
Effective Synthesis and Antifouling Activity of Dolastatin 16 Derivatives


Abstract
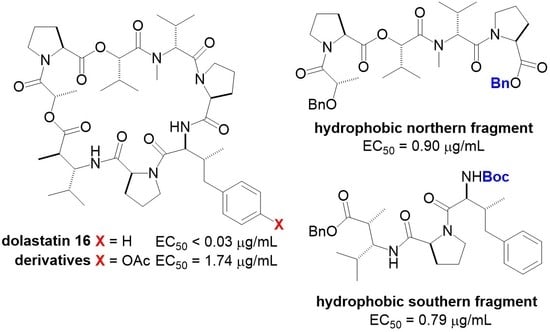
:
1. Introduction
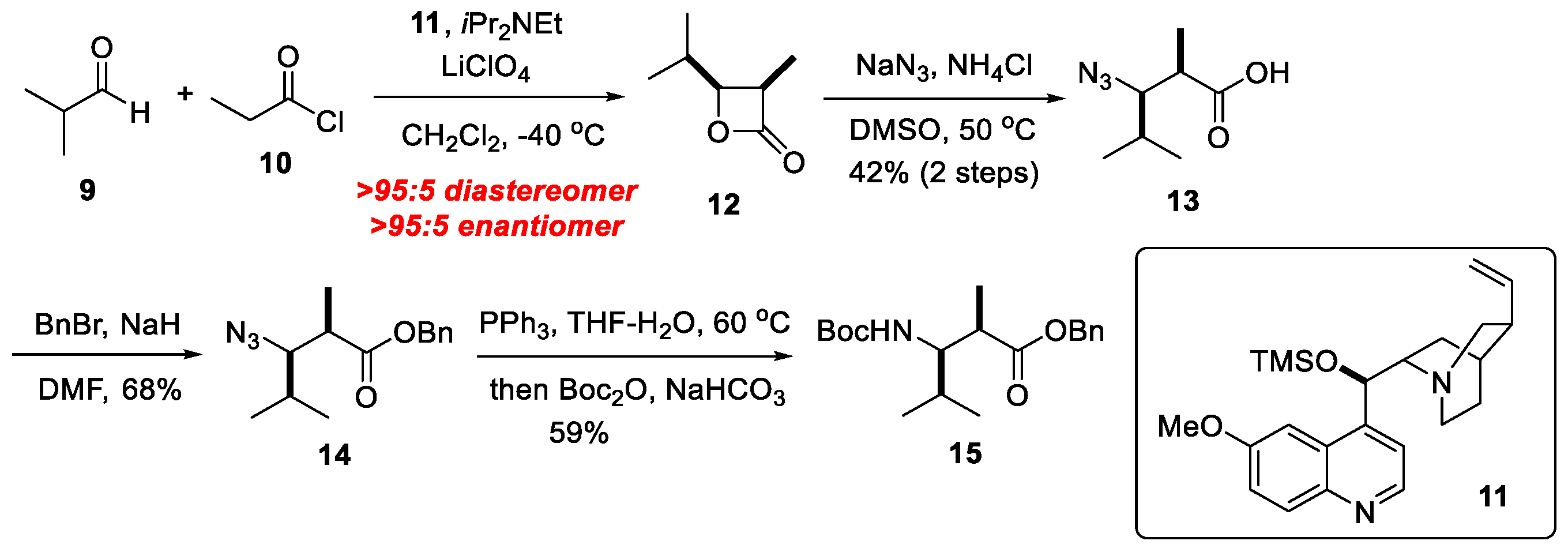
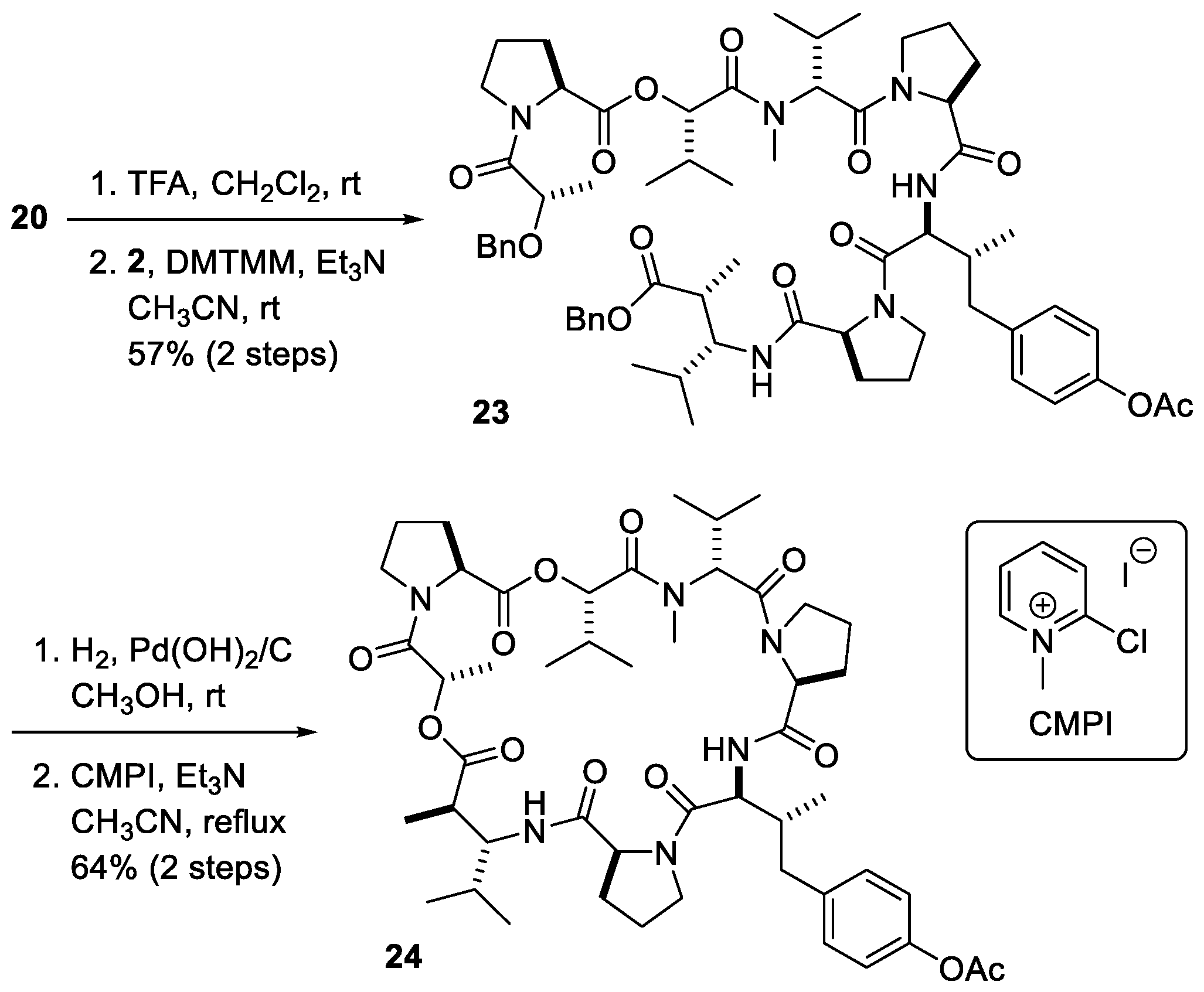
2. Results and Discussions
3. Materials and Methods
3.1. General Methods
3.2. Pht-Dpv-NH(8-quinoline) 5
3.3. Boc-Dpv-OH 7
3.4. Pht-Dpv(OTBS)-NH(8-quinoline) 6
3.5. Boc-Dpv(OH)-OH 8
3.6. N2-Dml-OH 13
3.7. N2-Dml-OBn 14
3.8. Boc-Dml-OBn 15
3.9. Boc-Dpv(OH)-Pro-OBn 16
3.10. Boc-Dpv(OAc)-Pro-OBn 17
3.11. Boc-Dpv(OAc)-Pro-Dml-OBn 20
3.12. Boc-Dpv(OH)-Pro-Dml-OBn 21
3.13. Boc-Dpv(OBn)-Pro-Dml-OBn 22
3.14. BnO-Lac-Pro-O-Hiv-D-MeVal-Pro-Dpv(OAc)-Pro-Dml-OBn 23
3.15. Dolastatin 16 Acetate 24
3.16. BnO-Lac-Pro-O-Hiv-D-MeVal-Pro-OBn 25
3.17. Boc-Pro-O-Hiv-D-MeVal-Pro-CH2OBn 29
3.18. Antifouling Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EDCI | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, |
| HOAt | 1-hydroxy-7-azabenzotriazole, |
| DMTMM | 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate, |
| PyBrop | bromotripyrrolidinophosphonium hexafluorophosphate. |
References
- Pettit, G.R.; Xu, J.-P.; Hogan, F.; Williams, M.D.; Doubek, D.L.; Schmidt, J.M.; Cerny, R.L.; Boyd, M.R. Isolation and Structure of the Human Cancer Cell Growth Inhibitory Cyclodepsipeptide Dolastatin 16. J. Nat. Prod. 1997, 60, 752–754. [Google Scholar] [CrossRef]
- Pettit, G.R.; Smith, T.H.; Xu, J.-P.; Herald, D.L.; Flahive, E.J.; Anderson, C.R.; Belcher, P.E.; Knight, J.C. Antineoplastic Agents. 590. X-ray Crystal Structure of Dolastatin 16 and Syntheses of the Dolamethylleuine and Dolaphenvaline Units. J. Nat. Prod. 2011, 74, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Smith, T.H.; Arce, P.M.; Flahive, E.J.; Anderson, C.R.; Chapuis, J.-C.; Xu, J.-P.; Groy, T.L.; Belcher, P.E.; Macdonald, C.B. Antineoplastic Agents. 599. Total Synthesis of Dolastatin 16. J. Nat. Prod. 2015, 78, 476–485. [Google Scholar] [CrossRef]
- Tan, L.K.; Goh, B.P.L.; Tripathi, A.; Lim, M.G.; Dickinson, G.H.; Lee, S.S.C.; Teo, S.L.M. Natural antifoulants from the marine cyanobacterium Lyngbya majuscule. Biofouling 2010, 26, 685–695. [Google Scholar] [CrossRef]
