
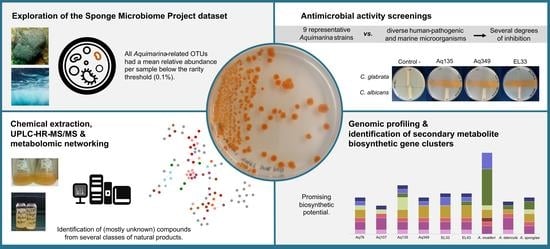
Insights into the Antimicrobial Activities and Metabolomes of Aquimarina (Flavobacteriaceae, Bacteroidetes) Species from the Rare Marine Biosphere

 ,
,  ,
,  ,
,  ,
,  , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
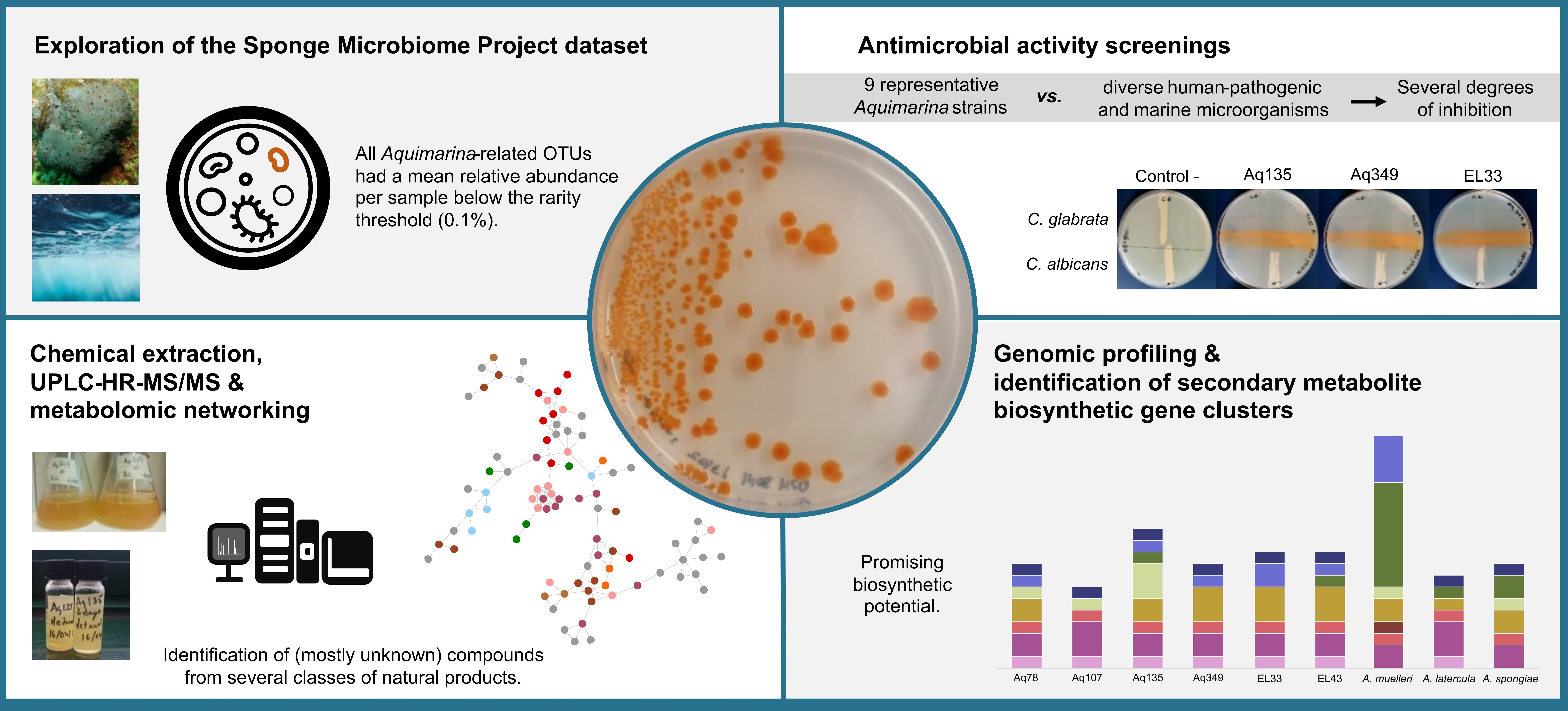
2.1. Abundance Distributions of Aquimarina spp. across Marine Biotopes
2.2. Diversity and Relatedness of Aquimarina OTUs
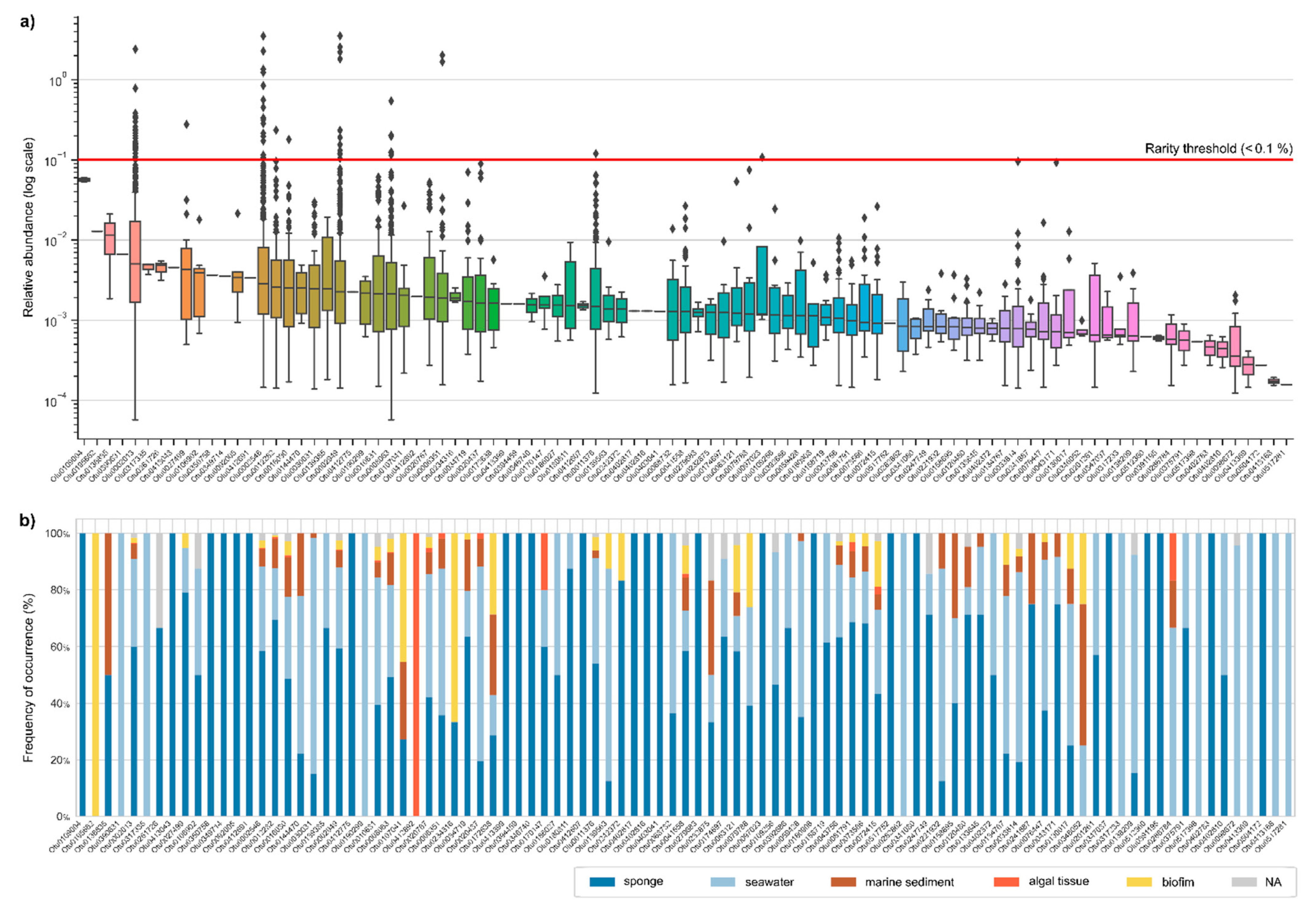
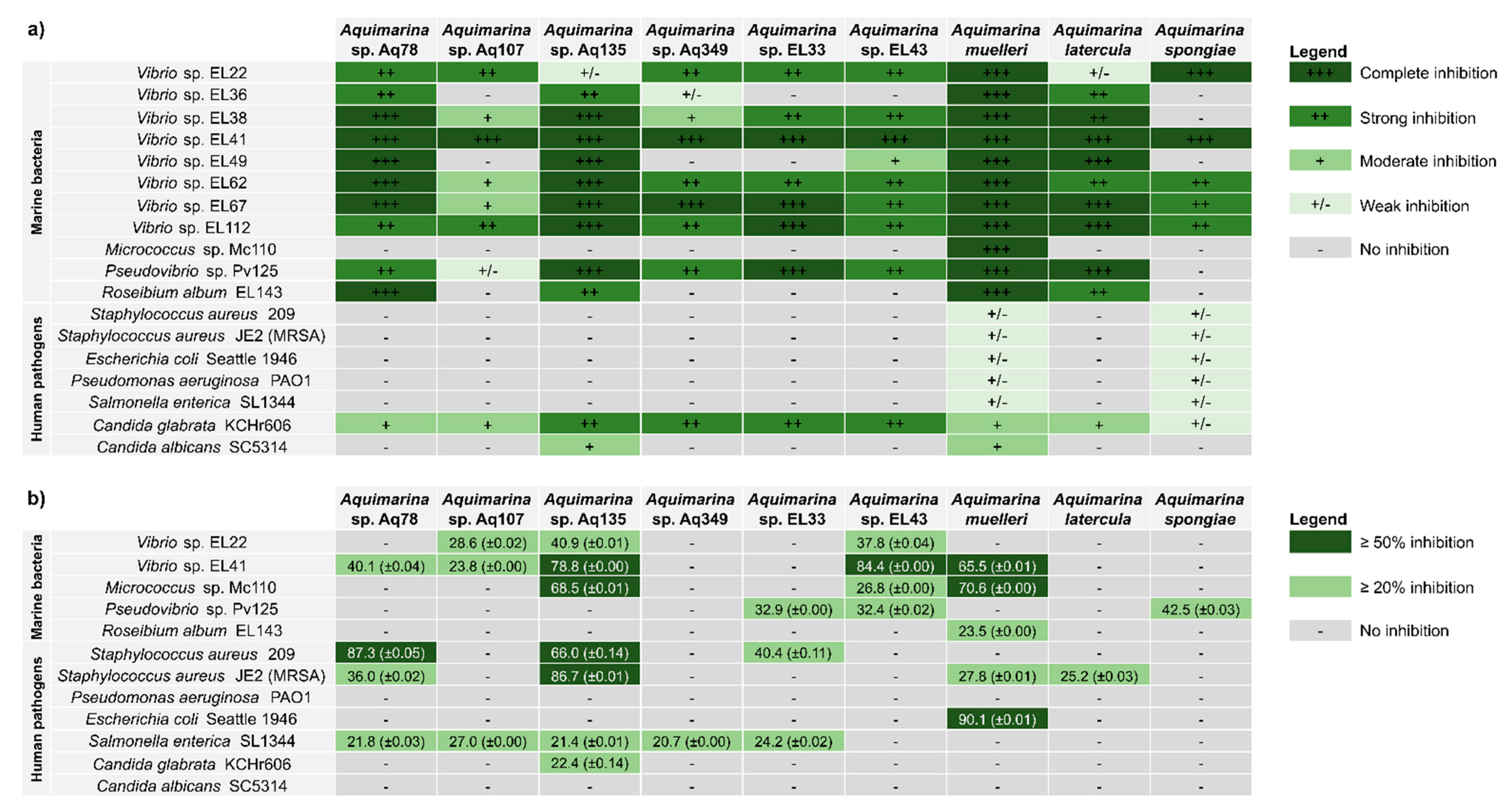
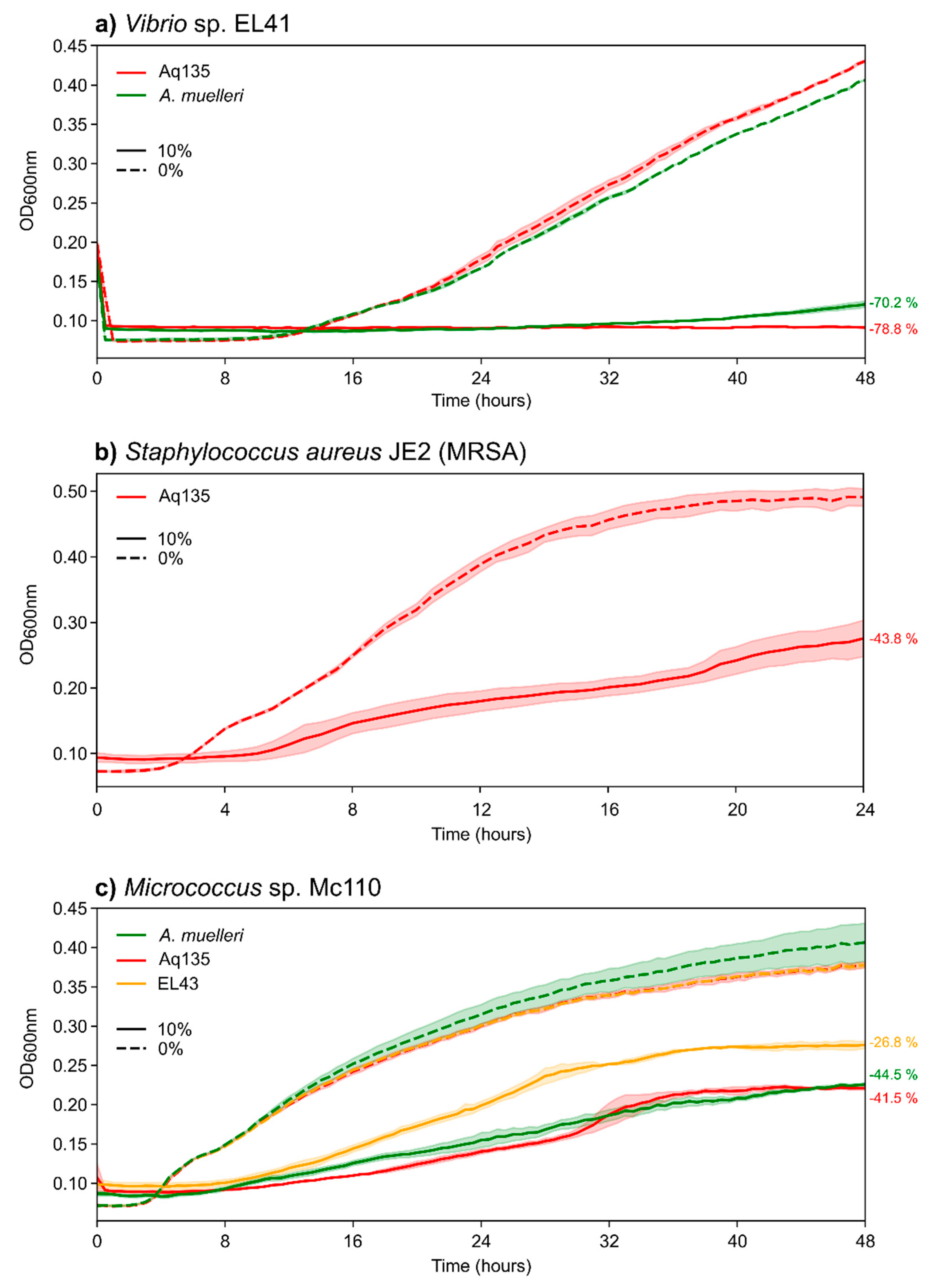
2.3. Antimicrobial Activities of Aquimarina spp.
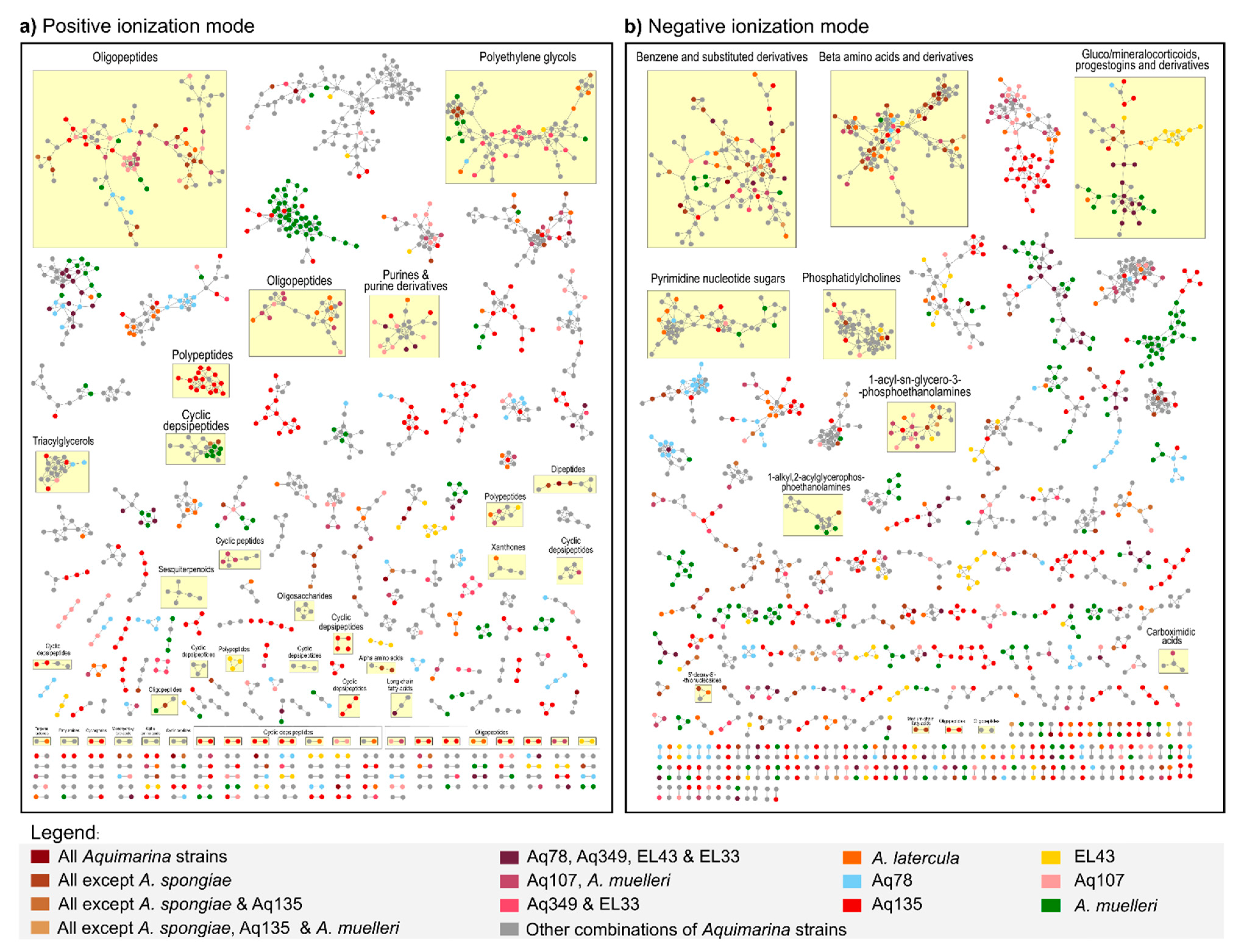
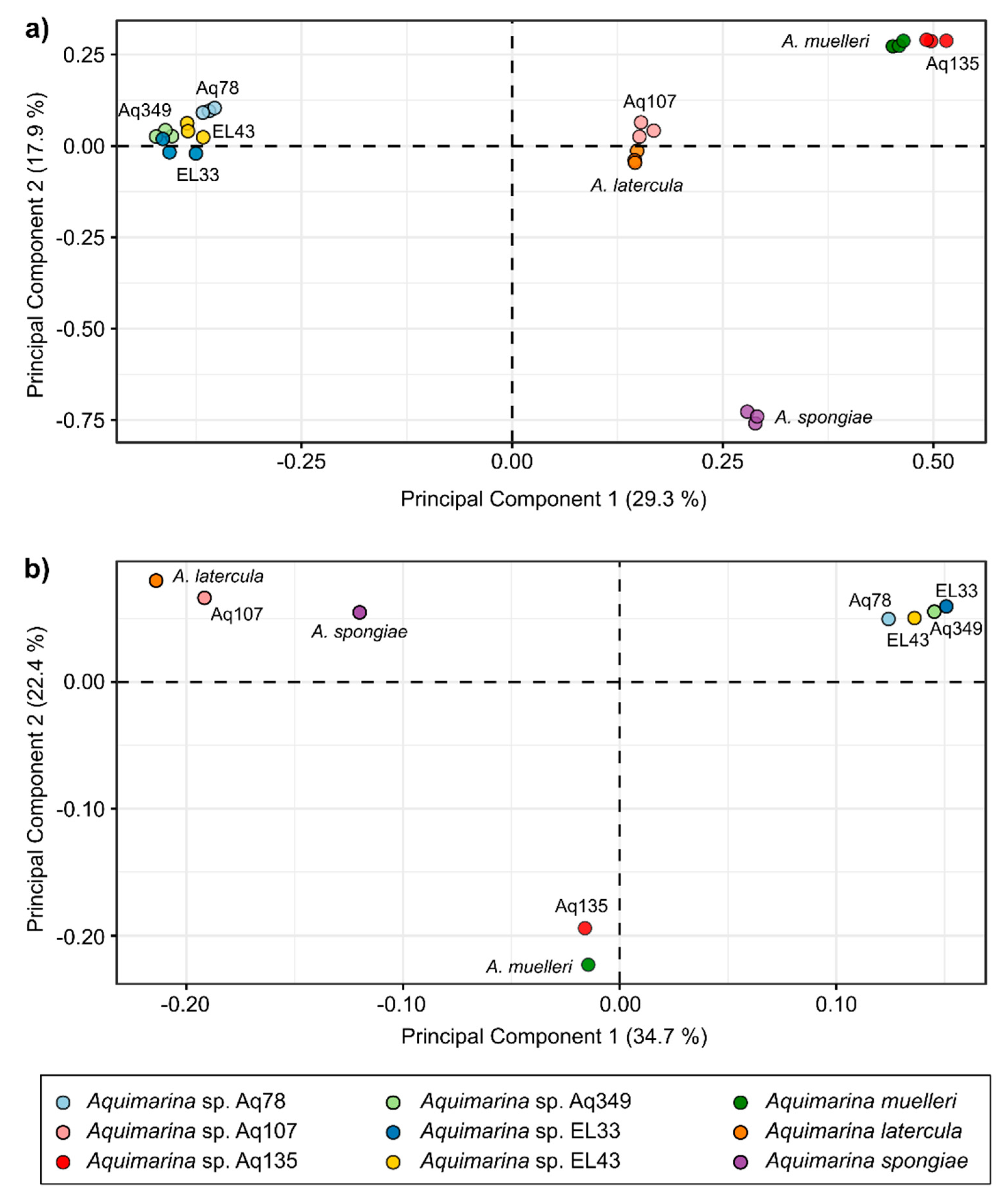
2.4. Liquid Chromatography-Mass Spectrometry (LC-MS)-Based Metabolomics Analysis of Aquimarina Extracts
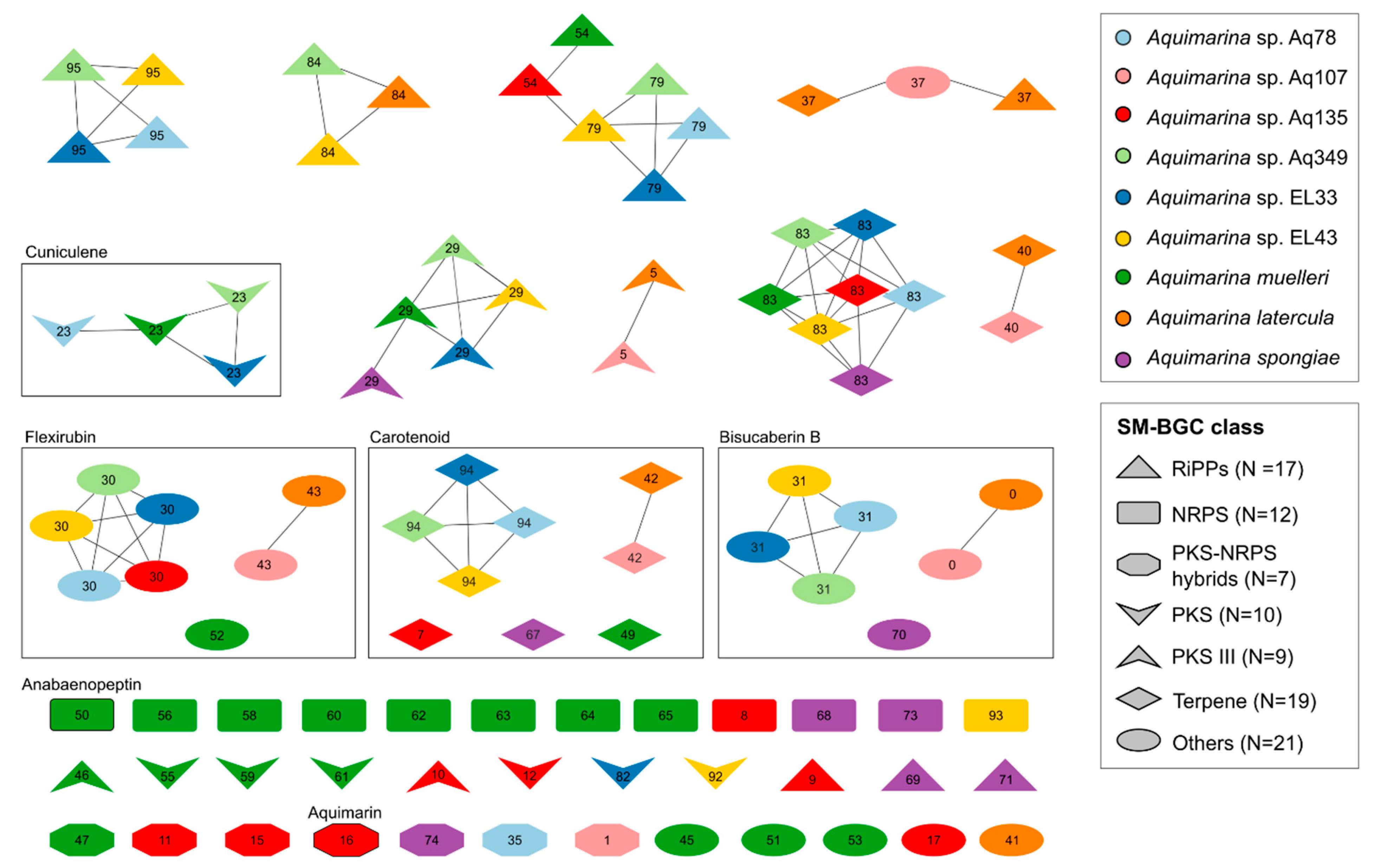
2.5. SM-BGC Identification on Aquimarina Genomes
3. Discussion
3.1. Aquimarina Is a Member of the Microbial Rare Biosphere
3.2. Aquimarina Strains Inhibit Other Marine Bacteria
3.3. Aquimarina as a Source of Novel Inhibitory Compounds against Human-Pathogenic Bacteria and Yeast
3.4. Aquimarina Bioactivity Profiles Change According to Experimental Conditions
3.5. Metabolomics Sheds Light on the Unknown Aquimarina Chemical Space and Indicates Presence of Novel, Cyclic Depsipeptide-Related Compounds
3.6. Metabolomics Analysis of Aquimarina Extracts Highlights Phylogenetic Relationships
3.7. Long-Read Sequencing of Aquimarina Genomes Reveals Full Biosynthetic Potential
4. Materials and Methods
4.1. Exploring Abundance Distributions of Aquimarina spp. in the Marine Environment
4.2. Strains and Cultivation Conditions
4.2.1. Aquimarina Strains
4.2.2. Test Strains Used in Antimicrobial Assays
4.3. Cross-Streak Assays
4.4. Preparation of Extracellular Metabolite (Crude) Extracts from Aquimarina Strains
4.5. Broth Microdilution Assays
