
Therapeutic Potential of Marine-Derived Cyclic Peptides as Antiparasitic Agents

 ,
,  , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Marine-Derived Cyclic Peptides in Drug Discovery
3. Marine-Derived Cyclic Peptides with Antiparasitic Effects
3.1. Malaria
3.2. Leishmaniasis
3.3. Trypanosomiasis
3.4. Final Remarks
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Renslo, A.R.; McKerrow, J.H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710. [Google Scholar] [CrossRef]
- Lee, S.-M.; Kim, M.-S.; Hayat, F.; Shin, D. Recent Advances in the Discovery of Novel Antiprotozoal Agents. Molecules 2019, 24, 3886. [Google Scholar] [CrossRef]
- Pink, R.; Hudson, A.; Mouriès, M.-A.; Bendig, M. Opportunities and Challenges in Antiparasitic Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Picot, S.; Beugnet, F.; Leboucher, G.; Bienvenu, A.-L. Drug resistant parasites and fungi from a one-health perspective: A global concern that needs transdisciplinary stewardship programs. One Health 2022, 14, 100368. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H. Designing Drugs for Parasitic Diseases of the Developing World. PLoS Med. 2005, 2, e210. [Google Scholar] [CrossRef] [PubMed]
- Goupil, L.S.; McKerrow, J.H. Introduction: Drug Discovery and Development for Neglected Diseases. Chem. Rev. 2014, 114, 11131–11137. [Google Scholar] [CrossRef]
- Cummings, R.D.; Hokke, C.H.; Haslam, S.M. Parasitic Infections. In Essentials of Glycobiology, 4th ed.; Harbor, C.S., Ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2022. [Google Scholar]
- Lacerda, A.F.; Pelegrini, P.B.; de Oliveira, D.M.; Vasconcelos, É.A.R.; Grossi-de-Sá, M.F. Anti-parasitic Peptides from Arthropods and their Application in Drug Therapy. Front. Microbiol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Requena-Méndez, A.; Cattaneo, P.; Bogale, R.T.; Marti-Soler, H.; Wångdahl, A.; Buonfrate, D.; Bisoffi, Z.; Färnert, A.; Rodríguez-Cuadrado, A.; Monge-Maillo, B.; et al. Malaria parasite prevalence among migrants: A systematic review and meta-analysis. Clin. Microbiol. Infect. 2023. [Google Scholar] [CrossRef]
- White, N.J.; Pukrittayakamee, S.; Hien, T.T.; Faiz, M.A.; Mokuolu, O.A.; Dondorp, A.M. Malaria. Lancet 2014, 383, 723–735. [Google Scholar] [CrossRef]
- WHO World Malaria Report 2022. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 (accessed on 5 October 2023).
- Bloland, P.B. Drug Resistance in Malaria; WHO: Chamblee, GA, USA, 2011; Volume WHO/CDS/CSR/DRS/2001.4. [Google Scholar]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 144. [Google Scholar] [CrossRef]
- Guo, Z. Artemisinin anti-malarial drugs in China. Acta Pharm. Sin. B 2016, 6, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Talapko, J.; Škrlec, I.; Alebić, T.; Jukić, M.; Včev, A. Malaria: The Past and the Present. Microorganisms 2019, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Menard, D.; Dondorp, A. Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harb. Perspect. Med. 2017, 7, a025619. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.K.; Anand, U.; Siddiqui, W.A.; Tripathi, R. Drug Development Strategies for Malaria: With the Hope for New Antimalarial Drug Discovery-An Update. Adv. Med. 2023, 2023, 5060665. [Google Scholar] [CrossRef]
- Wani, W.A.; Jameel, E.; Baig, U.; Mumtazuddin, S.; Hun, L.T. Ferroquine and its derivatives: New generation of antimalarial agents. Eur. J. Med. Chem. 2015, 101, 534–551. [Google Scholar] [CrossRef]
- Adoke, Y.; Zoleko-Manego, R.; Ouoba, S.; Tiono, A.B.; Kaguthi, G.; Bonzela, J.E.; Duong, T.T.; Nahum, A.; Bouyou-Akotet, M.; Ogutu, B.; et al. A randomized, double-blind, phase 2b study to investigate the efficacy, safety, tolerability and pharmacokinetics of a single-dose regimen of ferroquine with artefenomel in adults and children with uncomplicated Plasmodium falciparum malaria. Malar. J. 2021, 20, 222. [Google Scholar] [CrossRef]
- Tajuddeen, N.; Van Heerden, F.R. Antiplasmodial natural products: An update. Malar. J. 2019, 18, 404. [Google Scholar] [CrossRef]
- Ribeiro, G.D.J.G.; Rei Yan, S.L.; Palmisano, G.; Wrenger, C. Plant Extracts as a Source of Natural Products with Potential Antimalarial Effects: An Update from 2018 to 2022. Pharmaceutics 2023, 15, 1638. [Google Scholar] [CrossRef]
- Zahari, A.; Cheah, F.K.; Mohamad, J.; Sulaiman, S.N.; Litaudon, M.; Leong, K.H.; Awang, K. Antiplasmodial and antioxidant isoquinoline alkaloids from Dehaasia longipedicellata. Planta Med. 2014, 80, 599–603. [Google Scholar] [CrossRef]
- Claudino, V.D.; da Silva, K.C.; Cechinel Filho, V.; Yunes, R.A.; Delle Monache, F.; Giménez, A.; Salamanca, E.; Gutierrez-Yapu, D.; Malheiros, A. Drimanes from Drimys brasiliensis with leishmanicidal and antimalarial activity. Mem. Inst. Oswaldo Cruz 2013, 108, 140–144. [Google Scholar] [CrossRef]
- Konziase, B. Protective activity of biflavanones from Garcinia kola against Plasmodium infection. J. Ethnopharmacol. 2015, 172, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Abedi, A.K.; Valenciano, A.L.; Fernández-Murga, M.L.; Cassera, M.B.; Rasamison, V.E.; Applequist, W.L.; Miller, J.S.; Kingston, D.G.I. Isolation of the New Antiplasmodial Butanolide, Malleastrumolide A, from Malleastrum sp. (Meliaceae) from Madagascar. Chem. Biodivers. 2017, 14, e1700331. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.M.; Ghimire, B.K.; Kang, E.Y.; Moon, H.I. Antiplasmodial and cytotoxic activity of khellactone derivatives from Angelica purpuraefolia Chung. Phytother. Res. 2010, 24, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Upegui, Y.; Robledo, S.M.; Gil Romero, J.F.; Quiñones, W.; Archbold, R.; Torres, F.; Escobar, G.; Nariño, B.; Echeverri, F. In vivo Antimalarial Activity of α-Mangostin and the New Xanthone δ-Mangostin. Phytother. Res. 2015, 29, 1195–1201. [Google Scholar] [CrossRef]
- Dai, Y.; Harinantenaina, L.; Bowman, J.D.; Da Fonseca, I.O.; Brodie, P.J.; Goetz, M.; Cassera, M.B.; Kingston, D.G. Isolation of antiplasmodial anthraquinones from Kniphofia ensifolia, and synthesis and structure-activity relationships of related compounds. Bioorg. Med. Chem. 2014, 22, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Abdallah, H.M.; Elkhayat, E.S.; Al Musayeib, N.M.; Asfour, H.Z.; Zayed, M.F.; Mohamed, G.A. Fusaripeptide A: New antifungal and anti-malarial cyclodepsipeptide from the endophytic fungus Fusarium sp. J. Asian Nat. Prod. Res. 2018, 20, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Bracegirdle, J.; Casandra, D.; Rocca, J.R.; Adams, J.H.; Baker, B.J. Highly N-Methylated Peptides from the Antarctic Sponge Inflatella coelosphaeroides Are Active against Plasmodium falciparum. J. Nat. Prod. 2022, 85, 2454–2460. [Google Scholar] [CrossRef]
- Parquet, V.; Henry, M.; Wurtz, N.; Dormoi, J.; Briolant, S.; Gil, M.; Baret, E.; Amalvict, R.; Rogier, C.; Pradines, B. Atorvastatin as a potential anti-malarial drug: In vitro synergy in combinational therapy with quinine against Plasmodium falciparum. Malar. J. 2010, 9, 139. [Google Scholar] [CrossRef]
- Pongratz, P.; Kurth, F.; Ngoma, G.M.; Basra, A.; Ramharter, M. In vitro activity of antifungal drugs against Plasmodium falciparum field isolates. Wien. Klin. Wochenschr. 2011, 123 (Suppl. 1), 26–30. [Google Scholar] [CrossRef]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Piscopo, T.V.; Mallia Azzopardi, C. Leishmaniasis. Postgrad. Med. J. 2007, 83, 649–657. [Google Scholar] [CrossRef] [PubMed]
- WHO Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 13 October 2023).