- Brooks, S.J.; Waldock, M. Copper Biocides in the Marine Environment. In Ecotoxicology of Antifouling Biocides; Arai, T., Harino, H., Ohji, M., Langston, W.J., Eds.; Springer: Tokyo, Japan, 2009; p. 414. [Google Scholar]
- Shimasaki, Y.; Kitano, T.; Oshima, Y.; Inoue, S.; Imada, N.; Honjo, T. Tributyltin causes masculinization in fish. Environ. Toxicol. Chem. 2003, 22, 141–144. [Google Scholar] [CrossRef]
- McAllister, B.G.; Kime, D.E. Early life exposure to environmental levels of the aromatase inhibitor tributyltin causes masculinisation and irreversible sperm damage in zebrafish (Danio rerio). Aquat. Toxicol. 2003, 65, 309–316. [Google Scholar] [CrossRef]
- Weis, J.S.; Perlmutter, J. Effects of Tributyltin on Activity and Burrowing Behavior of the Fiddler Crab, Uca pugilator. Estuaries 1987, 10, 342–346. [Google Scholar] [CrossRef]
- Weis, J.S.; Kim, K. Tributyltin is a teratogen in producing deformities in limbs of the fiddler crab,Uca pugilator. Arch. Environ. Contam. Toxicol. 1988, 17, 583–587. [Google Scholar] [CrossRef]
- Horiguchi, T.; Shiraishi, H.; Shimizu, M.; Yamazaki, S.; Morita, M. Imposex in Japanese gastropods (neogastropoda and mesogastropoda): Effects of tributyltin and triphenyltin from antifouling paints. Mar. Pollut. Bull. 1995, 31, 402–405. [Google Scholar] [CrossRef]
- Terlizzi, A.; Delos, A.L.; Garaventa, F.; Faimali, M.; Gerace, S. Limited effectiveness of marine protected areas: Imposex in Hexaplex trunculus (Gastropoda, Muricidae) populations from Italian marine reserves. Mar. Pollut. Bull. 2004, 48, 188–192. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Bryan, G.W. Reproductive Failure in Populations of the Dog-Whelk, Nucella Lapillus, Caused by Imposex Induced by Tributyltin from Antifouling Paints. J. Mar. Biol. Assoc. UK 1986, 66, 767–777. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Bryan, G.W. TBT-Induced Imposex in Neogastropod Snails: Masculinization to Mass Extinction. In Tributyltin: Case Study of an Environmental Contaminant; de Mora, S.J., Ed.; Cambridge University Press: Cambridge, UK, 1996; pp. 212–236. [Google Scholar]