4.6. Metabolomic Analyses of Aquimarina spp.
4.6.1. UPLC-HR-MS/MS Profiling of Aquimarina Extracts
4.6.2. Metabolomic Data Processing and Molecular Network Analyses
4.7. PacBio Genome Sequencing of Aquimarina Strains
4.8. Genome Annotation and SM-BGC Identification
4.9. Statistical Analyses and Data Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lynch, M.D.J.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 2015, 13, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Hardoim, C.C.P.; Cardinale, M.; Cúcio, A.C.B.; Esteves, A.I.S.; Berg, G.; Xavier, J.R.; Cox, C.J.; Costa, R. Effects of sample handling and cultivation bias on the specificity of bacterial communities in keratose marine sponges. Front. Microbiol. 2014, 5, 611. [Google Scholar] [CrossRef]
- Shade, A.; Hogan, C.S.; Klimowicz, A.K.; Linske, M.; McManus, P.S.; Handelsman, J. Culturing captures members of the soil rare biosphere. Environ. Microbiol. 2012, 14, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Keller-Costa, T.; Eriksson, D.; Gonçalves, J.M.S.; Gomes, N.C.M.; Lago-Lestón, A.; Costa, R. The gorgonian coral Eunicella labiata hosts a distinct prokaryotic consortium amenable to cultivation. FEMS Microbiol. Ecol. 2017, 93, fix143. [Google Scholar] [CrossRef] [PubMed]
- Karimi, E.; Keller-Costa, T.; Slaby, B.M.; Cox, C.J.; da Rocha, U.N.; Hentschel, U.; Costa, R. Genomic blueprints of sponge-prokaryote symbiosis are shared by low abundant and cultivatable Alphaproteobacteria. Sci. Rep. 2019, 9, 1999. [Google Scholar] [CrossRef]
- Pascoal, F.; Costa, R.; Magalhaes, C. The microbial rare biosphere: Current concepts, methods and ecological principles. FEMS Microbiol. Ecol. 2021, 97, fiaa227. [Google Scholar] [CrossRef]
- Jensen, P.R.; Moore, B.S.; Fenical, W. The marine actinomycete genus Salinispora: A model organism for secondary metabolite discovery. Nat. Prod. Rep. 2015, 32, 738–751. [Google Scholar] [CrossRef] [Green Version]
