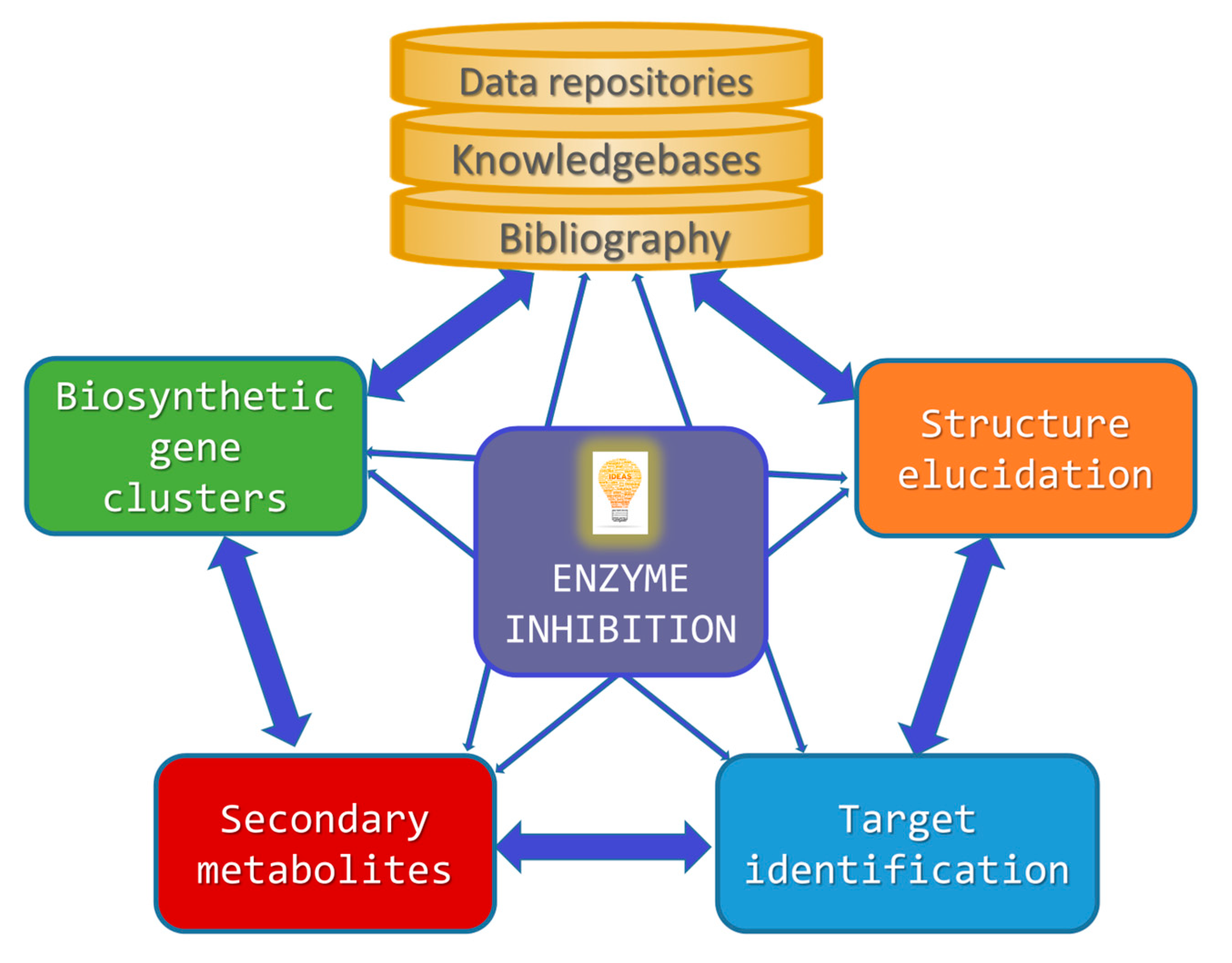
Computational Approaches to Enzyme Inhibition by Marine Natural Products in the Search for New Drugs


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
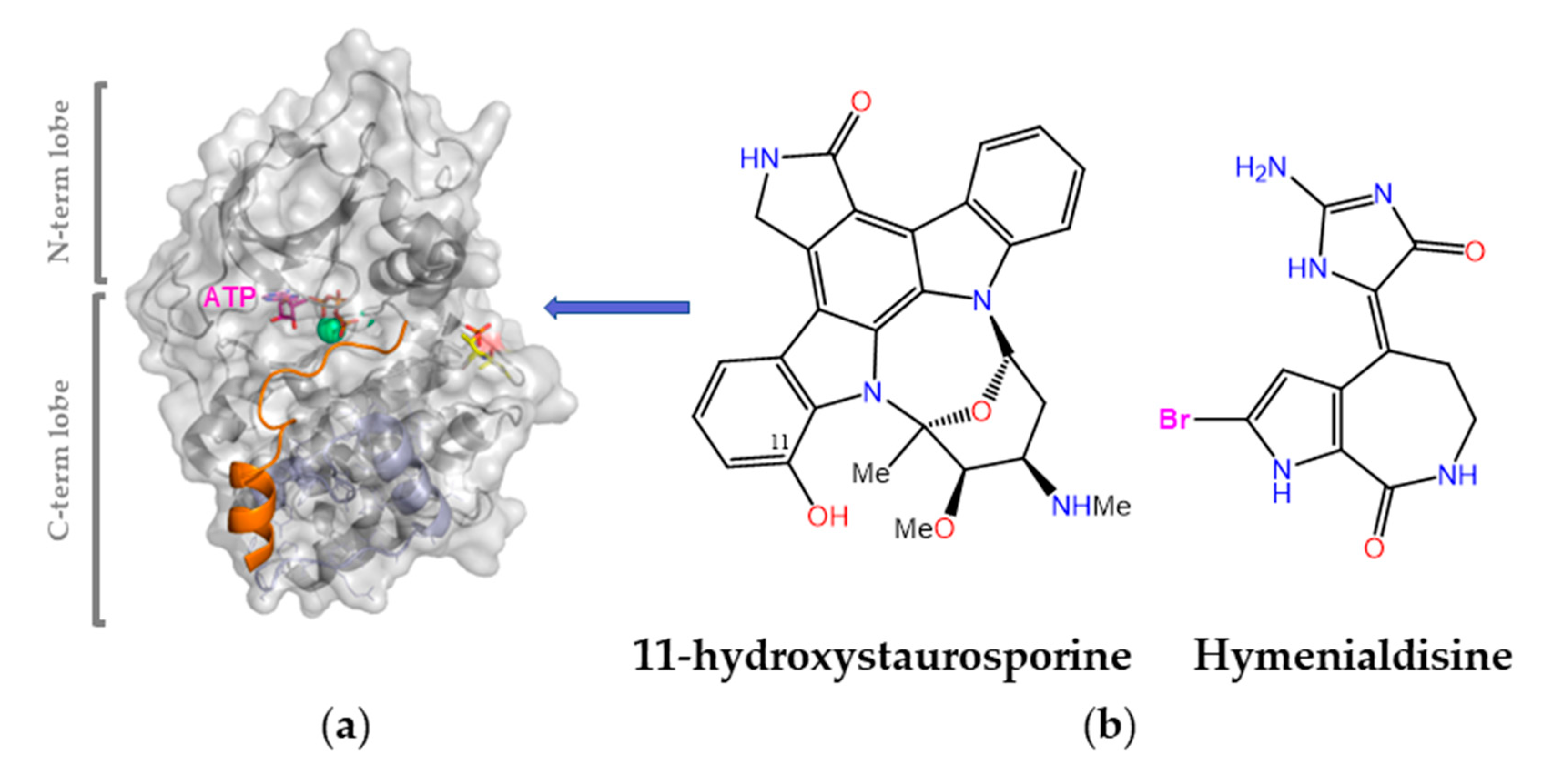{kind=link}
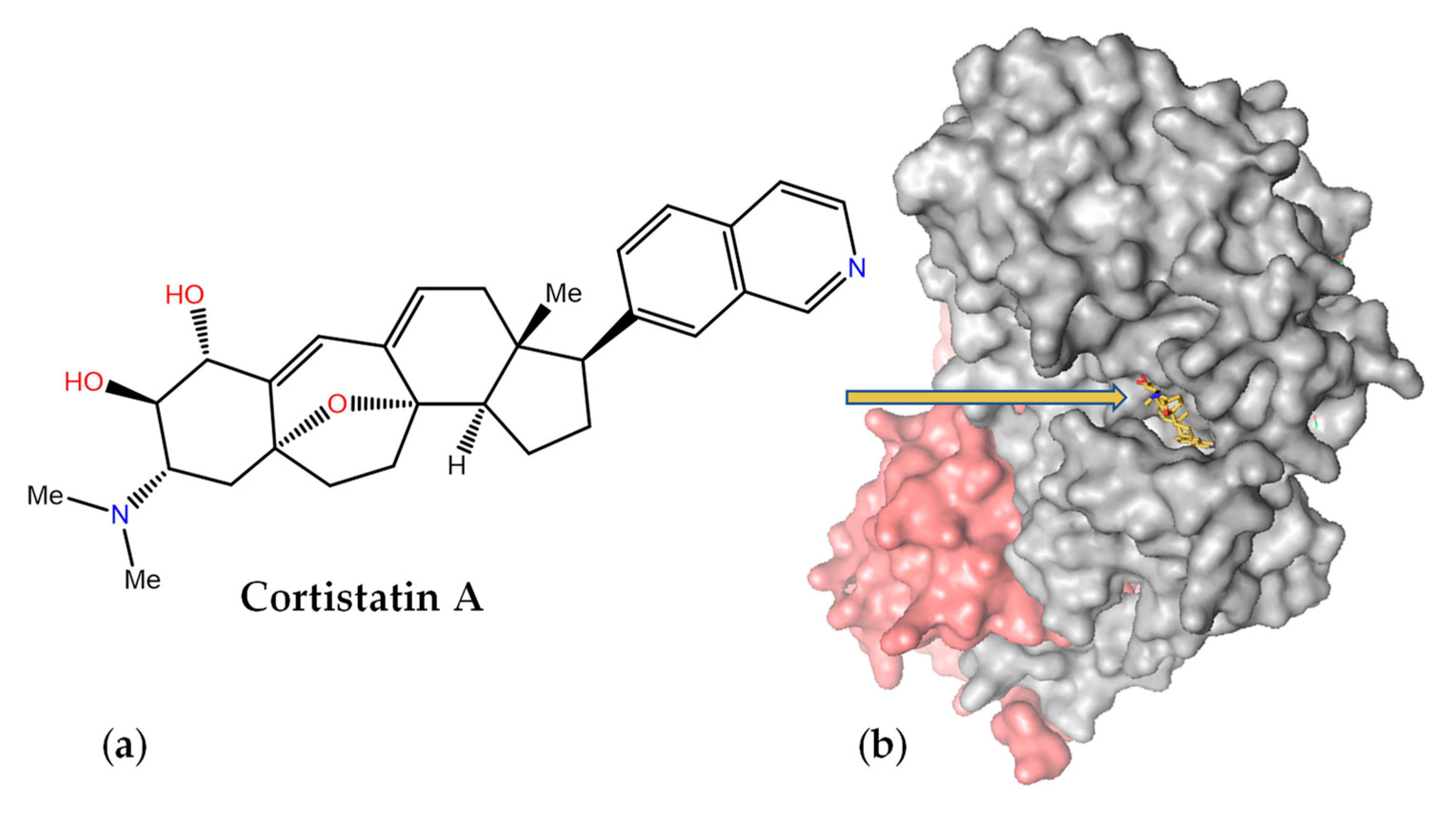{kind=link}
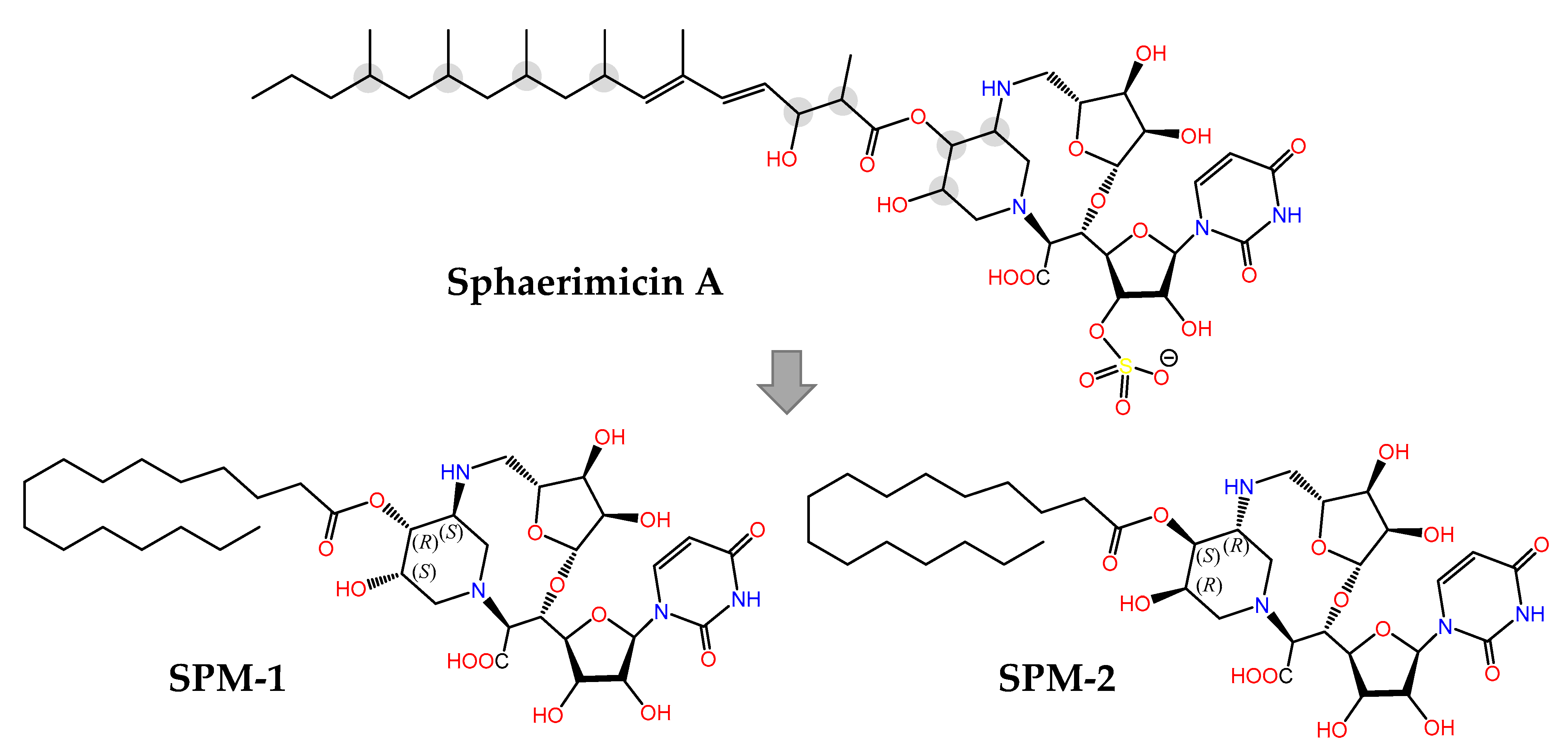{kind=link}
1. Overview
2. Bibliographical Sources and Virtual NP Databases
3. Linking Chemical Diversity of Secondary Metabolites to Biosynthetic Gene Clusters
4. Classification and Chemoinformatic Analyses of Natural Products
5. Linking NPs to Their Targets: Computational Methodologies for Building Global Networks
6. Selected Examples of MNPs Acting as Enzyme Inhibitors
7. Conclusions and Outlook
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Funayama, S.; Cordell, G.A. Alkaloids: A Treasury of Poisons and Medicines; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Mora, C.; Tittensor, D.P.; Adl, S.; Simpson, A.G.; Worm, B. How many species are there on Earth and in the ocean? PLoS Biol. 2011, 9, e1001127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Glaser, K.B.; Mayer, A.M. A renaissance in marine pharmacology: From preclinical curiosity to clinical reality. Biochem. Pharmacol. 2009, 78, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharate, S.B.; Sawant, S.D.; Singh, P.P.; Vishwakarma, R.A. Kinase inhibitors of marine origin. Chem. Rev. 2013, 113, 6761–6815. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M.; Battershill, C.N. Therapeutic agents from the sea: Biodiversity, chemo-evolutionary insight and advances to the end of Darwin’s 200th year. Diving Hyperb. Med. 2009, 39, 216–225. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [Green Version]
- Nakao, Y.; Fusetani, N. Enzyme Inhibitors from Marine Invertebrates. In Handbook of Marine Natural Products; Fattorusso, E., Gerwick, W.H., Taglialatela-Scafati, O., Eds.; Springer Science+Business Media B.V.: Dordrecht, The Netherlands, 2012; pp. 1145–1229. [Google Scholar]
- Duan, D.; Doak, A.K.; Nedyalkova, L.; Shoichet, B.K. Colloidal aggregation and the in vitro activity of traditional Chinese medicines. ACS Chem. Biol. 2015, 10, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Seidler, J.; McGovern, S.L.; Doman, T.N.; Shoichet, B.K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003, 46, 4477–4486. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Weissman, K.J. The structural biology of biosynthetic megaenzymes. Nat. Chem. Biol. 2015, 11, 660–670. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Biochemistry. Directing biosynthesis. Science 2006, 314, 603–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurjens, G.; Kirschning, A.; Candito, D.A. Lessons from the synthetic chemist nature. Nat. Prod. Rep. 2015, 32, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, D.H.; Rath, C.M.; Mortison, J.; Scaglione, J.B.; Kittendorf, J.D. Biosynthetic Principles in Marine Natural Product Systems. In Handbook of Marine Natural Products; Fattorusso, E., Gerwick, W.H., Taglialatela-Scafati, O., Eds.; Springer Nature Switzerland AG: Dordrecht, The Netherlands, 2012; pp. 947–976. [Google Scholar]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Wieland Brown, L.C.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Harizani, M.; Ioannou, E.; Roussis, V. The Laurencia paradox: An endless source of chemodiversity. Prog. Chem. Org. Nat. Prod. 2016, 102, 91–252. [Google Scholar] [CrossRef]
- van Santen, J.A.; Poynton, E.F.; Iskakova, D.; McMann, E.; Alsup, T.A.; Clark, T.N.; Fergusson, C.H.; Fewer, D.P.; Hughes, A.H.; McCadden, C.A.; et al. The Natural Products Atlas 2.0: A database of microbially-derived natural products. Nucleic Acids Res. 2022, 50, D1317–D1323. [Google Scholar] [CrossRef] [PubMed]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Sussmuth, R.D. Bioactive peptide natural products as lead structures for medicinal use. Acc. Chem. Res. 2017, 50, 1566–1576. [Google Scholar] [CrossRef]
- Korteling, J.E.; van de Boer-Visschedijk, G.C.; Blankendaal, R.A.M.; Boonekamp, R.C.; Eikelboom, A.R. Human-versus Artificial Intelligence. Front. Artif. Intell. 2021, 4, 622364. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Fabbro, D.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. The concise guide to pharmacology 2019/20: Enzymes. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S297–S396. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameduh, T.; Haddad, Y.; Adam, V.; Heger, Z. Homology modeling in the time of collective and artificial intelligence. Comput. Struct. Biotechnol. J. 2020, 18, 3494–3506. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic level protein structure with a language model. bioRxiv 2022. [Google Scholar] [CrossRef]
- Drysdale, R.; Cook, C.E.; Petryszak, R.; Baillie-Gerritsen, V.; Barlow, M.; Gasteiger, E.; Gruhl, F.; Haas, J.; Lanfear, J.; Lopez, R.; et al. The ELIXIR Core Data Resources: Fundamental infrastructure for the life sciences. Bioinformatics 2020, 36, 2636–2642. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef]
- Ribeiro, A.J.M.; Holliday, G.L.; Furnham, N.; Tyzack, J.D.; Ferris, K.; Thornton, J.M. Mechanism and Catalytic Site Atlas (M-CSA): A database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 2018, 46, D618–D623. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Pence, H.E.; Williams, A. Chemspider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2002, 19, 1R–49R. [Google Scholar] [CrossRef]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2022, 39, 1122–1171. [Google Scholar] [CrossRef]