- Horiguchi, T. Mechanism of Imposex Induced by Organotins in Gastropods. In Ecotoxicology of Antifouling Biocides; Arai, T., Harino, H., Ohji, M., Langston, W.J., Eds.; Springer: Tokyo, Japan, 2009; p. 111. [Google Scholar]
- Evans, S.M. TBT or not TBT?: That is the question. Biofouling 1999, 14, 117–129. [Google Scholar] [CrossRef]
- Konstantinou, I.K.; Albanis, T.A. Worldwide occurrence and effects of antifouling paint booster biocides in the aquatic environment: A review. Environ. Int. 2004, 30, 235–248. [Google Scholar] [CrossRef]
- Thomas, K.V.; Brooks, S. The Environmental Fate and Effects of Antifouling Paint Biocides. Biofouling 2010, 26, 73–88. [Google Scholar] [CrossRef]
- Kitano, Y.; Ito, T.; Suzuki, T.; Nogata, Y.; Shinshima, K.; Yoshimura, E.; Chiba, K.; Tada, M.; Sakaguchi, I. Synthesis and Antifouling Activity of 3-Isocyanotheonellin and Its Analogues. J. Chem. Soc. Perkin Trans. 2002, 1, 2251–2255. [Google Scholar] [CrossRef]
- Nogata, Y.; Kitano, Y.; Yoshimura, E.; Shinshima, K.; Sakaguchi, I. Antifouling Activity of Simple Synthetic Isocyanides Against Larvae of the Barnacle Balanus amphitrite. Biofouling 2004, 20, 87–91. [Google Scholar] [CrossRef]
- Kitano, Y.; Akima, C.; Yoshimura, E.; Nogata, Y. Anti-barnacle activity of novel simple alkyl isocyanides derived from citronellol. Biofouling 2011, 27, 201–205. [Google Scholar] [CrossRef]
- Kitano, Y.; Chiba, K.; Tada, M. Highly Efficient Conversion of Alcohols to Isocyanides. Synthesis 2001, 437–443. [Google Scholar] [CrossRef]
- Mihara, K.; Okada, I.; Chiba, K.; Kitano, Y. Facile Synthesis of N-Substituted Amides from Alkenes and Amides by a Brønsted Acid Mediated Electrophilic Addition Reaction. Synthesis 2014, 46, 1455–1462. [Google Scholar]
- Fukuda, T.; Wagatsuma, H.; Kominami, Y.; Nogata, Y.; Yoshimura, E.; Chiba, K.; Kitano, Y. Anti-barnacle Activity of Isocyanides Derived from Amino Acids. Chem. Biodivers. 2016, 13, 1502–1510. [Google Scholar] [CrossRef]
- Inoue, Y.; Takashima, S.; Nogata, Y.; Yoshimura, E.; Chiba, K.; Kitano, Y. Isocyanides Derived from α,α-Disubstituted Amino Acids: Synthesis and Antifouling Activity Assessment. Chem. Biodivers. 2018, 15, e1700571. [Google Scholar] [CrossRef]