- Okwor, I.; Uzonna, J. Social and Economic Burden of Human Leishmaniasis. Am. J. Trop. Med. Hyg. 2016, 94, 489–493. [Google Scholar] [CrossRef] [PubMed]
- WHO. Control of the Leishmaniases; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Pradhan, S.; Schwartz, R.A.; Patil, A.; Grabbe, S.; Goldust, M. Treatment options for leishmaniasis. Clin. Exp. Dermatol. 2022, 47, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Sangshetti, J.N.; Kalam Khan, F.A.; Kulkarni, A.A.; Arote, R.; Patil, R.H. Antileishmanial drug discovery: Comprehensive review of the last 10 years. RSC Adv. 2015, 5, 32376–32415. [Google Scholar] [CrossRef]
- Alvar, J.; Arana, B. Leishmaniasis, Impact and Therapeutic Needs. In Drug Discovery for Leishmaniasis; Rivas, L., Gil, C., Eds.; The Royal Society of Chemistry: London, UK, 2017; p. 402. [Google Scholar]
- De Luca, L.; Ferro, S.; Buemi, M.R.; Monforte, A.-M.; Gitto, R.; Schirmeister, T.; Maes, L.; Rescifina, A.; Micale, N. Discovery of benzimidazole-based Leishmania mexicana cysteine protease CPB2.8ΔCTE inhibitors as potential therapeutics for leishmaniasis. Chem. Biol. Drug Des. 2018, 92, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Stevanović, S.; Perdih, A.; Senćanski, M.; Glišić, S.; Duarte, M.; Tomás, A.M.; Sena, F.V.; Sousa, F.M.; Pereira, M.M.; Solmajer, T. In Silico Discovery of a Substituted 6-Methoxy-quinalidine with Leishmanicidal Activity in Leishmania infantum. Molecules 2018, 23, 772. [Google Scholar] [CrossRef] [PubMed]
- Villa-Pulgarín, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela, M.R.; Cuesta-Marbán, Á.; Müller, I.; Modolell, M.; Revuelta, J.L.; et al. Mitochondria and lipid raft-located FOF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef]
- Pierson, J.-T.; Dumètre, A.; Hutter, S.; Delmas, F.; Laget, M.; Finet, J.-P.; Azas, N.; Combes, S. Synthesis and antiprotozoal activity of 4-arylcoumarins. Eur. J. Med. Chem. 2010, 45, 864–869. [Google Scholar] [CrossRef]
- Zhai, L.; Blom, J.; Chen, M.; Christensen, S.B.; Kharazmi, A. The antileishmanial agent licochalcone A interferes with the function of parasite mitochondria. Antimicrob. Agents Chemother. 1995, 39, 2742–2748. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Shaikh, R.I.; Khan, F.A.K.; Patil, R.H.; Marathe, S.D.; Gade, W.N.; Shinde, D.B. Synthesis, antileishmanial activity and docking study of N’-substitutedbenzylidene-2-(6,7-dihydrothieno [3,2-c]pyridin-5(4H)-yl)acetohydrazides. Bioorg. Med. Chem. Lett. 2014, 24, 1605–1610. [Google Scholar] [CrossRef]
- Ruiz-Santaquiteria, M.; Sánchez-Murcia, P.A.; Toro, M.A.; de Lucio, H.; Gutiérrez, K.J.; de Castro, S.; Carneiro, F.A.C.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.-J.; et al. First example of peptides targeting the dimer interface of Leishmania infantum trypanothione reductase with potent in vitro antileishmanial activity. Eur. J. Med. Chem. 2017, 135, 49–59. [Google Scholar] [CrossRef]
- Kumar, V.; Chugh, A. Peptide-mediated leishmaniasis management strategy: Tachyplesin emerges as an effective anti-leishmanial peptide against Leishmania donovani. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183629. [Google Scholar] [CrossRef] [PubMed]
- Balunas, M.J.; Linington, R.G.; Tidgewell, K.; Fenner, A.M.; Ureña, L.D.; Togna, G.D.; Kyle, D.E.; Gerwick, W.H. Dragonamide E, a modified linear lipopeptide from Lyngbya majuscula with antileishmanial activity. J. Nat. Prod. 2010, 73, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Besednova, N.N.; Zaporozhets, T.S.; Andryukov, B.G.; Kryzhanovsky, S.P.; Ermakova, S.P.; Kuznetsova, T.A.; Voronova, A.N.; Shchelkanov, M.Y. Antiparasitic Effects of Sulfated Polysaccharides from Marine Hydrobionts. Mar. Drugs 2021, 19, 637. [Google Scholar] [CrossRef]
- WHO. Trypanosomiasis, Human African (Sleeping Sickness). Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 17 October 2023).
- WHO. Human African Trypanosomiasis (Sleeping Sickness). Available online: https://www.who.int/health-topics/human-african-trypanosomiasis#tab=tab_1 (accessed on 17 October 2023).
- Sutherland, C.S.; Yukich, J.; Goeree, R.; Tediosi, F. A Literature Review of Economic Evaluations for a Neglected Tropical Disease: Human African Trypanosomiasis (“Sleeping Sickness”). PLOS Negl. Trop. Dis. 2015, 9, e0003397. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Interim Guidelines for the Treatment of Gambiense Human African Trypanosomiasis. Available online: https://iris.who.int/bitstream/handle/10665/326178/9789241550567-eng.pdf (accessed on 17 October 2023).
- Kasozi, K.I.; MacLeod, E.T.; Ntulume, I.; Welburn, S.C. An Update on African Trypanocide Pharmaceutics and Resistance. Front. Vet. Sci. 2022, 9, 828111. [Google Scholar] [CrossRef]
- Deeks, E.D. Fexinidazole: First Global Approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef]
- Dickie, E.A.; Giordani, F.; Gould, M.K.; Mäser, P.; Burri, C.; Mottram, J.C.; Rao, S.P.S.; Barrett, M.P. New Drugs for Human African Trypanosomiasis: A Twenty First Century Success Story. Trop. Med. Int. Health 2020, 5, 29. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an Orally-Active Benzoxaborole for the Treatment of Stage 2 Human African Trypanosomiasis. PLOS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef]
- Tarral, A.; Hovsepian, L.; Duvauchelle, T.; Donazzolo, Y.; Latreille, M.; Felices, M.; Gualano, V.; Delhomme, S.; Valverde Mordt, O.; Blesson, S.; et al. Determination of the Optimal Single Dose Treatment for Acoziborole, a Novel Drug for the Treatment of Human African Trypanosomiasis: First-in-Human Study. Clin. Pharmacokinet. 2023, 62, 481–491. [Google Scholar] [CrossRef]
- Kumeso, V.K.B.; Kalonji, W.M.; Rembry, S.; Mordt, O.V.; Tete, D.N.; Prêtre, A.; Delhomme, S.; Kyhi, M.I.W.; Camara, M.; Catusse, J.; et al. Efficacy and safety of acoziborole in patients with human African trypanosomiasis caused by Trypanosoma brucei gambiense: A multicentre, open-label, single-arm, phase 2/3 trial. Lancet Infect. Dis. 2023, 23, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Kurisawa, N.; Iwasaki, A.; Jeelani, G.; Nozaki, T.; Suenaga, K. Iheyamides A-C, Antitrypanosomal Linear Peptides Isolated from a Marine Dapis sp. Cyanobacterium. J. Nat. Prod. 2020, 83, 1684–1690. [Google Scholar] [CrossRef] [PubMed]
- Hemmige, V.; Tanowitz, H.; Sethi, A. Trypanosoma cruzi infection: A review with emphasis on cutaneous manifestations. Int. J. Dermatol. 2012, 51, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Bern, C.; Montgomery, S.P.; Herwaldt, B.L.; Rassi, A.; Marin-Neto, J.A.; Dantas, R.O.; Maguire, J.H.; Acquatella, H.; Morillo, C.; Kirchhoff, L.V.; et al. Evaluation and Treatment of Chagas Disease in the United StatesA Systematic Review. JAMA 2007, 298, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- García-Huertas, P.; Cardona-Castro, N. Advances in the treatment of Chagas disease: Promising new drugs, plants and targets. Biomed. Pharmacother. 2021, 142, 112020. [Google Scholar] [CrossRef]