- Blunt, J.; Munro, M.H.G. Dictionary of Marine Natural Products, with CD-ROM; Chapman & Hall/CRC: Boca Raton, FA, USA, 2008. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20-A free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminform. 2020, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Kern, F.; Fehlmann, T.; Keller, A. On the lifetime of bioinformatics web services. Nucleic Acids Res. 2020, 48, 12523–12533. [Google Scholar] [CrossRef]
- Rutz, A.; Sorokina, M.; Galgonek, J.; Mietchen, D.; Willighagen, E.; Gaudry, A.; Graham, J.G.; Stephan, R.; Page, R.; Vondrasek, J.; et al. The LOTUS initiative for open knowledge management in natural products research. Elife 2022, 11, e70780. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Erehman, J.; Gohlke, B.O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II--a database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. J. Cheminform. 2021, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.; Munro, M.; Upjohn, M. The Role of Databases in Marine Natural Products Research. In Handbook of Marine Natural Products; Fattorusso, E., Gerwick, W.H., Taglialatela-Scafati, O., Eds.; Springer Science+Business Media B.V.: Dordrecht, The Netherlands, 2012; pp. 389–421. [Google Scholar]
- Shen, S.M.; Appendino, G.; Guo, Y.W. Pitfalls in the structural elucidation of small molecules. A critical analysis of a decade of structural misassignments of marine natural products. Nat. Prod. Rep. 2022, 39, 1803–1832. [Google Scholar] [CrossRef]
- Wishart, D.S. Computational strategies for metabolite identification in metabolomics. Bioanalysis 2009, 1, 1579–1596. [Google Scholar] [CrossRef]
- Burns, D.C.; Mazzola, E.P.; Reynolds, W.F. The role of computer-assisted structure elucidation (CASE) programs in the structure elucidation of complex natural products. Nat. Prod. Rep. 2019, 36, 919–933. [Google Scholar] [CrossRef]
- Urban, S.; Brkljaca, R.; Hoshino, M.; Lee, S.; Fujita, M. Determination of the absolute configuration of the pseudo-symmetric natural product elatenyne by the crystalline sponge method. Angew. Chem. Int. Ed. Engl. 2016, 55, 2678–2682. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef]
- Muchiri, R.N.; van Breemen, R.B. Affinity selection-mass spectrometry for the discovery of pharmacologically active compounds from combinatorial libraries and natural products. J. Mass Spectrom. 2021, 56, e4647. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [Green Version]
- Magrane, M.; UniProt, C. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, T.; Kim, H.U. The secondary metabolite bioinformatics portal: Computational tools to facilitate synthetic biology of secondary metabolite production. Synth. Syst. Biotechnol. 2016, 1, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Scherlach, K.; Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 2021, 12, 3864. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef] [Green Version]