- Subramani, R.; Sipkema, D. Marine rare actinomycetes: A promising source of structurally diverse and unique novel natural products. Mar. Drugs 2019, 17, 249. [Google Scholar] [CrossRef] [Green Version]
- Nedashkovskaya, O.I. Description of Aquimarina muelleri gen. nov., sp. nov., and proposal of the reclassification of [Cytophaga] latercula Lewin 1969 as Stanierella latercula gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2005, 55, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.-J.; You, H.-S.; Lee, D.-H.; Oh, D.-C. Aquimarina spongiae sp. nov., isolated from marine sponge Halichondria oshoro. Int. J. Syst. Evol. Microbiol. 2011, 61, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.; Margassery, L.M.; O’Leary, N.D.; O’Gara, F.; Morrissey, J.; Dobson, A.D.W. Aquimarina amphilecti sp. nov., isolated from the sponge Amphilectus fucorum. Int. J. Syst. Evol. Microbiol. 2014, 64, 501–505. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.-X.; Wang, C.; Du, Z.-J.; Chen, G.-J. Aquimarina agarivorans sp. nov., a genome-sequenced member of the class Flavobacteriia isolated from Gelidium amansii. Int. J. Syst. Evol. Microbiol. 2015, 65, 2684–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Lu, G.; Zheng, Y.; Xie, W.; Li, S.; Hu, Z. Aquimarina agarilytica sp. nov., an agarolytic species isolated from a red alga. Int. J. Syst. Evol. Microbiol. 2012, 62, 869–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller-Costa, T.; Silva, R.; Lago-Lestón, A.; Costa, R. Genomic insights into Aquimarina sp. strain EL33, a bacterial symbiont of the gorgonian coral Eunicella labiata. Genome Announc. 2016, 4, e00855-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.-S.; Kahng, H.-Y.; Lee, Y.S.; Yoon, B.-J.; Lim, S.-B.; Jung, J.S.; Oh, D.-C.; Lee, D.