- Takamura, H.; Ohashi, T.; Kikuchi, T.; Endo, N.; Fukuda, Y.; Kadota, I. Late-stage divergent synthesis and antifouling activity of geraniol–butenolide hybrid molecules. Org. Biomol. Chem. 2017, 15, 5549–5555. [Google Scholar] [CrossRef] [Green Version]
- Takamura, H.; Kikuchi, T.; Endo, N.; Fukuda, Y.; Kadota, I. Total Synthesis of Sarcophytonolide H and Isosarcophytonolide D: Structural Revision of Isosarcophytonolide D and Structure−Antifouling Activity Relationship of Sarcophytonolide H. Org. Lett. 2016, 18, 2110–2113. [Google Scholar] [CrossRef]
- Sjögren, M.; Johnson, A.-L.; Hedner, E.; Dahlström, M.; Göransson, U.; Shirani, H.; Bergman, J.; Jonsson, P.R.; Bohlin, L. Antifouling Activity of Synthesized Peptide Analogs of the Sponge Metabolite Barettin. Peptides 2006, 27, 2058–2064. [Google Scholar] [CrossRef]
- Qian, P.-Y.; Chen, L.; Xu, Y. Mini-review: Molecular Mechanisms of Antifouling Compounds. Biofouling 2013, 29, 381–400. [Google Scholar] [CrossRef]
- Liao, S.; Xu, Y.; Tang, Y.; Wang, J.; Zhou, X.; Xu, L.; Liu, Y. Design, Synthesis and Biological Evaluation of Soluble 2,5-Diketopiperazines Derivatives as Potential Antifouling Agents. RSC Adv. 2015, 5, 51020–51026. [Google Scholar] [CrossRef]
- Fusetani, N. Antifouling marine natural products. Nat. Prod. Rep. 2011, 28, 400–410. [Google Scholar] [CrossRef]
- Nishikawa, K.; Nakahara, H.; Shirokura, Y.; Nogata, Y.; Yoshimura, E.; Umezawa, T.; Okino, T.; Matsuda, F. Total Synthesis of 10-Isocyano-4-cadinene and Determination of Its Absolute Configuration. Org. Lett. 2010, 12, 904–907. [Google Scholar] [CrossRef]
- Nishikawa, K.; Nakahara, H.; Shirokura, Y.; Nogata, Y.; Yoshimura, E.; Umezawa, T.; Okino, T.; Matsuda, F. Total Synthesis of 10-Isocyano-4-cadinene and Its Stereoisomers and Evaluation of Antifouling Actitities. J. Org. Chem. 2011, 76, 6558–6573. [Google Scholar] [CrossRef]
- Nishikawa, K.; Umezawa, T.; Garson, M.J.; Matsuda, F. Confirmation of the Configurations of 10-Isothiocyano-4-cadinene through Synthesis. J. Nat. Prod. 2012, 75, 2232–2235. [Google Scholar] [CrossRef]