- Klau, L.J.; Podell, S.; Creamer, K.E.; Demko, A.M.; Singh, H.W.; Allen, E.E.; Moore, B.S.; Ziemert, N.; Letzel, A.C.; Jensen, P.R. The Natural Product Domain Seeker version 2 (NaPDoS2) webtool relates ketosynthase phylogeny to biosynthetic function. J. Biol. Chem. 2022, 298, 102480. [Google Scholar] [CrossRef]
- Albarano, L.; Esposito, R.; Ruocco, N.; Costantini, M. Genome mining as new challenge in natural products discovery. Mar. Drugs 2020, 18, 199. [Google Scholar] [CrossRef] [Green Version]
- Medema, M.H.; Fischbach, M.A. Computational approaches to natural product discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef]
- Winter, J.M.; Behnken, S.; Hertweck, C. Genomics-inspired discovery of natural products. Curr. Opin. Chem. Biol. 2011, 15, 22–31. [Google Scholar] [CrossRef]
- Lane, A.L.; Moore, B.S. A sea of biosynthesis: Marine natural products meet the molecular age. Nat. Prod. Rep. 2011, 28, 411–428. [Google Scholar] [CrossRef] [Green Version]
- Bonet, B.; Teufel, R.; Crusemann, M.; Ziemert, N.; Moore, B.S. Direct capture and heterologous expression of Salinispora natural product genes for the biosynthesis of enterocin. J. Nat. Prod. 2015, 78, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, P.R. Natural products and the gene cluster revolution. Trends Microbiol. 2016, 24, 968–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medema, M.H. The year 2020 in natural product bioinformatics: An overview of the latest tools and databases. Nat. Prod. Rep. 2021, 38, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Doroghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K.S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014, 10, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; de Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Masso-Silva, J.A.; Diamond, G. Antimicrobial peptides from fish. Pharmaceuticals 2014, 7, 265–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barroso, C.; Carvalho, P.; Goncalves, J.F.M.; Rodrigues, P.N.S.; Neves, J.V. Antimicrobial peptides: Identification of two b-defensins in a teleost fish, the european sea bass (Dicentrarchus labrax). Pharmaceuticals 2021, 14, 566. [Google Scholar] [CrossRef]
- Tincu, J.A.; Taylor, S.W. Antimicrobial peptides from marine invertebrates. Antimicrob. Agents Chemother. 2004, 48, 3645–3654. [Google Scholar] [CrossRef] [Green Version]
- Sychev, S.V.; Sukhanov, S.V.; Panteleev, P.V.; Shenkarev, Z.O.; Ovchinnikova, T.V. Marine antimicrobial peptide arenicin adopts a monomeric twisted beta-hairpin structure and forms low conductivity pores in zwitterionic lipid bilayers. Pept. Sci. 2017, 110, e23093. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekman, D.C.; Zenz, A.; Gudmundsdottir, B.K.; Lohner, K.; Maier, V.H.; Gudmundsson, G.H. Functional characterization of codCath, the mature cathelicidin antimicrobial peptide from Atlantic cod (Gadus morhua). Peptides 2011, 32, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Lazzarini, A.; Carrano, L.; Corti, E.; Ciciliato, I.; Gastaldo, L.; Candiani, P.; Losi, D.; Marinelli, F.; Selva, E.; et al. Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chem. Biol. 2008, 15, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Chen, Q.; Chen, S.; Xu, B.; Ju, J.; Wang, H. Mathermycin, a lantibiotic from the marine actinomycete Marinactinospora thermotolerans SCSIO 00652. Appl. Environ. Microbiol. 2017, 83, e00926-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G. The antimicrobial peptide database provides a platform for decoding the design principles of naturally occurring antimicrobial peptides. Protein Sci. 2020, 29, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef]
- Waghu, F.H.; Idicula-Thomas, S. Collection of antimicrobial peptides database and its derivatives: Applications and beyond. Protein Sci. 2020, 29, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Gawde, U.; Chakraborty, S.; Waghu, F.H.; Barai, R.S.; Khanderkar, A.; Indraguru, R.; Shirsat, T.; Idicula-Thomas, S. CAMPR4: A database of natural and synthetic antimicrobial peptides. Nucleic Acids Res. 2022, 51, D377–D383. [Google Scholar] [CrossRef]
- van Santen, J.A.; Jacob, G.; Singh, A.L.; Aniebok, V.; Balunas, M.J.; Bunsko, D.; Neto, F.C.; Castano-Espriu, L.; Chang, C.; Clark, T.N.; et al. The Natural Products Atlas: An open access knowledge base for microbial natural products discovery. ACS Cent. Sci. 2019, 5, 1824–1833. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.R.; Pinto, E.; Torres, M.A.; Dorr, F.; Mazur-Marzec, H.; Szubert, K.; Tartaglione, L.; Dell’Aversano, C.; Miles, C.O.; Beach, D.G.; et al. CyanoMetDB, a comprehensive public database of secondary metabolites from cyanobacteria. Water Res. 2021, 196, 117017. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Wang, M.; Leber, C.A.; Nothias, L.F.; Reher, R.; Kang, K.B.; van der Hooft, J.J.J.; Dorrestein, P.C.; Gerwick, W.H.; Cottrell, G.W. NPClassifier: A deep neural network-based structural classification tool for natural products. J. Nat. Prod. 2021, 84, 2795–2807. [Google Scholar] [CrossRef]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Ocean plankton. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Zhang, W.; Ding, W.; Wang, M.; Fan, S.; Yang, B.; McMinn, A.; Wang, M.; Xie, B.B.; Qin, Q.L.; et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome 2020, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology, C. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Wishart, D.S.; Girod, S.; Peters, H.; Oler, E.; Jovel, J.; Budinski, Z.; Milford, R.; Lui, V.W.; Sayeeda, Z.; Mah, R.; et al. ChemFOnt: The chemical functional ontology resource. Nucleic Acids Res. 2022, 51, D1220–D1229. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. Properties of known drugs. 2. Side chains. J. Med. Chem. 1999, 42, 5095–5099. [Google Scholar] [CrossRef]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008, 48, 68–74. [Google Scholar] [CrossRef]