-H. Aquimarina litoralis sp. nov., isolated from a coastal seawater. J. Microbiol. 2010, 48, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Choi, B.G.; Kim, J.Y.; Roh, S.W.; Lee, S.J. Aquimarina seongsanensis sp. nov., isolated from sea water. Antonie Van Leeuwenhoek 2017, 110, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zhang, Z.; Fan, X.; Shi, X.; Zhang, X.-H. Aquimarina megaterium sp. nov., isolated from seawater. Int. J. Syst. Evol. Microbiol. 2014, 64, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.-N.; Zhou, L.-Y.; Li, Y.-X.; Du, Z.-J. Aquimarina sediminis sp. nov., isolated from coastal sediment. Antonie Van Leeuwenhoek 2018, 111, 2257–2265. [Google Scholar] [CrossRef]
- Miyazaki, M.; Nagano, Y.; Fujiwara, Y.; Hatada, Y.; Nogi, Y. Aquimarina macrocephali sp. nov., isolated from sediment adjacent to sperm whale carcasses. Int. J. Syst. Evol. Microbiol. 2010, 60, 2298–2302. [Google Scholar] [CrossRef]
- Esteves, A.I.S.; Hardoim, C.C.P.; Xavier, J.R.; Gonçalves, J.M.S.; Costa, R. Molecular richness and biotechnological potential of bacteria cultured from Irciniidae sponges in the north-east Atlantic. FEMS Microbiol. Ecol. 2013, 85, 519–536. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez Jimenez, A.; Dechamps, E.; Giaux, A.; Goetghebuer, L.; Bauwens, M.; Willenz, P.; Flahaut, S.; Laport, M.S.; George, I.F. The sponges Hymeniacidon perlevis and Halichondria panicea are reservoirs of antibiotic-producing bacteria against multi-drug resistant Staphylococcus aureus. J. Appl. Microbiol. 2021, 131, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, E.J.N.; Ueoka, R.; Dolev, A.; Rust, M.; Meoded, R.A.; Bhushan, A.; Califano, G.; Costa, R.; Gugger, M.; Steinbeck, C.; et al. Automated structure prediction of trans-acyltransferase polyketide synthase products. Nat. Chem. Biol. 2019, 15, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, C.L.; Probst, S.I.; Ueoka, R.; Sandu, I.; Schäfle, D.; Molin, M.D.; Minas, H.A.; Costa, R.; Oxenius, A.; Sander, P.; et al. Aquimarins, peptide antibiotics with amino-modified C-termini from a sponge-derived Aquimarina sp. bacterium. Angew. Chem. Int. Ed. 2021, 61, e202115802. [Google Scholar] [CrossRef]
- Silva, S.G.; Blom, J.; Keller-Costa, T.; Costa, R. Comparative genomics reveals complex natural product biosynthesis capacities and carbon metabolism across host-associated and free-living Aquimarina (Bacteroidetes, Flavobacteriaceae) species. Environ. Microbiol. 2019, 21, 4002–4019. [Google Scholar] [CrossRef]
- Moitinho-Silva, L.; Nielsen, S.; Amir, A.; Gonzalez, A.; Ackermann, G.L.; Cerrano, C.; Astudillo-Garcia, C.; Easson, C.; Sipkema, D.; Liu, F.; et al. The sponge microbiome project. GigaScience 2017, 6, gix077. [Google Scholar] [CrossRef]
- Nedashkovskaya, O.I.; Vancanneyt, M.; Christiaens, L.; Kalinovskaya, N.I.; Mikhailov, V.V.; Swings, J. Aquimarina intermedia sp. nov., reclassification of Stanierella latercula (Lewin 1969) as Aquimarina latercula comb. nov. and Gaetbulimicrobium brevivitae Yoon et al. 2006 as Aquimarina brevivitae comb. nov. and emended description of the genus Aquimarina. Int. J. Syst. Evol. Microbiol. 2006, 56, 2037–2041. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [Green Version]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Ernst, M.; Kang, K.B.; Caraballo-Rodríguez, A.M.; Nothias, L.-F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. MolNetEnhancer: Enhanced molecular networks by integrating metabolome mining and annotation tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef] [Green Version]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Navarro-Munoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Pita, L.; Fraune, S.; Hentschel, U. Emerging sponge models of animal-microbe symbioses. Front. Microbiol. 2016, 7, 2102. [Google Scholar] [CrossRef] [PubMed]