- Umezawa, T.; Oguri, Y.; Matsuura, H.; Yamazaki, S.; Suzuki, M.; Yoshimura, E.; Furuta, T.; Nogata, Y.; Serisawa, Y.; Matsuyama-Serisawa, K.; et al. Omaezallene from Red Alga Laurencia sp.: Structure Elucidation, Total Synthesis and Antifouling Activity. Angew. Chem. Int. Ed. 2014, 53, 3909–3912. [Google Scholar] [CrossRef]
- Umezawa, T.; Prakoso, N.I.; Kannaka, M.; Nogata, Y.; Yoshimura, E.; Okino, T.; Matsuda, F. Synthesis and Structure-Activity Relationship of Omaezallene Derivatives. Chem. Biodivers. 2019, 16, e1800451. [Google Scholar] [CrossRef]
- Umezawa, T.; Mizutani, N.; Matsuo, K.; Tokunaga, Y.; Matsuda, F.; Nehira, T. Assignment of Absolute Configuration of Bromoallenes by Vacuum-Ultraviolet Circular Dichroism (VUVCD). Molecules 2021, 26, 1296. [Google Scholar] [CrossRef]
- Umezawa, T.; Sato, A.; Ameda, Y.; Casalme, L.O.; Matsuda, F. Synthetic Study on Dolastatin 16: Concise and Scalable Synthesis of Two Unusual Amino Acid Units. Tetrahedron Lett. 2015, 56, 168–171. [Google Scholar] [CrossRef]
- Casalme, L.O.; Yamauchi, A.; Sato, A.; Petitbois, J.G.; Nogata, Y.; Yoshimura, E.; Okino, T.; Umezawa, T.; Matsuda, F. Total Synthesis and Biological Activity of Dolastatin 16. Org. Biomol. Chem. 2017, 15, 1140–1150. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Reddy, L.R.; Corey, E.J. Novel Acetoxylation and C−C Coupling Reactions at Unactivated Positions in α-Amino Acid Derivatives. Org. Lett. 2006, 8, 3391–3394. [Google Scholar] [CrossRef]
- Zhu, C.; Shen, X.; Nelson, S.G. Cinchona Alkaloid-Lewis Acid Catalyst Systems for Enantioselective Ketene−Aldehyde Cycloadditions. J. Am. Chem. Soc. 2004, 126, 5352–5353. [Google Scholar] [CrossRef]
- Shiina, I.; Kubota, M.; Ibuka, R. A Novel and Efficient Macrolactonization of ω-Hydroxycarboxylic Acids Using 2-Methyl-6-nitrobenzoic Anhydride (MNBA). Tetrahedron Lett. 2002, 43, 7535–7539. [Google Scholar] [CrossRef]
- Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. An Effective Use of Benzoic Anhydride and Its Derivatives for the Synthesis of Carboxylic Esters and Lactones: A Powerful and Convenient Mixed Anhydride Method Promoted by Basic Catalysts. J. Org. Chem. 2004, 69, 1822–1830. [Google Scholar] [CrossRef]
