hz, H-16), 5.49 (1 H, dq, J = 15.0, 6.5 Hz, H-17), 6.97 (1 H, s, H-21), 7.02–7.09 (1 H, m, H-25), 7.06 (1 H, d, J = 8.5 Hz, H-38), 7.18 (1 H, dd, J = 7.3, 7.3 Hz, H-24), 7.36 (1 H, d, J = 8.2 Hz, H-23), 7.41 (1 H, dd, J = 8.6, 2.1 Hz, H-39), 7.52 (1 H, d, J = 7.9 Hz, H-26), 7.78 (1 H, d, J = 2.1 Hz, H-35); Minor rotamer: 1H-NMR (CD3OD, 500 MHz) δ 0.56 (3 H, d, J = 6.6 Hz, H-32), 0.64 (3 H, d, J = 6.6 Hz, H-32’), 0.90 (3 H, d, J = 5.7 Hz, H-48’), 0.92 (3 H, d, J = 5.8 Hz, H-48), 1.36 (3 H, d, J = 6.9 Hz, H-40), 1.52 (3 H, d, J = 6.0 Hz, H-18), 2.77 (3 H, s, H-29), 3.77 (3 H, s, H-28), 7.62 (1 H, d, J = 7.9 Hz, H-26), 7.90 (1 H, d, J = 2.0 Hz, H-35); Major rotamer: 13C-NMR (CD3OD, 125 MHz) δ 17.1 (q, C-40), 18.7 (q, C-18), 19.3 (q, C-45), 21.4 (q, C-32), 22.2 (q, C-48), 23.3 (q, C-48’), 23.6 (q, C-32’), 25.6 (d, C-31), 26.1 (d, C-47), 28.6 (t, C-19), 30.0 (q, C-41), 30.0 (q, C-29), 33.0 (q, C-28), 35.2 (t, C-42), 36.2 (t, C-15), 38.2 (t, C-30), 38.4 (t, C-33), 39.0 (d, C-43), 45.1 (t, C-46), 47.0 (d, C-6), 52.5 (d, C-12), 53.5 (d, C-14), 55.0 (d, C-2), 55.7 (d, C-8), 60.1 (d, C-10), 60.2 (d, C-4), 110.0 (s, C-20), 110.8 (d, C-23), 119.8 (d, C-26), 120.4 (d, C-38), 121.3 (d, C-25), 123.1 (d, C-24), 125.5 (d, C-16), 126.9 (d, C-35), 129.2 (s, C-27), 129.3 (d, C-21), 129.8 (s, C-34), 131.4 (d, C-17), 135.7 (s, C-36), 138.7 (s, C-22), 139.3 (d, C-39), 154.8 (s, C-37), 169.9 (s, C-9), 171.1 (s, C-3), 172.5 (s, C-7), 173.2 (s, C-13), 174.0 (s, C-1), 174.2 (s, C-11), 175.1 (s, C-5), 181.0 (s, C-44); Minor rotamer: 13C-NMR (CD3OD, 125 MHz) δ 16.8 (q, C-40), 18.4 (q, C-18), 21.6 (q, C-32), 23.4 (q, C-32’); HRMS (ESI) m/z calculated for C50H70N9O12 [M + H]+ 988.5138, found 988.5139.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}