- Apt, W.; Arribada, A.; Zulantay, I.; Rodríguez, J.; Saavedra, M.; Muñoz, A. Treatment of Chagas’ disease with itraconazole: Electrocardiographic and parasitological conditions after 20 years of follow-up. J. Antimicrob. Chemother. 2013, 68, 2164–2169. [Google Scholar] [CrossRef]
- Gulin, J.E.N.; Eagleson, M.A.; Postan, M.; Cutrullis, R.A.; Freilij, H.; Bournissen, F.G.; Petray, P.B.; Altcheh, J. Efficacy of voriconazole in a murine model of acute Trypanosoma cruzi infection. J. Antimicrob. Chemother. 2012, 68, 888–894. [Google Scholar] [CrossRef]
- Urbina, J.A.; Payares, G.; Sanoja, C.; Lira, R.; Romanha, A.J. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int. J. Antimicrob. Agents 2003, 21, 27–38. [Google Scholar] [CrossRef]
- Padilla, A.M.; Wang, W.; Akama, T.; Carter, D.S.; Easom, E.; Freund, Y.; Halladay, J.S.; Liu, Y.; Hamer, S.A.; Hodo, C.L.; et al. Discovery of an orally active benzoxaborole prodrug effective in the treatment of Chagas disease in non-human primates. Nat. Microbiol. 2022, 7, 1536–1546. [Google Scholar] [CrossRef]
- Furtado, J.M.; Smith, J.R.; Belfort, R.J.; Gattey, D.; Winthrop, K.L. Toxoplasmosis: A Global Threat. J. Glob. Infect. Dis. 2011, 3, 281–284. [Google Scholar] [CrossRef]
- Hajj, R.E.; Tawk, L.; Itani, S.; Hamie, M.; Ezzeddine, J.; El Sabban, M.; El Hajj, H. Toxoplasmosis: Current and Emerging Parasite Druggable Targets. Microorganisms 2021, 9, 2531. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, J.G.; Bordón, C.; Posner, G.H.; Yolken, R.; Jones-Brando, L. Artemisinin derivatives inhibit Toxoplasma gondii in vitro at multiple steps in the lytic cycle. J. Antimicrob. Chemother. 2008, 63, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Mui, E.J.; Schiehser, G.A.; Milhous, W.K.; Hsu, H.; Roberts, C.W.; Kirisits, M.; Muench, S.; Rice, D.; Dubey, J.P.; Fowble, J.W.; et al. Novel Triazine JPC-2067-B Inhibits Toxoplasma gondii In Vitro and In Vivo. PLOS Negl. Trop. Dis. 2008, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, B.F.; Gomes, A.O.; Ferro, E.A.V.; Napolitano, D.R.; Mineo, J.R.; Silva, N.M. Enrofloxacin is able to control Toxoplasma gondii infection in both in vitro and in vivo experimental models. Vet. Parasitol. 2012, 187, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Fomovska, A.; Huang, Q.; Bissati, K.E.; Mui, E.J.; Witola, W.H.; Cheng, G.; Zhou, Y.; Sommerville, C.; Roberts, C.W.; Bettis, S.; et al. Novel N-Benzoyl-2-Hydroxybenzamide Disrupts Unique Parasite Secretory Pathway. Antimicrob. Agents Chemother. 2012, 56, 2666–2682. [Google Scholar] [CrossRef] [PubMed]
- Hegewald, J.; Gross, U.; Bohne, W. Identification of dihydroorotate dehydrogenase as a relevant drug target for 1-hydroxyquinolones in Toxoplasma gondii. Mol. Biochem. Parasitol. 2013, 190, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Tenório, R.P.; Carvalho, C.S.; Pessanha, C.S.; de Lima, J.G.; de Faria, A.R.; Alves, A.J.; de Melo, E.J.T.; Góes, A.J.S. Synthesis of thiosemicarbazone and 4-thiazolidinone derivatives and their in vitro anti-Toxoplasma gondii activity. Bioorganic Med. Chem. Lett. 2005, 15, 2575–2578. [Google Scholar] [CrossRef]
- Xing, L.; Wang, Z.; Hao, Y.; Zhang, W. Marine Products as a Promising Resource of Bioactive Peptides: Update of Extraction Strategies and Their Physiological Regulatory Effects. J. Agric. Food Chem. 2022, 70, 3081–3095. [Google Scholar] [CrossRef]
- Nweze, J.A.; Mbaoji, F.N.; Li, Y.M.; Yang, L.Y.; Huang, S.S.; Chigor, V.N.; Eze, E.A.; Pan, L.X.; Zhang, T.; Yang, D.F. Potentials of marine natural products against malaria, leishmaniasis, and trypanosomiasis parasites: A review of recent articles. Infect. Dis. Pover. 2021, 10, 9. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Q.; Zhang, Q.; Cui, X.; Zhu, L. Promising Antiparasitic Natural and Synthetic Products from Marine Invertebrates and Microorganisms. Mar. Drugs 2023, 21, 84. [Google Scholar] [CrossRef]
- Karthikeyan, A.; Joseph, A.; Nair, B.G. Promising bioactive compounds from the marine environment and their potential effects on various diseases. J. Genet. Eng. Biotechnol. 2022, 20, 14. [Google Scholar] [CrossRef] [PubMed]
- Shinde, P.; Banerjee, P.; Mandhare, A. Marine natural products as source of new drugs: A patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 283–309. [Google Scholar] [CrossRef]
- Banerjee, P.; Mandhare, A.; Bagalkote, V. Marine natural products as source of new drugs: An updated patent review (July 2018–July 2021). Expert Opin. Ther. Pat. 2022, 32, 317–363. [Google Scholar] [CrossRef] [PubMed]
- Macedo, M.W.F.S.; da Cunha, N.B.; Carneiro, J.A.; da Costa, R.A.; de Alencar, S.A.; Cardoso, M.H.; Franco, O.L.; Dias, S.C. Marine organisms as a rich source of biologically active peptides. Front. Mar. Sci. 2021, 8, 667764. [Google Scholar] [CrossRef]
- Bhandari, D.; Rafiq, S.; Gat, Y.; Gat, P.; Waghmare, R.; Kumar, V. A Review on Bioactive Peptides: Physiological Functions, Bioavailability and Safety. Int. J. Pept. Res. Ther. 2020, 26, 139–150. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.E.; Deer, T.R. Ziconotide: A clinical update and pharmacologic review. Expert Opin. Pharmacother. 2013, 14, 957–966. [Google Scholar] [CrossRef]
- McIntosh, M.; Cruz, L.J.; Hunkapiller, M.W.; Gray, W.R.; Olivera, B.M. Isolation and structure of a peptide toxin from the marine snail Conus magus. Arch. Biochem. Biophys. 1982, 218, 329–334. [Google Scholar] [CrossRef]
- Ghoran, S.H.; Taktaz, F.; Sousa, E.; Fernandes, C.; Kijjoa, A. Peptides from Marine-Derived Fungi: Chemistry and Biological Activities. Mar. Drugs 2023, 21, 510. [Google Scholar] [CrossRef]
- Rangel, M.; José Correia de Santana, C.; Pinheiro, A.; Dos Anjos, L.; Barth, T.; Rodrigues Pires Júnior, O.; Fontes, W.; Castro, M.S. Marine depsipeptides as promising pharmacotherapeutic agents. Curr. Protein Pept. Sci. 2017, 18, 72–91. [Google Scholar] [CrossRef] [PubMed]
- Anjum, K.; Abbas, S.Q.; Akhter, N.; Shagufta, B.I.; Shah, S.A.A.; Hassan, S.S.U. Emerging biopharmaceuticals from bioactive peptides derived from marine organisms. Chem. Biol. Drug Des. 2017, 90, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.; Anil, S.; Kim, S.-K.; Shim, M.S. Marine fish proteins and peptides for cosmeceuticals: A review. Mar. Drugs 2017, 15, 143. [Google Scholar] [CrossRef] [PubMed]
- Corinaldesi, C.; Barone, G.; Marcellini, F.; Dell’Anno, A.; Danovaro, R. Marine microbial-derived molecules and their potential use in cosmeceutical and cosmetic products. Mar. Drugs 2017, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Charoensiddhi, S.; Conlon, M.A.; Franco, C.M.; Zhang, W. The development of seaweed-derived bioactive compounds for use as prebiotics and nutraceuticals using enzyme technologies. Trends Food Sci. Technol. 2017, 70, 20–33. [Google Scholar] [CrossRef]
- Ghosh, S.; Sarkar, T.; Pati, S.; Kari, Z.A.; Edinur, H.A.; Chakraborty, R. Novel Bioactive Compounds from Marine Sources as a Tool for Functional Food Development. Front. Mar. Sci. 2022, 9, 832957. [Google Scholar] [CrossRef]