- Shiina, I.; Fukui, H.; Sasaki, A. Synthesis of Lactones Using Substituted Benzoic Anhydride as a Coupling Reagent. Nat. Protoc. 2007, 2, 2312–2317. [Google Scholar] [CrossRef]
- Narasaka, K.; Maruyama, K.; Mukaiyama, T. A Useful Method for the Synthesis of Macrocyclic Lactone. Chem. Lett. 1978, 7, 885–888. [Google Scholar] [CrossRef]
- Aurelio, L.; Brownlee, R.T.C.; Hughes, A.B. Solution-Phase Peptide Synthesis; Synthesis of ‘North-Western’ and ‘South-Eastern’ Fragments of the Antifungal Cyclodepsipeptide Petriellin A. Aust. J. Chem. 2008, 61, 615–629. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC50 (μg/mL) 1 | EC50 (μM) | LC50 (μg/mL) 2 |
|---|---|---|---|
| 1 3 | <0.03 | <0.03 | >10 |
| 2 3 | >10 | >17.0 | >10 |
| 3 3 | 1.17 | 1.92 | >10 |
| 24 | 1.74 | 1.86 | >10 |
| Boc-3 | 0.79 | 1.30 | >10 |
| 21 | 0.60 | 0.96 | >10 |
| 22 | 4.62 | 6.47 | >10 |
| 25 | 0.90 | 1.32 | >10 |
| 29 | 3.27 | 6.52 | >10 |
| CuSO4 3 | 0.10 | 0.63 | >10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casalme, L.O.; Katayama, K.; Hayakawa, Y.; Nakamura, K.; Yamauchi, A.; Nogata, Y.; Yoshimura, E.; Matsuda, F.; Umezawa, T. Effective Synthesis and Antifouling Activity of Dolastatin 16 Derivatives. Mar. Drugs 2022, 20, 124. https://doi.org/10.3390/md20020124
Casalme LO, Katayama K, Hayakawa Y, Nakamura K, Yamauchi A, Nogata Y, Yoshimura E, Matsuda F, Umezawa T. Effective Synthesis and Antifouling Activity of Dolastatin 16 Derivatives. Marine Drugs. 2022; 20(2):124. https://doi.org/10.3390/md20020124
Chicago/Turabian StyleCasalme, Loida O., Keisuke Katayama, Yoshiki Hayakawa, Kensuke Nakamura, Arisa Yamauchi, Yasuyuki Nogata, Erina Yoshimura, Fuyuhiko Matsuda, and Taiki Umezawa. 2022. "Effective Synthesis and Antifouling Activity of Dolastatin 16 Derivatives" Marine Drugs 20, no. 2: 124. https://doi.org/10.3390/md20020124
APA StyleCasalme, L. O., Katayama, K., Hayakawa, Y., Nakamura, K., Yamauchi, A., Nogata, Y., Yoshimura, E., Matsuda, F., & Umezawa, T. (2022). Effective Synthesis and Antifouling Activity of Dolastatin 16 Derivatives. Marine Drugs, 20(2), 124. https://doi.org/10.3390/md20020124