- de Voogd, N.J.; Alvarez, B.; Boury-Esnault, N.; Carballo, J.L.; Cárdenas, P.; Díaz, M.-C.; Dohrmann, M.; Downey, R.; Hajdu, E.; Hooper, J.N.A.; et al. World Porifera Database. Sarcotragus spinosulus Schmidt. 1862. Available online: https://www.marinespecies.org/aphia.php?p=taxdetails&id=165086 (accessed on 22 March 2022).
- de Voogd, N.J.; Alvarez, B.; Boury-Esnault, N.; Carballo, J.L.; Cárdenas, P.; Díaz, M.-C.; Dohrmann, M.; Downey, R.; Hajdu, E.; Hooper, J.N.A.; et al. World Porifera Database. Ircinia variabilis var. typica Nardo. 1847. Available online: https://www.marinespecies.org/aphia.php?p=taxdetails&id=174753 (accessed on 22 March 2022).
- Mysara, M.; Vandamme, P.; Props, R.; Kerckhof, F.-M.; Leys, N.; Boon, N.; Raes, J.; Monsieurs, P. Reconciliation between operational taxonomic units and species boundaries. FEMS Microbiol. Ecol. 2017, 93, fix029. [Google Scholar] [CrossRef] [PubMed]
- Ooi, M.C.; Goulden, E.F.; Trotter, A.J.; Smith, G.G.; Bridle, A.R. Aquimarina sp. associated with a cuticular disease of cultured larval Palinurid and Scyllarid lobsters. Front. Microbiol. 2020, 11, 573588. [Google Scholar] [CrossRef]
- Tully, B.J.; Graham, E.D.; Heidelberg, J.F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 2018, 5, 170203. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Mao, Y.; Du, G.; Li, X.; Tang, X. On microbial community of Pyropia haitanensis by metagenomic analysis. J. Oceanol. Limnol. 2020, 39, 1091–1102. [Google Scholar] [CrossRef]
- Keller-Costa, T.; Kozma, L.; Silva, S.G.; Toscan, R.; Gonçalves, J.; Lago-Lestón, A.; Kyrpides, N.C.; Rocha, U.N.; Costa, R. Evidence for Cross-Feeding, Metabolic Specialization, and Niche Partitioning in the Octocoral Holobiont. 2022. Available online: https://www.researchsquare.com/article/rs-1630933/v1 (accessed on 10 May 2022).