- Wibowo, J.T.; Bayu, A.; Aryati, W.D.; Fernandes, C.; Yanuar, A.; Kijjoa, A.; Putra, M.Y. Secondary Metabolites from Marine-Derived Bacteria with Antibiotic and Antibiofilm Activities against Drug-Resistant Pathogens. Mar. Drugs 2023, 21, 50. [Google Scholar] [CrossRef]
- Milligan, K.E.; Marquez, B.L.; Williamson, R.T.; Gerwick, W.H. Lyngbyabellin B, a toxic and antifungal secondary metabolite from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 1440–1443. [Google Scholar] [CrossRef]
- de Sá, J.D.M.; Kumla, D.; Dethoup, T.; Kijjoa, A. Bioactive Compounds from Terrestrial and Marine-Derived Fungi of the Genus Neosartorya. Molecules 2022, 27, 2351. [Google Scholar] [CrossRef]
- Daletos, G.; Kalscheuer, R.; Koliwer-Brandl, H.; Hartmann, R.; De Voogd, N.J.; Wray, V.; Lin, W.; Proksch, P. Callyaerins from the Marine Sponge Callyspongia aerizusa: Cyclic Peptides with Antitubercular Activity. J. Nat. Prod. 2015, 78, 1910–1925. [Google Scholar] [CrossRef]
- O’connor, J.; Garcia-Vaquero, M.; Meaney, S.; Tiwari, B.K. Bioactive Peptides from Algae: Traditional and Novel Generation Strategies, Structure-Function Relationships, and Bioinformatics as Predictive Tools for Bioactivity. Mar. Drugs 2022, 20, 317. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Adholeya, A.; Deshmukh, S.K. The pharmacological potential of non-ribosomal peptides from marine sponge and tunicates. Front. Pharmacol. 2016, 7, 333. [Google Scholar] [CrossRef] [PubMed]
- Amelia, T.S.M.; Suaberon, F.A.C.; Vad, J.; Fahmi, A.D.M.; Saludes, J.P.; Bhubalan, K. Recent Advances of Marine Sponge-Associated Microorganisms as a Source of Commercially Viable Natural Products. Mar. Biotechnol. 2022, 24, 492–512. [Google Scholar] [CrossRef] [PubMed]
- Prompanya, C.; Fernandes, C.; Cravo, S.; Pinto, M.M.M.; Dethoup, T.; Silva, A.M.S.; Kijjoa, A. A new cyclic hexapeptide and a new isocoumarin derivative from the marine sponge-associated fungus Aspergillus similanensis KUFA 0013. Mar. Drugs 2015, 13, 1432–1450. [Google Scholar] [CrossRef] [PubMed]
- Zin, W.W.M.; Buttachon, S.; Dethoup, T.; Fernandes, C.; Cravo, S.; Pinto, M.M.M.; Gales, L.; Pereira, J.A.; Silva, A.M.S.; Sekeroglu, N.; et al. New cyclotetrapeptides and a new diketopiperzine derivative from the marine sponge-associated fungus Neosartorya glabra KUFA 0702. Mar. Drugs 2016, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.K.; Seo, C.H.; Park, Y. Marine peptides and their anti-infective activities. Mar. Drugs 2015, 13, 618–654. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Akbarian, M.; Khani, A.; Eghbalpour, S.; Uversky, V.N. Bioactive Peptides: Synthesis, Sources, Applications, and Proposed Mechanisms of Action. Int. J. Mol. Sci. 2022, 23, 1445. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef]
- Choi, J.S.; Joo, S.H. Recent Trends in Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2020, 28, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, V.; Nicholson, G.J.; Ries, J.; Recktenwald, J.; Schefer, A.B.; Shawky, R.M.; Schröder, J.; Wohlleben, W.; Pelzer, S. A polyketide synthase in glycopeptide biosynthesis: The biosynthesis of the non-proteinogenic amino acid (S)-3, 5-dihydroxyphenylglycine. J. Biol. Chem. 2001, 276, 38370–38377. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.P.; Nogawa, T.; Futamura, Y.; Shimizu, T.; Hashizume, D.; Takahashi, S.; Jang, J.H.; Ahn, J.S.; Osada, H. Octaminomycins A and B, cyclic octadepsipeptides active against Plasmodium falciparum. J. Nat. Prod. 2017, 80, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, K.; Takada, K.; Okada, S.; Matsunaga, S. Nazumazoles A-C, cyclic pentapeptides dimerized through a disulfide bond from the marine sponge theonella swinhoei. Org. Lett. 2015, 17, 2646–2648. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Krunic, A.; Kang, H.S.; Chen, W.L.; Woodard, J.L.; Fuchs, J.R.; Swanson, S.M.; Orjala, J. Trichormamides A and B with antiproliferative activity from the cultured freshwater cyanobacterium Trichormus sp. UIC 10339. J. Nat. Prod. 2014, 77, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.M.; de Bruijn, I.; Nybroe, O.; Ongena, M. Natural functions of lipopeptides from Bacillus and Pseudomonas: More than surfactants and antibiotics. FEMS Microbiol. Rev. 2010, 34, 1037–1062. [Google Scholar] [CrossRef] [PubMed]
- Sieber, S.A.; Marahiel, M.A. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 2005, 105, 715–738. [Google Scholar] [CrossRef]
- Kohli, R.M.; Walsh, C.T.; Burkart, M.D. Biomimetic synthesis and optimization of cyclic peptide antibiotics. Nature 2002, 418, 658–661. [Google Scholar] [CrossRef]
- Hamada, Y.; Shioiri, T. Recent Progress of the Synthetic Studies of Biologically Active Marine Cyclic Peptides and Depsipeptides. Chem. Rev. 2005, 105, 4441–4482. [Google Scholar] [CrossRef]
- Jing, X.; Jin, K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev. 2020, 40, 753–810. [Google Scholar] [CrossRef]
- Gentilucci, L.; de Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, R. Review on the Bioactive Peptides from Marine Sources: Indication for Health Effects. Int. J. Pept. Res. Ther. 2019, 25, 1187–1199. [Google Scholar] [CrossRef]
- Cunha, S.A.; Pintado, M.E. Bioactive peptides derived from marine sources: Biological and functional properties. Trends Food Sci. Technol. 2022, 119, 348–370. [Google Scholar] [CrossRef]
- Kang, H.K.; Choi, M.-C.; Seo, C.H.; Park, Y. Therapeutic properties and biological benefits of marine-derived anticancer peptides. Int. J. Mol. Sci. 2018, 19, 919. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.; Pinto, E.; Fernandes, C.; Sousa, E. Marine Cyclic Peptides: Antimicrobial Activity and Synthetic Strategies. Mar. Drugs 2022, 20, 397. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Ribeiro, R.; Pinto, M.; Kijjoa, A. Absolute Stereochemistry Determination of Bioactive Marine-Derived Cyclopeptides by Liquid Chromatography Methods: An Update Review (2018–2022). Molecules 2023, 28, 615. [Google Scholar] [CrossRef] [PubMed]
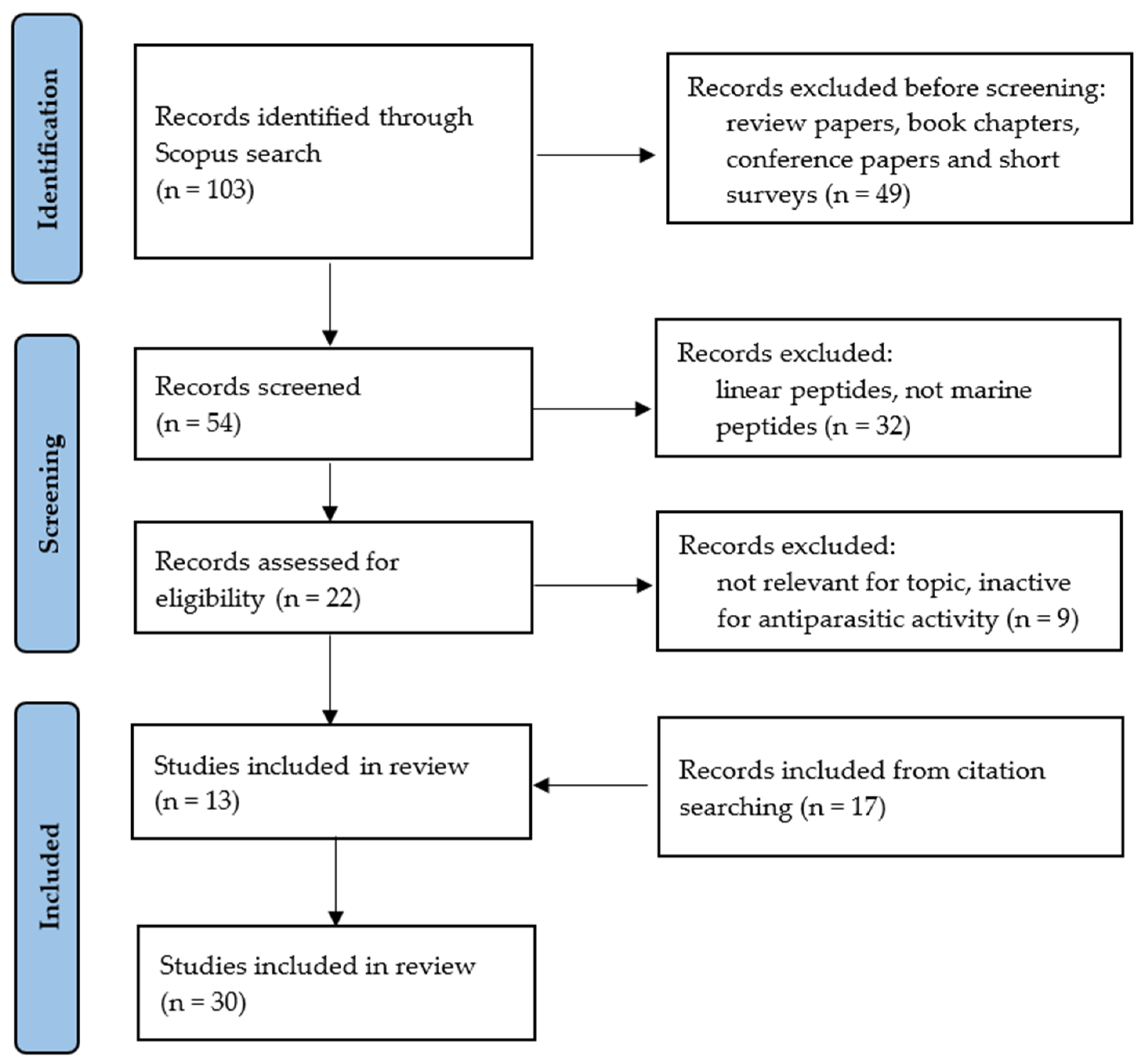
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Syst. Rev. 2021, 10, 89. [Google Scholar] [CrossRef]
- Crews, P.; Manes, L.V.; Boehler, M. Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis sp. Tetrahedron Lett. 1986, 27, 2797–2800. [Google Scholar] [CrossRef]
- Mizuno, Y.; Makioka, A.; Kawazu, S.I.; Kano, S.; Kawai, S.; Akaki, M.; Aikawa, M.; Ohtomo, H. Effect of jasplakinolide on the growth, invasion, and actin cytoskeleton of Plasmodium falciparum. Parasitol. Res. 2002, 88, 844–848. [Google Scholar] [CrossRef]
- Poupel, O.; Tardieux, I. Toxoplasma gondii motility and host cell invasiveness are drastically impaired by jasplakinolide, a cyclic peptide stabilizing F-actin. Microbes Infect. 1999, 1, 653–662. [Google Scholar] [CrossRef]