- Chen, Y.; García de Lomana, M.; Friedrich, N.O.; Kirchmair, J. Characterization of the chemical space of known and readily obtainable natural products. J. Chem. Inf. Model. 2018, 58, 1518–1532. [Google Scholar] [CrossRef]
- Stone, S.; Newman, D.J.; Colletti, S.L.; Tan, D.S. Cheminformatic analysis of natural product-based drugs and chemical probes. Nat. Prod. Rep. 2022, 39, 20–32. [Google Scholar] [CrossRef]
- Zhang, M.Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef]
- Oprea, T.I.; Gottfries, J. Chemography: The art of navigating in chemical space. J. Comb. Chem. 2001, 3, 157–166. [Google Scholar] [CrossRef]
- Larsson, J.; Gottfries, J.; Bohlin, L.; Backlund, A. Expanding the ChemGPS chemical space with natural products. J. Nat. Prod. 2005, 68, 985–991. [Google Scholar] [CrossRef]
- Larsson, J.; Gottfries, J.; Muresan, S.; Backlund, A. ChemGPS-NP: Tuned for navigation in biologically relevant chemical space. J. Nat. Prod. 2007, 70, 789–794. [Google Scholar] [CrossRef]
- Rosén, J.; Lövgren, A.; Kogej, T.; Muresan, S.; Gottfries, J.; Backlund, A. ChemGPS-NP(Web): Chemical space navigation online. J. Comput. Aided Mol. Des. 2009, 23, 253–259. [Google Scholar] [CrossRef]
- Koch, M.A.; Schuffenhauer, A.; Scheck, M.; Wetzel, S.; Casaulta, M.; Odermatt, A.; Ertl, P.; Waldmann, H. Charting biologically relevant chemical space: A structural classification of natural products (SCONP). Proc. Natl. Acad. Sci. USA 2005, 102, 17272–17277. [Google Scholar] [CrossRef] [Green Version]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Ertl, P.; Schuhmann, T. Cheminformatics analysis of natural product scaffolds: Comparison of scaffolds produced by animals, plants, fungi and bacteria. Mol. Inform. 2020, 39, e2000017. [Google Scholar] [CrossRef]
- Schafer, T.; Kriege, N.; Humbeck, L.; Klein, K.; Koch, O.; Mutzel, P. Scaffold Hunter: A comprehensive visual analytics framework for drug discovery. J. Cheminform. 2017, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Voser, T.M.; Campbell, M.D.; Carroll, A.R. How different are marine microbial natural products compared to their terrestrial counterparts? Nat. Prod. Rep. 2022, 39, 7–19. [Google Scholar] [CrossRef]
- Over, B.; Wetzel, S.; Grutter, C.; Nakai, Y.; Renner, S.; Rauh, D.; Waldmann, H. Natural-product-derived fragments for fragment-based ligand discovery. Nat. Chem. 2013, 5, 21–28. [Google Scholar] [CrossRef]
- Elion, G.B.; Hitchings, G.H. The synthesis of 6-thioguanine. J. Am. Chem. Soc. 2002, 77, 1676. [Google Scholar] [CrossRef]