- Kumar, V.; Zozaya-Valdes, E.; Kjelleberg, S.; Thomas, T.; Egan, S. Multiple opportunistic pathogens can cause a bleaching disease in the red seaweed Delisea pulchra. Environ. Microbiol. 2016, 18, 3962–3975. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Shimizu, T.; Sanda, T.; Hamasaki, K.; Dan, S.; Lal, M.T.B.M.; Kato, G.; Sano, M. Characterization of Aquimarina hainanensis isolated from diseased mud crab Scylla serrata larvae in a hatchery. J. Fish Dis. 2020, 43, 541–549. [Google Scholar] [CrossRef]
- Keller-Costa, T.; Lago-Lestón, A.; Saraiva, J.P.; Toscan, R.; Silva, S.G.; Gonçalves, J.; Cox, C.J.; Kyrpides, N.; Nunes da Rocha, U.; Costa, R. Metagenomic insights into the taxonomy, function, and dysbiosis of prokaryotic communities in octocorals. Microbiome 2021, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, I.; Silva, R.; Meunier, L.; Valente, S.M.; Lago-Lestón, A.; Keller-Costa, T.; Costa, R. Functional metagenomics reveals differential chitin degradation and utilization features across free-living and host-associated marine microbiomes. Microbiome 2021, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Baker-Austin, C.; Oliver, J.D. Vibrio vulnificus: New insights into a deadly opportunistic pathogen. Environ. Microbiol. 2018, 20, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza Valente, C.; Wan, A.H.L. Vibrio and major commercially important vibriosis diseases in decapod crustaceans. J. Invertebr. Pathol. 2021, 181, 107527. [Google Scholar] [CrossRef] [PubMed]
- Ina-Salwany, M.Y.; Al-saari, N.; Mohamad, A.; Mursidi, F.-A.; Mohd-Aris, A.; Amal, M.N.A.; Kasai, H.; Mino, S.; Sawabe, T.; Zamri-Saad, M. Vibriosis in fish: A review on disease development and prevention. J. Aquat. Anim. Health 2018, 31, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Corzett, C.H.; Elsherbini, J.; Chien, D.M.; Hehemann, J.-H.; Henschel, A.; Preheim, S.P.; Yu, X.; Alm, E.J.; Polz, M.F.; DiRita, V.J. Evolution of a vegetarian Vibrio: Metabolic specialization of Vibrio breoganii to macroalgal substrates. J. Bacteriol. 2018, 200, e00020-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.S.; Gigante, V.; Sati, H.; Paulin, S.; Al-Sulaiman, L.; Rex, J.H.; Fernandes, P.; Arias, C.A.; Paul, M.; Thwaites, G.E.; et al. Analysis of the clinical pipeline of treatments for drug-resistant bacterial infections: Despite progress, more action is needed. Antimicrob. Agents Chemother. 2022, 66, e01991-21. [Google Scholar] [CrossRef]
- Olson, M.L.; Jayaraman, A.; Kao, K.C.; McBain, A.J. Relative abundances of Candida albicans and Candida glabrata in in vitro coculture biofilms impact biofilm structure and formation. Appl. Environ. Microbiol. 2018, 84, e02769-17. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.D.; Iqbal, N.; Bolden, C.B.; Kuykendall, R.J.; Harrison, L.H.; Farley, M.M.; Schaffner, W.; Beldavs, Z.G.; Chiller, T.M.; Park, B.J.; et al. Role of FKS mutations in Candida glabrata: MIC values, echinocandin resistance, and multidrug resistance. Antimicrob. Agents Chemother. 2014, 58, 4690–4696. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 2022. [Google Scholar] [CrossRef]
- Ksiezopolska, E.; Gabaldón, T. Evolutionary emergence of drug resistance in Candida opportunistic pathogens. Genes 2018, 9, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore Nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Mouton, J.W.; Meletiadis, J.; Voss, A.; Turnidge, J. Variation of MIC measurements: The contribution of strain and laboratory variability to measurement precision. J. Antimicrob. Chemother. 2018, 73, 2374–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombach, M.; Ochoa, C.; Maurer, F.P.; Pfiffner, T.; Böttger, E.C.; Furrer, R. Relative contribution of biological variation and technical variables to zone diameter variations of disc diffusion susceptibility testing. J. Antimicrob. Chemother. 2016, 71, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.; Süssmuth, R.D. Bioactive peptide natural products as lead structures for medicinal use. Acc. Chem. Res. 2017, 50, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Hosotani, N.; Kumagai, K.; Nakagawa, H.; Shimatani, T.; Saji, I. Antimycins A10∼A16, seven new antimycin antibiotics produced by Streptomyces spp. SPA-10191 and SPA-8893. J. Antibiot. 2005, 58, 460–467. [Google Scholar] [CrossRef]
- Dose, B.; Niehs, S.P.; Scherlach, K.; Flórez, L.V.; Kaltenpoth, M.; Hertweck, C. Unexpected bacterial origin of the antibiotic icosalide: Two-tailed depsipeptide assembly in multifarious Burkholderia symbionts. ACS Chem. Biol. 2018, 13, 2414–2420. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, X.; Kim, S.J.; Zhang, W. Antimycin-type depsipeptides: Discovery, biosynthesis, chemical synthesis, and bioactivities. Nat. Prod. Rep. 2016, 33, 1146–1165. [Google Scholar] [CrossRef]
- Challis, G.L.; Hopwood, D.A. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc. Natl. Acad. Sci. USA 2003, 100, 14555–14561. [Google Scholar] [CrossRef] [Green Version]
- Mrak, P.; Krastel, P.; Pivk Lukančič, P.; Tao, J.; Pistorius, D.; Moore, C.M. Discovery of the actinoplanic acid pathway in Streptomyces rapamycinicus reveals a genetically conserved synergism with rapamycin. J. Biol. Chem. 2018, 293, 19982–19995. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Wang, M.; Leber, C.A.; Nothias, L.-F.; Reher, R.; Kang, K.B.; van der Hooft, J.J.J.; Dorrestein, P.C.; Gerwick, W.H.; Cottrell, G.W. NPClassifier: A deep neural network-based structural classification tool for natural products. J. Nat. Prod. 2021, 84, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminf. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, P.R.; do Amaral, S.C.; Siqueira, A.S.; Xavier, L.P.; Santos, A.V. Anabaenopeptins: What we know so far. Toxins 2021, 13, 522. [Google Scholar] [CrossRef]
- Entfellner, E.; Frei, M.; Christiansen, G.; Deng, L.; Blom, J.; Kurmayer, R. Evolution of anabaenopeptin peptide structural variability in the Cyanobacterium Planktothrix. Front. Microbiol. 2017, 8, 219. [Google Scholar] [CrossRef] [Green Version]
- Klassen, J.L.; Currie, C.R. Gene fragmentation in bacterial draft genomes: Extent, consequences and mitigation. BMC Genom. 2012, 13, 14. [Google Scholar] [CrossRef] [Green Version]