- Makioka, A.; Kumagai, M.; Ohtomo, H.; Kobayashi, S.; Takeuchi, T. Effect of jasplakinolide on the growth, encystation, and actin cytoskeleton of Entamoeba histolytica and Entamoeba invadens. J. Parasitol. 2001, 87, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Makioka, A.; Kumagai, M.; Ohtomo, H.; Kobayashi, S.; Takeuchi, T. Growth inhibition and actin aggregate formation of Entamoeba histolytica by jasplakinolide. Arch. Med. Res. 2000, 31, S145–S146. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.K.; Shen, Y.C.; Cheng, X.C.; Jensen, P.R.; Frankmoelle, W.; Kauffman, C.A.; Fenical, W.; Lobkovsky, E.; Clardy, J. Cyclomarins A-C, new antiinflammatory cyclic peptides produced by a marine bacterium (Streptomyces sp.). J. Am. Chem. Soc. 1999, 121, 11273–11276. [Google Scholar] [CrossRef]
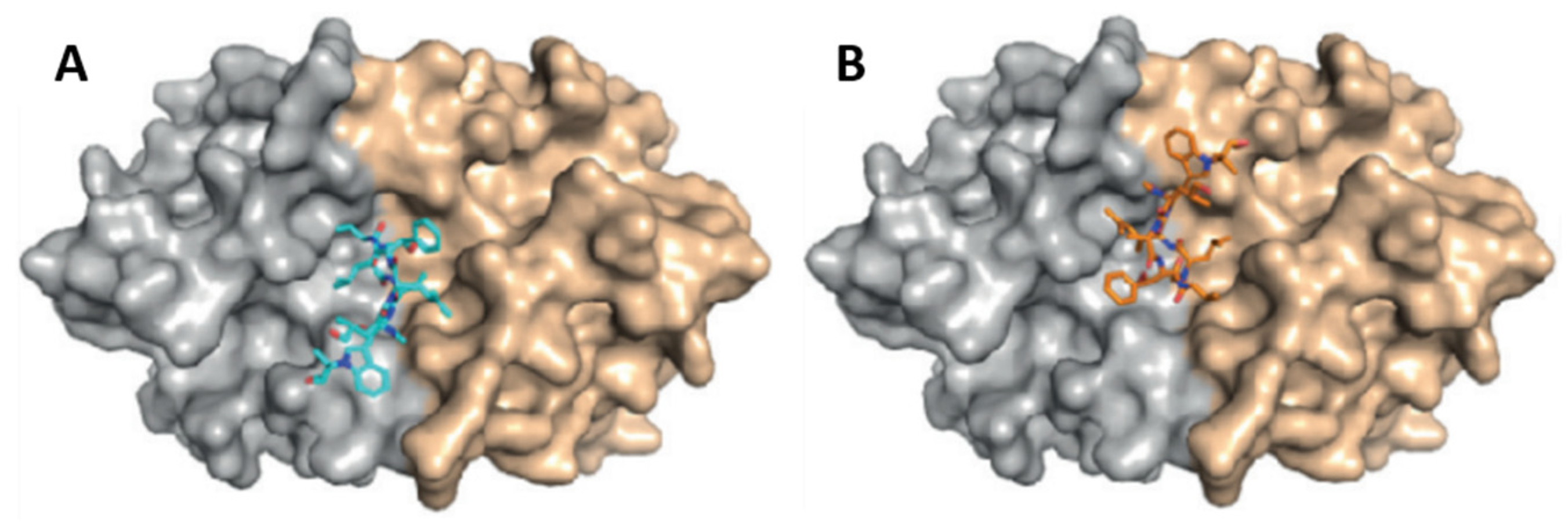
- Bürstner, N.; Roggo, S.; Ostermann, N.; Blank, J.; Delmas, C.; Freuler, F.; Gerhartz, B.; Hinniger, A.; Hoepfner, D.; Liechty, B.; et al. Gift from Nature: Cyclomarin A Kills Mycobacteria and Malaria Parasites by Distinct Modes of Action. ChemBioChem 2015, 16, 2433–2436. [Google Scholar] [CrossRef] [PubMed]
- Intaraudom, C.; Rachtawee, P.; Suvannakad, R.; Pittayakhajonwut, P. Antimalarial and antituberculosis substances from Streptomyces sp. BCC26924. Tetrahedron 2011, 67, 7593–7597. [Google Scholar] [CrossRef]
- Linington, R.G.; González, J.; Ureña, L.D.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H. Venturamides A and B: Antimalarial constituents of the Panamanian marine cyanobacterium Oscillatoria sp. J. Nat. Prod. 2007, 70, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; Edwards, D.J.; Shuman, C.F.; McPhail, K.L.; Matainaho, T.; Gerwick, W.H. Symplocamide A, a potent cytotoxin and chymotrypsin inhibitor from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2008, 71, 22–27. [Google Scholar] [CrossRef]
- Donia, M.S.; Wang, B.; Dunbar, D.C.; Desai, P.V.; Patny, A.; Avery, M.; Hamann, M.T. Mollamides B and C, Cyclic hexapeptides from the indonesian tunicate Didemnum molle. J. Nat. Prod. 2008, 71, 941–945. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Tan, L.T. Lagunamides A and B: Cytotoxic and antimalarial cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 1810–1814. [Google Scholar] [CrossRef]
- Gumila, C.; Ancelin, M.L.; Jeminet, G.; Delort, A.M.; Miquel, G.; Vial, H.J. Differential in vitro activities of ionophore compounds against Plasmodium falciparum and mammalian cells. Antimicrob. Agents Chemother. 1996, 40, 602–608. [Google Scholar] [CrossRef]
- Pimentel-Elardo, S.M.; Kozytska, S.; Bugni, T.S.; Ireland, C.M.; Moll, H.; Hentschel, U. Anti-parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar. Drugs 2010, 8, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.; Khalil, Z.G.; Piggott, A.M.; Blumenthal, A.; Gardiner, D.L.; Skinner-Adams, T.S.; Capon, R.J. Mollemycin A: An antimalarial and antibacterial glyco-hexadepsipeptide- polyketide from an Australian marine-derived Streptomyces sp. (CMB-M0244). Org. Lett. 2014, 16, 1716–1719. [Google Scholar] [CrossRef] [PubMed]
- Vining, O.B.; Medina, R.A.; Mitchell, E.A.; Videau, P.; Li, D.; Serrill, J.D.; Kelly, J.X.; Gerwick, W.H.; Proteau, P.J.; Ishmael, J.E. Depsipeptide companeramides from a panamanian marine cyanobacterium associated with the coibamide producer. J. Nat. Prod. 2015, 78, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Almaliti, J.; Malloy, K.L.; Glukhov, E.; Spadafora, C.; Gutiérrez, M.; Gerwick, W.H. Dudawalamides A-D, Antiparasitic Cyclic Depsipeptides from the Marine Cyanobacterium Moorea producens. J. Nat. Prod. 2017, 80, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Sweeney-Jones, A.M.; Gagaring, K.; Antonova-Koch, J.; Zhou, H.; Mojib, N.; Soapi, K.; Skolnick, J.; McNamara, C.W.; Kubanek, J. Antimalarial peptide and polyketide natural products from the Fijian marine cyanobacterium Moorea producens. Mar. Drugs 2020, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.T.; Otto, C.S.; Scheuer, P.J.; Dunbar, D.C. Kahalalides: Bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Bryopsis sp. J. Org. Chem. 1996, 61, 6594–6600. [Google Scholar] [CrossRef]
- Hamann, M.T.; Scheuer, P.J. Kahalalide F: A Bioactive Depsipeptide from the Sacoglossan Mollusk Elysia rufescens and the Green Alga Bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Cruz, L.J.; Luque-Ortega, J.R.; Rivas, L.; Albericio, F. Kahalalide F, an antitumor depsipeptide in clinical trials, and its analogues as effective antileishmanial agents. Mol. Pharm. 2009, 6, 813–824. [Google Scholar] [CrossRef]
- Cruz, L.J.; Martínez Insua, M.; Pérez Baz, J.; Trujillo, M.; Rodriguez-Mias, R.A.; Oliveira, E.; Giralt, E.; Albericio, F.; Cañedo, L.M. IB-01212, a new cytotoxic cyclodepsipeptide isolated from the marine fungus Clonostachys sp. ESNA-A009. J. Org. Chem. 2006, 71, 3335–3338. [Google Scholar] [CrossRef]
- Luque-Ortega, J.R.; Cruz, L.J.; Albericio, F.; Rivas, L. The antitumoral depsipeptide IB-01212 kills Leishmania through an apoptosis-like process involving intracellular targets. Mol. Pharm. 2010, 7, 1608–1617. [Google Scholar] [CrossRef]
- Ogawa, H.; Iwasaki, A.; Sumimoto, S.; Kanamori, Y.; Ohno, O.; Iwatsuki, M.; Ishiyama, A.; Hokari, R.; Otoguro, K.; Omura, S.; et al. Janadolide, a Cyclic Polyketide-Peptide Hybrid Possessing a tert-Butyl Group from an Okeania sp. Marine Cyanobacterium. J. Nat. Prod. 2016, 79, 1862–1866. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Tang, A.H.; Geraghty, K.; Corcilius, L.; Kaiser, M.; Payne, R.J. Total Synthesis and Antitrypanosomal Activity of Janadolide and Simplified Analogues. Org. Lett. 2020, 22, 3089–3093. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Iwasaki, A.; Kurisawa, N.; Suzuki, R.; Jeelani, G.; Matsubara, T.; Sato, T.; Nozaki, T.; Suenaga, K. Motobamide, an Antitrypanosomal Cyclic Peptide from a Leptolyngbya sp. Marine Cyanobacterium. J. Nat. Prod. 2021, 84, 1649–1655. [Google Scholar] [CrossRef] [PubMed]
- Fotie, J. The potential of peptides and depsipeptides from terrestrial and marine organisms in the fight against human protozoan diseases. In Bioactive Natural Products: Chemistry and Biology; Wiley Blackwell: Hoboken, NJ, USA, 2015; pp. 279–320. [Google Scholar]
- Xu, Y.Y.; Liu, C.; Liu, Z.P. Advances in the total synthesis of cyclodepsipeptide (+)-jasplakinolide (jaspamide) and its analogs. Curr. Org. Synth. 2013, 10, 67–89. [Google Scholar]
- Scott, V.R.; Boehme, R.; Matthews, T.R. New class of antifungal agents: Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis species. Antimicrob. Agents Chemother. 1988, 32, 1154–1157. [Google Scholar] [CrossRef]
- Zabriskie, T.M.; Ireland, C.M.; Klocke, J.A.; Marcus, A.H.; Molinski, T.F.; Faulkner, D.J.; Xu, C.; Clardy, J.C. Jaspamide, a Modified Peptide from a Jaspis Sponge, with Insecticidal and Antifungal Activity. J. Am. Chem. Soc. 1986, 108, 3123–3124. [Google Scholar] [CrossRef]
- Yang, K.; Luo, M.; Li, H.; Abdulrehman, G.; Kang, L. Effects of jasplakinolide on cytotoxicity, cytoskeleton and apoptosis in two different colon cancer cell lines treated with m-THPC-PDT. Photodiagn. Photodyn. Ther. 2021, 35, 102425. [Google Scholar] [CrossRef]