- Coyne, S.; Chizzali, C.; Khalil, M.N.; Litomska, A.; Richter, K.; Beerhues, L.; Hertweck, C. Biosynthesis of the antimetabolite 6-thioguanine in Erwinia amylovora plays a key role in fire blight pathogenesis. Angew. Chem. Int. Ed. Engl. 2013, 52, 10564–10568. [Google Scholar] [CrossRef]
- Grigalunas, M.; Brakmann, S.; Waldmann, H. Chemical evolution of natural product structure. J. Am. Chem. Soc. 2022, 144, 3314–3329. [Google Scholar] [CrossRef]
- Müller, G. Medicinal chemistry of target family-directed masterkeys. Drug Discov. Today 2003, 8, 681–691. [Google Scholar] [CrossRef]
- Bon, R.S.; Waldmann, H. Bioactivity-guided navigation of chemical space. Acc. Chem. Res. 2010, 43, 1103–1114. [Google Scholar] [CrossRef]
- Wetzel, S.; Bon, R.S.; Kumar, K.; Waldmann, H. Biology-oriented synthesis. Angew. Chem. Int. Ed. Engl. 2011, 50, 10800–10826. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Seiple, I.B.; Zhang, Z.; Jakubec, P.; Langlois-Mercier, A.; Wright, P.M.; Hog, D.T.; Yabu, K.; Allu, S.R.; Fukuzaki, T.; Carlsen, P.N.; et al. A platform for the discovery of new macrolide antibiotics. Nature 2016, 533, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Könst, Z.A.; Szklarski, A.R.; Pellegrino, S.; Michalak, S.E.; Meyer, M.; Zanette, C.; Cencic, R.; Nam, S.; Voora, V.K.; Horne, D.A.; et al. Synthesis facilitates an understanding of the structural basis for translation inhibition by the lissoclimides. Nat. Chem. 2017, 9, 1140–1149. [Google Scholar] [CrossRef]
- Tan, D.S.; Foley, M.A.; Shair, M.D.; Schreiber, S.L. Stereoselective synthesis of over two million compounds having structural features both reminiscent of natural products and compatible with miniaturized cell-based assays. J. Am. Chem. Soc. 1998, 120, 8565–8566. [Google Scholar] [CrossRef]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [Green Version]
- Wender, P.A. Toward the ideal synthesis and molecular function through synthesis-informed design. Nat. Prod. Rep. 2014, 31, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremosnik, G.S.; Liu, J.; Waldmann, H. Guided by evolution: From biology oriented synthesis to pseudo natural products. Nat. Prod. Rep. 2020, 37, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [Google Scholar] [CrossRef]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Brakmann, S.; Waldmann, H. Pseudo natural products-chemical evolution of natural product structure. Angew. Chem. Int. Ed. Engl. 2021, 60, 15705–15723. [Google Scholar] [CrossRef]
- van Hattum, H.; Waldmann, H. Biology-oriented synthesis: Harnessing the power of evolution. J. Am. Chem. Soc. 2014, 136, 11853–11859. [Google Scholar] [CrossRef]
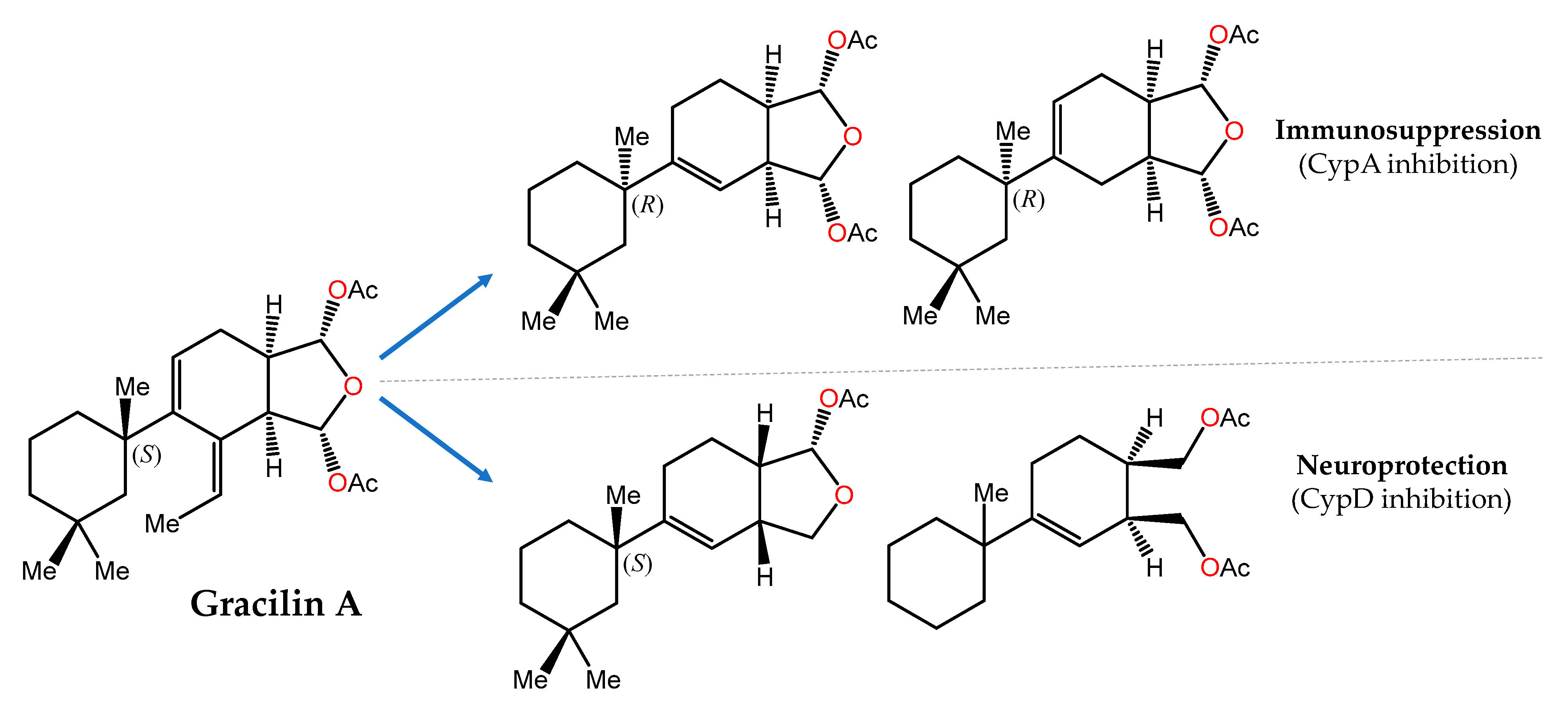