- Chase, A.B.; Sweeney, D.; Muskat, M.N.; Guillén-Matus, D.G.; Jensen, P.R.; Ravel, J. Vertical inheritance facilitates interspecies diversification in biosynthetic gene clusters and specialized metabolites. mBio 2021, 12, e02700-21. [Google Scholar] [CrossRef]
- Meleshko, D.; Mohimani, H.; Tracanna, V.; Hajirasouliha, I.; Medema, M.H.; Korobeynikov, A.; Pevzner, P.A. BiosyntheticSPAdes: Reconstructing biosynthetic gene clusters from assembly graphs. Genome Res. 2019, 29, 1352–1362. [Google Scholar] [CrossRef]
- Thomas, T.; Moitinho-Silva, L.; Lurgi, M.; Björk, J.R.; Easson, C.; Astudillo-García, C.; Olson, J.B.; Erwin, P.M.; López-Legentil, S.; Luter, H.; et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 2016, 7, 11870. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity George, M.; Tiedje James, M.; Cole James, R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Battistuzzi, F.U. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T.-a. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Ueno, K.; Uno, J.; Nakayama, H.; Sasamoto, K.; Mikami, Y.; Chibana, H. Development of a highly efficient gene targeting system induced by transient repression of YKU80 expression in Candida glabrata. Eukaryot. Cell 2007, 6, 1239–1247. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.; Cavalheiro, M.; Costa, C.; Takahashi-Nakaguchi, A.; Okamoto, M.; Chibana, H.; Teixeira, M.C. Screening the drug: H+ antiporter family for a role in biofilm formation in Candida glabrata. Front. Cell. Infect. Microbiol. 2020, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Keller-Costa, T.; Hubbard, P.C.; Paetz, C.; Nakamura, Y.; da Silva, J.P.; Rato, A.; Barata, E.N.; Schneider, B.; Canario, A.V.M. Identity of a tilapia pheromone released by dominant males that primes females for reproduction. Curr. Biol. 2014, 24, 2130–2135. [Google Scholar] [CrossRef] [Green Version]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; CLSI Standard M07; Clinical and Laboratory Standard Institute: Wayne, PA, USA, 2018; Volume 90, pp. 6314–6322. [Google Scholar]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Klepser, M.E.; Ernst, E.J.; Lewis, R.E.; Ernst, M.E.; Pfaller, M.A. Influence of test conditions on antifungal time-kill curve results: Proposal for standardized methods. Antimicrob. Agents Chemother. 1998, 42, 1207–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, C.Q.V.; Afonso, I.I.; Lage, S.; Costa, P.R.; Canário, A.V.M.; Da Silva, J.P. Quantitation overcoming matrix effects of lipophilic toxins in Mytilus galloprovincialis by liquid chromatography-full scan high resolution mass spectrometry analysis (LC-HR-MS). Mar. Drugs 2022, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Mohimani, H.; Gurevich, A.; Mikheenko, A.; Garg, N.; Nothias, L.-F.; Ninomiya, A.; Takada, K.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem. Biol. 2016, 13, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Mikheenko, A.; Shlemov, A.; Korobeynikov, A.; Mohimani, H.; Pevzner, P.A. Increased diversity of peptidic natural products revealed by modification-tolerant database search of mass spectra. Nat. Microbiol. 2018, 3, 319–327. [Google Scholar] [CrossRef]
- Mohimani, H.; Gurevich, A.; Shlemov, A.; Mikheenko, A.; Korobeynikov, A.; Cao, L.; Shcherbin, E.; Nothias, L.-F.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of microbial metabolites through database search of mass spectra. Nat. Commun. 2018, 9, 4035. [Google Scholar] [CrossRef] [Green Version]
- van der Hooft, J.J.J.; Wandy, J.; Barrett, M.P.; Burgess, K.E.V.; Rogers, S. Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. USA 2016, 113, 13738–13743. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kucera, M.; Isserlin, R.; Arkhangorodsky, A.; Bader, G.D. AutoAnnotate: A Cytoscape app for summarizing networks with semantic annotations. F1000Research 2016, 5, 1717. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K.; et al. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2017, 15, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen rearrangement rules: Computational MS/MS fragmentation and structure elucidation using MS-FINDER software. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef] [PubMed]
- Fraisier-Vannier, O.; Chervin, J.; Cabanac, G.; Puech, V.; Fournier, S.; Durand, V.; Amiel, A.; André, O.; Benamar, O.A.; Dumas, B.; et al. MS-CleanR: A feature-filtering workflow for untargeted LC–MS based metabolomics. Anal. Chem. 2020, 92, 9971–9981. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. BBMap: A fast, accurate, splice-aware aligner. In Proceedings of the 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, USA, 17–20 March 2014. [Google Scholar]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.R.; et al. Vegan: Community Ecology Package. Available online: https://CRAN.R-project.org/package=vegan (accessed on 12 January 2022).
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 12 January 2022).
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, S.G.; Paula, P.; da Silva, J.P.; Mil-Homens, D.; Teixeira, M.C.; Fialho, A.M.; Costa, R.; Keller-Costa, T. Insights into the Antimicrobial Activities and Metabolomes of Aquimarina (Flavobacteriaceae, Bacteroidetes) Species from the Rare Marine Biosphere. Mar. Drugs 2022, 20, 423. https://doi.org/10.3390/md20070423
Silva SG, Paula P, da Silva JP, Mil-Homens D, Teixeira MC, Fialho AM, Costa R, Keller-Costa T. Insights into the Antimicrobial Activities and Metabolomes of Aquimarina (Flavobacteriaceae, Bacteroidetes) Species from the Rare Marine Biosphere. Marine Drugs. 2022; 20(7):423. https://doi.org/10.3390/md20070423
Chicago/Turabian StyleSilva, Sandra Godinho, Patrícia Paula, José Paulo da Silva, Dalila Mil-Homens, Miguel Cacho Teixeira, Arsénio Mendes Fialho, Rodrigo Costa, and Tina Keller-Costa. 2022. "Insights into the Antimicrobial Activities and Metabolomes of Aquimarina (Flavobacteriaceae, Bacteroidetes) Species from the Rare Marine Biosphere" Marine Drugs 20, no. 7: 423. https://doi.org/10.3390/md20070423
APA StyleSilva, S. G., Paula, P., da Silva, J. P., Mil-Homens, D., Teixeira, M. C., Fialho, A. M., Costa, R., & Keller-Costa, T. (2022). Insights into the Antimicrobial Activities and Metabolomes of Aquimarina (Flavobacteriaceae, Bacteroidetes) Species from the Rare Marine Biosphere. Marine Drugs, 20(7), 423. https://doi.org/10.3390/md20070423