- Ali, R.; Mir, H.A.; Hamid, R.; Shah, R.A.; Khanday, F.A.; Bhat, S.S. Jasplakinolide Attenuates Cell Migration by Impeding Alpha-1-syntrophin Protein Phosphorylation in Breast Cancer Cells. Protein J. 2021, 40, 234–244. [Google Scholar] [CrossRef]
- Odaka, C.; Sanders, M.L.; Crews, P. Jasplakinolide induces apoptosis in various transformed cell lines by a caspase-3-like protease-dependent pathway. Clin. Diagn. Lab. Immunol. 2000, 7, 947–952. [Google Scholar] [CrossRef]
- Takeuchi, H.; Ara, G.; Sausville, E.A.; Teicher, B. Jasplakinolide: Interaction with radiation and hyperthermia in human prostate carcinoma and Lewis lung carcinoma. Cancer Chemother. Pharmacol. 1998, 42, 491–496. [Google Scholar] [CrossRef]
- Senderowicz, A.M.J.; Kaur, G.; Sainz, E.; Laing, C.; Inman, W.D.; Rodriguez, J.; Crews, P.; Malspeis, L.; Grever, M.R.; Sausville, E.A.; et al. Jasplakinolide’s inhibition of the growth of prostate carcinoma cells in vitro with disruption of the actin cytoskeleton. J. Natl. Cancer Inst. 1995, 87, 46–51. [Google Scholar] [CrossRef]
- Alvarinõ, R.; Alonso, E.; Tabudravu, J.N.; Pérez-Fuentes, N.; Alfonso, A.; Botana, L.M. Tavarua Deoxyriboside A and Jasplakinolide as Potential Neuroprotective Agents: Effects on Cellular Models of Oxidative Stress and Neuroinflammation. ACS Chem. Neurosci. 2021, 12, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Bubb, M.R.; Senderowicz, A.M.J.; Sausville, E.A.; Duncan, K.L.K.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871. [Google Scholar] [CrossRef]
- Holzinger, A. Jasplakinolide: An actin-specific reagent that promotes actin polymerization. Methods Mol. Biol. 2009, 586, 71–87. [Google Scholar]
- Pospich, S.; Merino, F.; Raunser, S. Structural Effects and Functional Implications of Phalloidin and Jasplakinolide Binding to Actin Filaments. Structure 2020, 28, 437–449.e5. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, X.; Hu, L.; Wang, X.; Sun, Z.; Zhao, E.; Wang, J.; Wei, F. F-actin microfilament polymerized by jasplakinolide affects the expression of aquaporin-4 in astrocytic swelling after oxygen-glucose deprivation and reoxygenation. Mater. Express 2020, 10, 563–570. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, Y.; Pan, D.; Sun, Y.; Cao, J. The effect of Cytochalasin B and Jasplakinolide on depolymerization of actin filaments in goose muscles during postmortem conditioning. Food Res. Int. 2016, 90, 1–7. [Google Scholar] [CrossRef]
- Zhang, X.; Cui, X.; Cheng, L.; Guan, X.; Li, H.; Li, X.; Cheng, M. Actin Stabilization by Jasplakinolide Affects the Function of Bone Marrow-Derived Late Endothelial Progenitor Cells. PLoS ONE 2012, 7, e50899. [Google Scholar] [CrossRef]
- Jing, X.; Sun, J.L.; Zhang, X.Y.; Tang, K.X.; Yin, Q.L.; Wu, H.Y.; Wang, J.H.; Wang, J.W.; Cheng, M. Jasplakinolide affects the functions of HUVECs via actin stabilization. Chin. Pharmacol. Bull. 2013, 29, 1079–1083. [Google Scholar]
- Bubb, M.R.; Spector, I.; Beyer, B.B.; Fosen, K.M. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 2000, 275, 5163–5170. [Google Scholar] [CrossRef]
- Skillman, K.M.; Diraviyam, K.; Khan, A.; Tang, K.; Sept, D.; Sibley, L.D. Evolutionarily divergent, unstable filamentous actin is essential for gliding motility in apicomplexan parasites. PLoS Pathog. 2011, 7, e1002280. [Google Scholar] [CrossRef] [PubMed]
- Drewry, L.L.; Sibley, L.D. Toxoplasma actin is required for efficient host cell invasion. mBio 2015, 6, 00557-15. [Google Scholar] [CrossRef] [PubMed]
- Pospich, S.; Kumpula, E.P.; Von Der Ecken, J.; Vahokoski, J.; Kursula, I.; Raunser, S. Near-atomic structure of jasplakinolide-stabilized malaria parasite F-actin reveals the structural basis of filament instability. Proc. Natl. Acad. Sci. USA 2017, 114, 10636–10641. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.K.; Tilney, L.G. Induction of an acrosomal process in Toxoplasma gondii: Visualization of actin filaments in a protozoan parasite. Proc. Natl. Acad. Sci. USA 1999, 96, 9095–9099. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, D.; Rao, S.P.S.; Noble, C.G. Structural basis of mycobacterial inhibition by Cyclomarin A. J. Biol. Chem. 2013, 288, 30883–30891. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.K.; Riwanto, M.; Sambandamurthy, V.; Roggo, S.; Miault, C.; Zwingelstein, C.; Krastel, P.; Noble, C.; Beer, D.; Rao, S.P.S.; et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew. Chem. Int. Ed. 2011, 50, 5889–5891. [Google Scholar] [CrossRef]
- Kazmaier, U.; Junk, L. Recent developments on the synthesis and bioactivity of ilamycins/rufomycins and cyclomarins, marine cyclopeptides that demonstrate anti-malaria and anti-tuberculosis activity. Mar. Drugs 2021, 19, 446. [Google Scholar] [CrossRef]
- Kiefer, A.; Kazmaier, U. Syntheses of Cyclomarins—Interesting Marine Natural Products with Distinct Mode of Action towards Malaria and Tuberculosis. Synthesis 2019, 51, 107–121. [Google Scholar] [CrossRef]
- Sugiyama, H.; Shioiri, T.; Yokokawa, F. Syntheses of four unusual amino acids, constituents of cyclomarin A. Tetrahedron Lett. 2002, 43, 3489–3492. [Google Scholar] [CrossRef]
- Wen, S.J.; Hu, T.S.; Yao, Z.J. Macrocyclization studies and total synthesis of cyclomarin C, an anti-inflammatory marine cyclopeptide. Tetrahedron 2005, 61, 4931–4938. [Google Scholar] [CrossRef]
- Wen, S.J.; Yao, Z.J. Total synthesis of cyclomarin C. Org. Lett. 2004, 6, 2721–2724. [Google Scholar] [CrossRef]
- Barbie, P.; Kazmaier, U. Total synthesis of cyclomarins A, C and D, marine cyclic peptides with interesting anti-tuberculosis and anti-malaria activities. Org. Biomol. Chem. 2016, 14, 6036–6054. [Google Scholar] [CrossRef]
- Barbie, P.; Kazmaier, U. Total Synthesis of Cyclomarin A, a Marine Cycloheptapeptide with Anti-Tuberculosis and Anti-Malaria Activity. Org. Lett. 2016, 18, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, A.; Bader, C.D.; Held, J.; Esser, A.; Rybniker, J.; Empting, M.; Müller, R.; Kazmaier, U. Synthesis of New Cyclomarin Derivatives and Their Biological Evaluation towards Mycobacterium tuberculosis and Plasmodium falciparum. Chem. Eur. J. 2019, 25, 8894–8902. [Google Scholar] [CrossRef] [PubMed]
- Barbie, P.; Kazmaier, U. Total synthesis of desoxycyclomarin C and the cyclomarazines A and B. Org. Biomol. Chem. 2016, 14, 6055–6064. [Google Scholar] [CrossRef]
- Liu, Y.; He, P.; Zhang, Y.; Zhang, X.; Liu, J.; Du, Y. One-pot enantiomeric synthesis of thiazole-containing amino acids: Total synthesis of venturamides A and B. J. Org. Chem. 2018, 83, 3897–3905. [Google Scholar] [CrossRef] [PubMed]
- Stolze, S.C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Solid phase total synthesis of the 3-amino-6-hydroxy-2-piperidone (Ahp) cyclodepsipeptide and protease inhibitor Symplocamide A. Chem. Commun. 2010, 46, 8857–8859. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Fang, W.; Leong, D.T.; Tan, L.T. Biochemical studies of the lagunamides, potent cytotoxic cyclic depsipeptides from the marine cyanobacterium Lyngbya majuscula. Mar. Drugs 2012, 10, 1126–1137. [Google Scholar] [CrossRef]
- Huang, X.; Huang, W.; Li, L.; Sun, X.; Song, S.; Xu, Q.; Zhang, L.; Wei, B.G.; Deng, X. Structure Determinants of Lagunamide A for Anticancer Activity and Its Molecular Mechanism of Mitochondrial Apoptosis. Mol. Pharm. 2016, 13, 3756–3763. [Google Scholar] [CrossRef]
- Dai, L.; Chen, B.; Lei, H.; Wang, Z.; Liu, Y.; Xu, Z.; Ye, T. Total synthesis and stereochemical revision of lagunamide A. Chem. Commun. 2012, 48, 8697–8699. [Google Scholar] [CrossRef]
- Huang, W.; Ren, R.G.; Dong, H.Q.; Wei, B.G.; Lin, G.Q. Diverse synthesis of marine cyclic depsipeptide lagunamide A and its analogues. J. Org. Chem. 2013, 78, 10747–10762. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Chakraborty, T.K. Toward the total synthesis of a lagunamide B analogue. Tetrahedron Lett. 2014, 55, 3469–3472. [Google Scholar] [CrossRef]
- Brockmann, H.; Schmidt-Kastner, G. Valinomycin I, XXVII. Mitteil. über Antibiotica aus Actinomyceten. Chem. Berichte 1955, 88, 57–61. [Google Scholar] [CrossRef]
- Huang, S.; Liu, Y.; Liu, W.; Neubauer, P.; Li, J. The nonribosomal peptide valinomycin: From discovery to bioactivity and biosynthesis. Microorganisms 2021, 9, 780. [Google Scholar] [CrossRef] [PubMed]
- Heisey, R.M.; Mishra, S.K.; Keller, J.E.; Miller, J.R.; Putnam, A.R.; Huang, J.; D’Silva, T.D.J. Production of valinomycin, an insecticidal antibiotic, by Streptomyces griseus var. flexipertum var. nov. J. Agric. Food Chem. 1988, 36, 1283–1286. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Z.; Chen, H.; Lu, Y.; Chen, X. Valinomycin as a potential antiviral agent against coronaviruses: A review. Biomed. J. 2020, 43, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, J.T.; Kellermann, M.Y.; Köck, M.; Putra, M.Y.; Murniasih, T.; Mohr, K.I.; Wink, J.; Praditya, D.F.; Steinmann, E.; Schupp, P.J. Anti-Infective and Antiviral Activity of Valinomycin and Its Analogues from a Sea Cucumber-Associated Bacterium, Streptomyces sp. SV 21. Mar. Drugs 2021, 19, 81. [Google Scholar] [CrossRef]
- Jeon, C.W.; Kim, D.R.; Kwak, Y.S. Valinomycin, produced by Streptomyces sp. S8, a key antifungal metabolite in large patch disease suppressiveness. World J. Microbiol. Biotechnol. 2019, 35, 128. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Baig, M.M.F.A.; Zhang, T.Q.; Zhai, T.T.; Qin, X.; Xia, X.H. Liposomal valinomycin mediated cellular K+ leak promoting apoptosis of liver cancer cells. J. Control. Release 2021, 337, 317–328. [Google Scholar] [CrossRef]
- Rakovic, A.; Ziegler, J.; Mårtensson, C.U.; Prasuhn, J.; Shurkewitsch, K.; König, P.; Paulson, H.L.; Klein, C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 2019, 26, 1428–1441. [Google Scholar] [CrossRef]
- Xiong, X.; Li, S.; Han, T.L.; Zhou, F.; Zhang, X.; Tian, M.; Tang, L.; Li, Y. Study of mitophagy and ATP-related metabolomics based on β-amyloid levels in Alzheimer’s disease. Exp. Cell Res. 2020, 396, 112266. [Google Scholar] [CrossRef]
- Luesch, H.; Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Ulongamides A-F, new β-amino acid-containing cyclodepsipeptides from Palauan collections of the marine cyanobacterium Lyngbya sp. J. Nat. Prod. 2002, 65, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, structure determination, and biological activity of lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Hamann, M.T. Chemistry and biology of kahalalides. Chem. Rev. 2011, 111, 3208–3235. [Google Scholar] [CrossRef] [PubMed]
- Goetz, G.; Yoshida, W.Y.; Scheuer, P.J. The absolute stereochemistry of kahalalide F. Tetrahedron 1999, 55, 7739–7746. [Google Scholar] [CrossRef]
- López-Macià, A.; Jiménez, J.C.; Royo, M.; Giralt, E.; Albericio, F. Synthesis and structure determination of kahalalide F. J. Am. Chem. Soc. 2001, 123, 11398–11401. [Google Scholar] [CrossRef]
- Bonnard, I.; Manzanares, I.; Rinehartt, K.L. Stereochemistry of Kahalalide F. J. Nat. Prod. 2003, 66, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.C.; López-Macià, A.; Gracia, C.; Varón, S.; Carrascal, M.; Caba, J.M.; Royo, M.; Francesch, A.M.; Cuevas, C.; Giralt, E.; et al. Structure-activity relationship of Kahalalide F synthetic analogues. J. Med. Chem. 2008, 51, 4920–4931. [Google Scholar] [CrossRef]
- Gracia, C.; Isidro-Llobet, A.; Cruz, L.J.; Acosta, G.A.; Álvarez, M.; Cuevas, C.; Giralt, E.; Albericio, F. Convergent approaches for the synthesis of the antitumoral peptide, Kahalalide F. study of orthogonal protecting groups. J. Org. Chem. 2006, 71, 7196–7204. [Google Scholar] [CrossRef]
- Wang, B.; Waters, A.L.; Valeriote, F.A.; Hamann, M.T. An efficient and cost-effective approach to kahalalide F N-terminal modifications using a nuisance algal bloom of Bryopsis pennata. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1849–1854. [Google Scholar] [CrossRef]
- Faircloth, G.; Cuevas, C. Kahalalide F and ES285: Potent anticancer agents from marine molluscs. Prog. Mol. Subcell. Biol. 2006, 43, 363–379. [Google Scholar] [PubMed]
- García-Rocha, M.; Bonay, P.; Avila, J. The antitumoral compound Kahalalide F acts on cell lysosomes. Cancer Lett. 1996, 99, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Shilabin, A.G.; Kasanah, N.; Wedge, D.E.; Hamann, M.T. Lysosome and HER3 (ErbB3) selective anticancer agent Kahalalide F: Semisynthetic modifications and antifungal lead-exploration studies. J. Med. Chem. 2007, 50, 4340–4350. [Google Scholar] [CrossRef] [PubMed]
- Janmaat, M.L.; Rodriguez, J.A.; Jimeno, J.; Kruyt, F.A.E.; Giaccone, G. Kahalalide F induces necrosis-like cell death that involves depletion of ErbB3 and inhibition of Akt signaling. Mol. Pharmacol. 2005, 68, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Piggott, A.M.; Karuso, P. Rapid identification of a protein binding partner for the marine natural product kahalalide F by using reverse chemical proteomics. ChemBioChem 2008, 9, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Suárez, Y.; González, L.; Cuadrado, A.; Berciano, M.; Lafarga, M.; Muñoz, A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003, 2, 863–872. [Google Scholar] [PubMed]
- Sewell, J.M.; Mayer, I.; Langdon, S.P.; Smyth, J.F.; Jodrell, D.I.; Guichard, S.M. The mechanism of action of Kahalalide F: Variable cell permeability in human hepatoma cell lines. Eur. J. Cancer 2005, 41, 1637–1644. [Google Scholar] [CrossRef]
- Rawat, D.S.; Joshi, M.C.; Joshi, P.; Atheaya, H. Marine peptides and related compounds in clinical trial+. Anti-Cancer Agents Med. Chem. 2006, 6, 33–40. [Google Scholar] [CrossRef]
- Pardo, B.; Paz-Ares, L.; Tabernero, J.; Ciruelos, E.; García, M.; Salazar, R.; López, A.; Blanco, M.; Nieto, A.; Jimeno, J.; et al. Phase I clinical and pharmacokinetic study of kahalalide F administered weekly as a 1-hour infusion to patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 1116–1123. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; Horenblas, S.; Meinhardt, W.; Stokvis, E.; De Reijke, T.M.; Jimeno, J.M.; Lopez-Lazaro, L.; Lopez Martin, J.A.; Beijnen, J.H.; Schellens, J.H.M. Phase I clinical and pharmacokinetic study of Kahalalide F in patients with advanced androgen refractory prostate cancer. Clin. Cancer Res. 2005, 11, 1854–1862. [Google Scholar] [CrossRef]
- Salazar, R.; Cortés-Funes, H.; Casado, E.; Pardo, B.; López-Martín, A.; Cuadra, C.; Tabernero, J.; Coronado, C.; García, M.; Soto Matos-Pita, A.; et al. Phase i study of weekly kahalalide F as prolonged infusion in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 72, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Martín-Algarra, S.; Espinosa, E.; Rubió, J.; López, J.J.L.; Manzano, J.L.; Carrión, L.A.; Plazaola, A.; Tanovic, A.; Paz-Ares, L. Phase II study of weekly Kahalalide F in patients with advanced malignant melanoma. Eur. J. Cancer 2009, 45, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Wyer, S.; Townsend, D.M.; Ye, Z.; Kourtidis, A.; Choo, Y.M.; de Barros, A.L.B.; Donia, M.S.; Hamann, M.T. Recent advances and limitations in the application of kahalalides for the control of cancer. Biomed. Pharmacother. 2022, 148, 112676. [Google Scholar] [CrossRef]
- Hosta, L.; Pla-Roca, M.; Arbiol, J.; López-Iglesias, C.; Samitier, J.; Cruz, L.J.; Kogan, M.J.; Albericio, F. Conjugation of Kahalalide F with gold nanoparticles to enhance in vitro antitumoral activity. Bioconjugate Chem. 2009, 20, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Shilabin, A.G.; Hamann, M.T. In vitro and in vivo evaluation of select kahalalide F analogs with antitumor and antifungal activities. Bioorg. Med. Chem. 2011, 19, 6628–6632. [Google Scholar] [CrossRef] [PubMed]
- Ciavatta, M.L.; Devi, P.; Carbone, M.; Mathieu, V.; Kiss, R.; Casapullo, A.; Gavagnin, M. Kahalalide F analogues from the mucous secretion of Indian sacoglossan mollusc Elysia ornata. Tetrahedron 2016, 72, 625–631. [Google Scholar] [CrossRef]
- Cruz, L.J.; Cuevas, C.; Cañedo, L.M.; Giralt, E.; Albericio, F. Total solid-phase synthesis of marine cyclodepsipeptide IB-01212. J. Org. Chem. 2006, 71, 3339–3344. [Google Scholar] [CrossRef]
- Cruz, L.J.; Francesch, A.; Cuevas, C.; Albericio, F. Synthesis and structure-activity relationship of cytotoxic marine cyclodepsipeptide IB-01212 analogues. ChemMedChem 2007, 2, 1076–1084. [Google Scholar] [CrossRef]
- Nabika, R.; Oishi, S.; Misu, R.; Ohno, H.; Fujii, N. Synthesis of IB-01212 by multiple N-methylations of peptide bonds. Bioorg. Med. Chem. 2014, 22, 6156–6162. [Google Scholar] [CrossRef]
- Ojima, D.; Mine, H.; Iwasaki, A.; Suenaga, K. Total synthesis of janadolide. Tetrahedron Lett. 2018, 59, 1360–1362. [Google Scholar] [CrossRef]
- Athawale, P.R.; Jachak, G.R.; Shukla, A.; Shanmugam, D.; Reddy, D.S. Efforts to Access the Potent Antitrypanosomal Marine Natural Product Janadolide: Synthesis of Des-tert-butyl Janadolide and Its Biological Evaluation. ACS Omega 2018, 3, 2383–2389. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | Types | Sources | Organisms | Country | Antiparasitic Activities | Refs. |
|---|---|---|---|---|---|---|
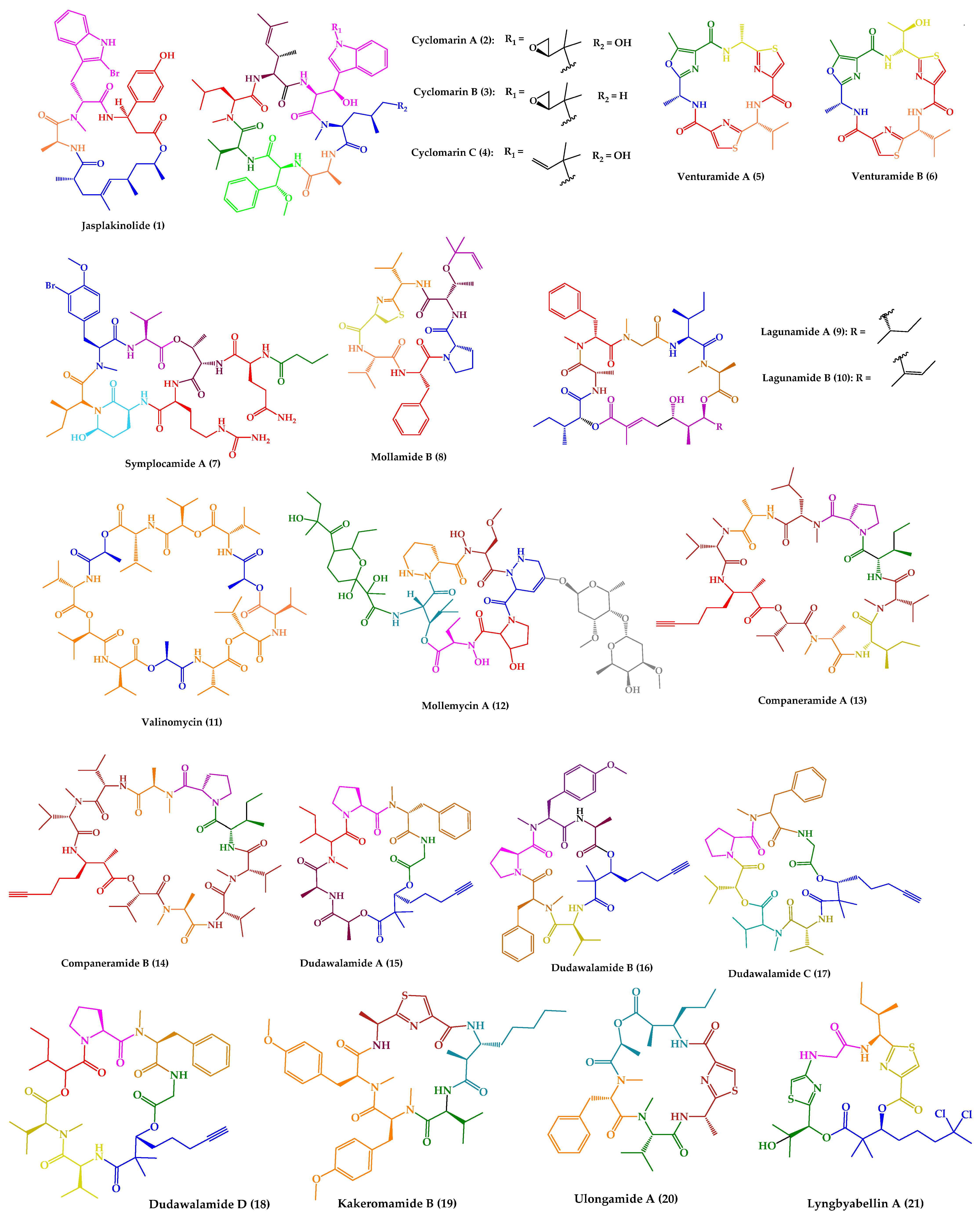
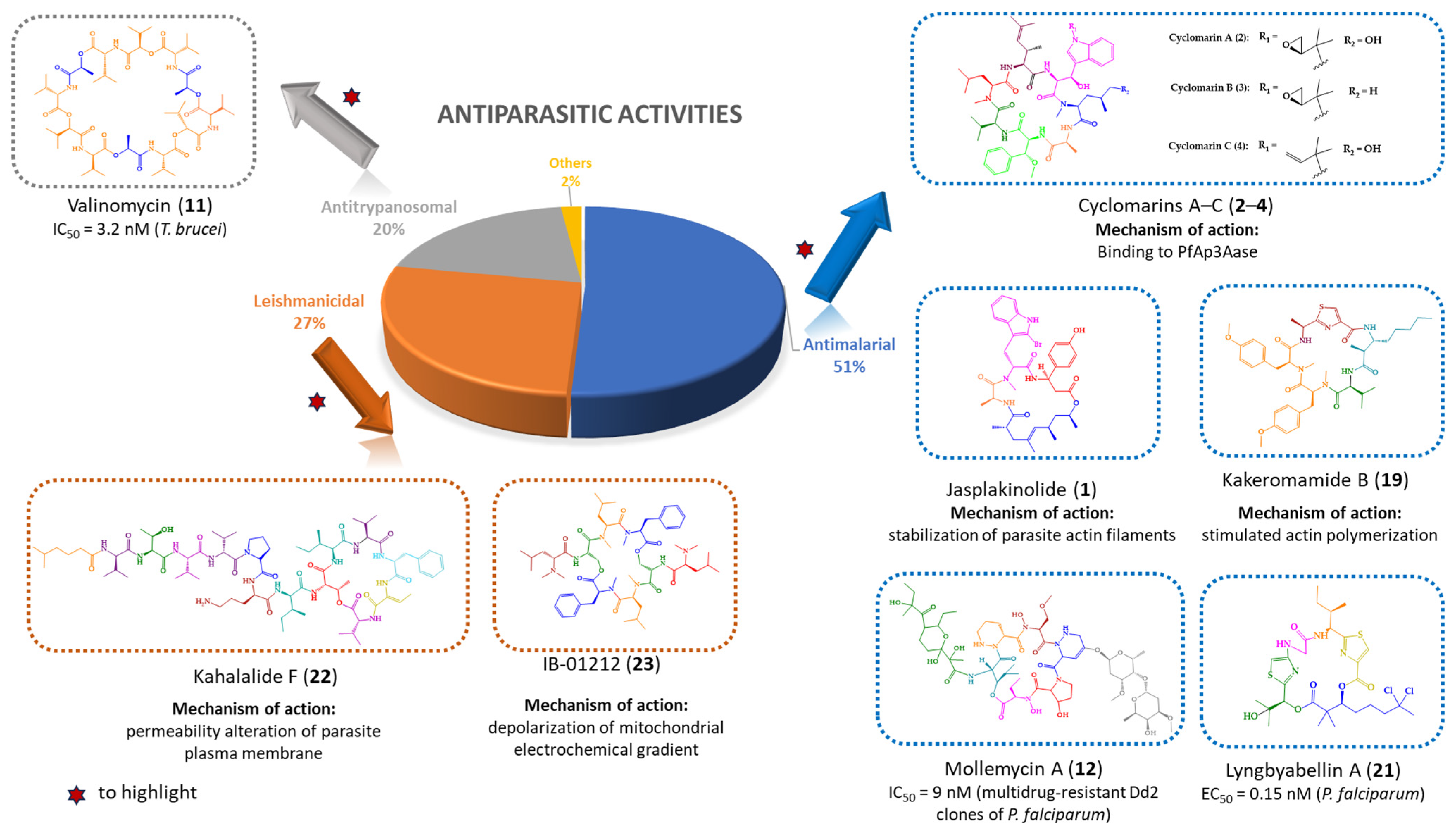
| Jasplakinolide (1) | Cyclic tridepsipeptide | Sponge | Jaspis sp. | Island of Benga, Fiji | active against P. falciparum, T. gondii, E. histolytica and E. invadens | [127,128,129,130,131] |
| Cyclomarins A–C (2–4) | Cyclic heptapeptides | Bacteria | Streptomyces sp. | United States (San Diego, CA) | 2: active against P. falciparum 4: active against multidrug-resistant P. falciparum strains | [132,133,134] |
| Venturamides A–B (5–6) | Cyclic hexapeptides | Cyanobacteria | Oscillatoria sp. | Panama | active against P. falciparum, T. cruzi, and L. donovani | [135] |
| Symplocamide A (7) | Cyclic lipohexadepsipeptide | Cyanobacteria | Symploca sp. | Papua New Guinea | active against P. falciparum, T. cruzi, L. donovani | [136] |
| Mollamide B (8) | Cyclic hexapeptide | Tunicate | Didemnum molle | Manado Bay, Indonesia | active against P. falciparum clones and L. donovani | [137] |
| Lagunamides A and B (9–10) | Cyclic pentadepsipeptides | Cyanobacteria | Lyngbya majuscula | Pulau Hantu Besar, Singapore | active against P. falciparum | [138] |
| Valinomycin (11) | Cyclic dodecadepsipeptide | Bacteria | Streptomyces sp. | Croatia | active against P. falciparum, T. brucei and L. major | [139,140] |
| Mollemycin A (12) | Cyclic glyco-hexadepsipeptide-polyketide | Bacteria | Streptomyces sp. | Australia, South Molle Island, Queensland | active against P. falciparum | [141] |
| Companeramides A and B (13–14) | Cyclic octadepsipeptides | Cyanobacteria | Leptolyngbya sp. | Coiba Island, Panama | active against P. falciparum | [142] |
| Dudawalamides A–D (15–18) | Cyclic hexadepsipeptides | Cyanobacteria | Moorea producens | Papua New Guinean | 15–18: active against P. falciparum and L. donovani 18: active against T. cruzi | [143] |
| Kakeromamide B (19) | Cyclic pentapeptide | Cyanobacteria | Moorea producens | Northern Lau Islands of Fiji | active against P. falciparum blood-stage and against P. berghei liver schizonts | [144] |
| Ulongamide A (20) | Cyclic tetradepsipeptide | Cyanobacteria | (a) Lyngbya sp. (b) Moorea producens | (a) and (b) Northern Lau Islands of Fiji | active against P. falciparum blood-stages | [144] |
| Lyngbyabellin A (21) | Cyclic tetradepsipeptide | Cyanobacteria | (a) Lyngbya majuscula (b) Moorea producens | (a) Northern Lau Islands of Fiji (b) Finger’s Reef, Guam | active against P. falciparum blood-stages | [144] |
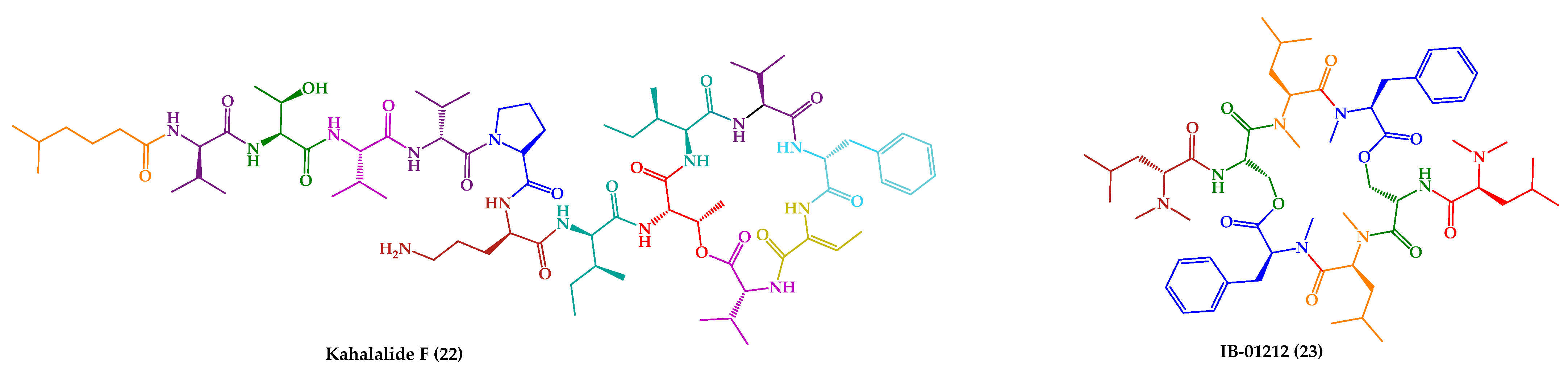
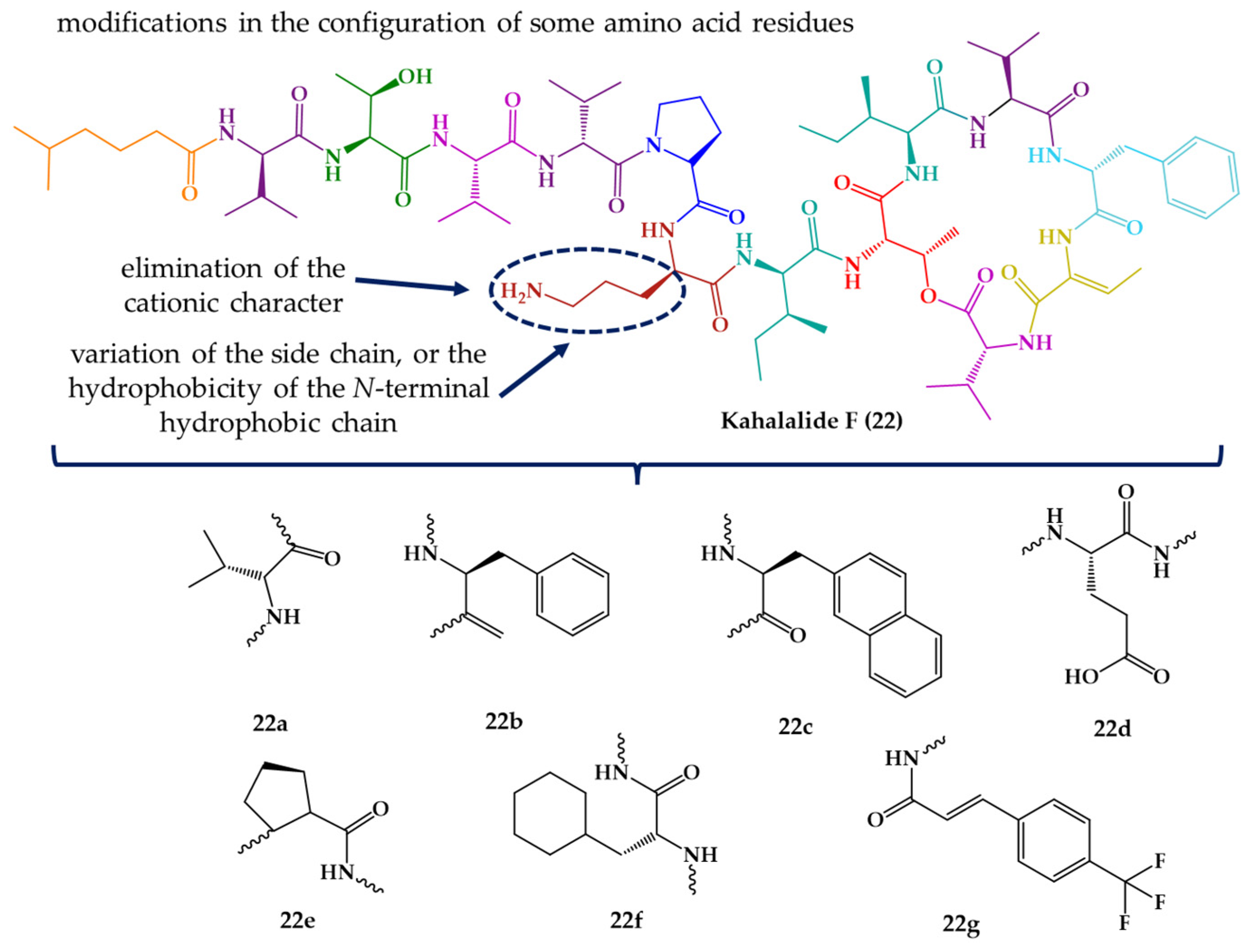
| Kahalalide F (22) | Cyclic dodecadepsipeptide | Mollusk | Elysia rufescens | O’ahu, Hawaii | active against L. donovani promastigotes, L. pifanoi promastigotes and amastigotes | [145,146,147] |
| IB-01212 (23) | Cyclic octadepsipeptide | Fungus | Clonostachys sp. ESNA-A009 | Japan | active against L. donovani promastigotes and L. pifanoi amastigotes | [148,149] |
| Janadolide (24) | Cyclic pentapolyketidepsipeptide hybrid | Cyanobacteria | Okeania sp. | Janado, Okinawa | active against T. b. brucei, T. b. rhodesiense and T. cruzi | [150,151] |
| Motobamide (25) | Cyclic decapeptide | Cyanobacteria | Leptolyngbya sp. | Okinawa Island, Japan | active against bloodstream forms of T. b. rhodesiense | [152] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, R.; Costa, L.; Pinto, E.; Sousa, E.; Fernandes, C. Therapeutic Potential of Marine-Derived Cyclic Peptides as Antiparasitic Agents. Mar. Drugs 2023, 21, 609. https://doi.org/10.3390/md21120609
Ribeiro R, Costa L, Pinto E, Sousa E, Fernandes C. Therapeutic Potential of Marine-Derived Cyclic Peptides as Antiparasitic Agents. Marine Drugs. 2023; 21(12):609. https://doi.org/10.3390/md21120609
Chicago/Turabian StyleRibeiro, Ricardo, Lia Costa, Eugénia Pinto, Emília Sousa, and Carla Fernandes. 2023. "Therapeutic Potential of Marine-Derived Cyclic Peptides as Antiparasitic Agents" Marine Drugs 21, no. 12: 609. https://doi.org/10.3390/md21120609
APA StyleRibeiro, R., Costa, L., Pinto, E., Sousa, E., & Fernandes, C. (2023). Therapeutic Potential of Marine-Derived Cyclic Peptides as Antiparasitic Agents. Marine Drugs, 21(12), 609. https://doi.org/10.3390/md21120609