- Abbasov, M.E.; Alvariño, R.; Chaheine, C.M.; Alonso, E.; Sánchez, J.A.; Conner, M.L.; Alfonso, A.; Jaspars, M.; Botana, L.M.; Romo, D. Simplified immunosuppressive and neuroprotective agents based on gracilin A. Nat. Chem. 2019, 11, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wu, Z.; Cai, C.; Wang, Q.; Tang, Y.; Cheng, F. Quantitative and systems pharmacology. 1. In silico prediction of drug-target interactions of natural products enables new targeted cancer therapy. J. Chem. Inf. Model. 2017, 57, 2657–2671. [Google Scholar] [CrossRef] [PubMed]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Tang, J.; Tanoli, Z.U.; Ravikumar, B.; Alam, Z.; Rebane, A.; Vaha-Koskela, M.; Peddinti, G.; van Adrichem, A.J.; Wakkinen, J.; Jaiswal, A.; et al. Drug Target Commons: A community effort to build a consensus knowledge base for drug-target interactions. Cell Chem. Biol. 2018, 25, 224–229.e2. [Google Scholar] [CrossRef] [Green Version]
- Pillich, R.T.; Chen, J.; Churas, C.; Liu, S.; Ono, K.; Otasek, D.; Pratt, D. NDEx: Accessing network models and streamlining network biology workflows. Curr. Protoc. 2021, 1, e258. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, F.; Li, J.; Li, W.; Liu, G.; Tang, Y. SDTNBI: An integrated network and chemoinformatics tool for systematic prediction of drug-target interactions and drug repositioning. Brief. Bioinform. 2017, 18, 333–347. [Google Scholar] [CrossRef]
- Wu, Z.; Ma, H.; Liu, Z.; Zheng, L.; Yu, Z.; Cao, S.; Fang, W.; Wu, L.; Li, W.; Liu, G.; et al. wSDTNBI: A novel network-based inference method for virtual screening. Chem. Sci. 2022, 13, 1060–1079. [Google Scholar] [CrossRef]
- Wu, Z.; Li, W.; Liu, G.; Tang, Y. Network-based methods for prediction of drug-target interactions. Front. Pharmacol. 2018, 9, 1134. [Google Scholar] [CrossRef] [Green Version]
- Gfeller, D.; Michielin, O.; Zoete, V. Shaping the interaction landscape of bioactive molecules. Bioinformatics 2013, 29, 3073–3079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Yaacoub, J.C.; Gleave, J.; Fernandez, M.; Ton, A.T.; Ban, F.; Stern, A.; Cherkasov, A. Artificial intelligence-enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 2022, 17, 672–697. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Cote, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef] [Green Version]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B. Feeling Nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- Bisson, J.; McAlpine, J.B.; Friesen, J.B.; Chen, S.N.; Graham, J.; Pauli, G.F. Can invalid bioactives undermine natural product-based drug discovery? J. Med. Chem. 2016, 59, 1671–1690. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Nissink, J.W.M. Seven year itch: Pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Stork, C.; Chen, Y.; Sicho, M.; Kirchmair, J. Hit Dexter 2.0: Machine-learning models for the prediction of frequent hitters. J. Chem. Inf. Model. 2019, 59, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
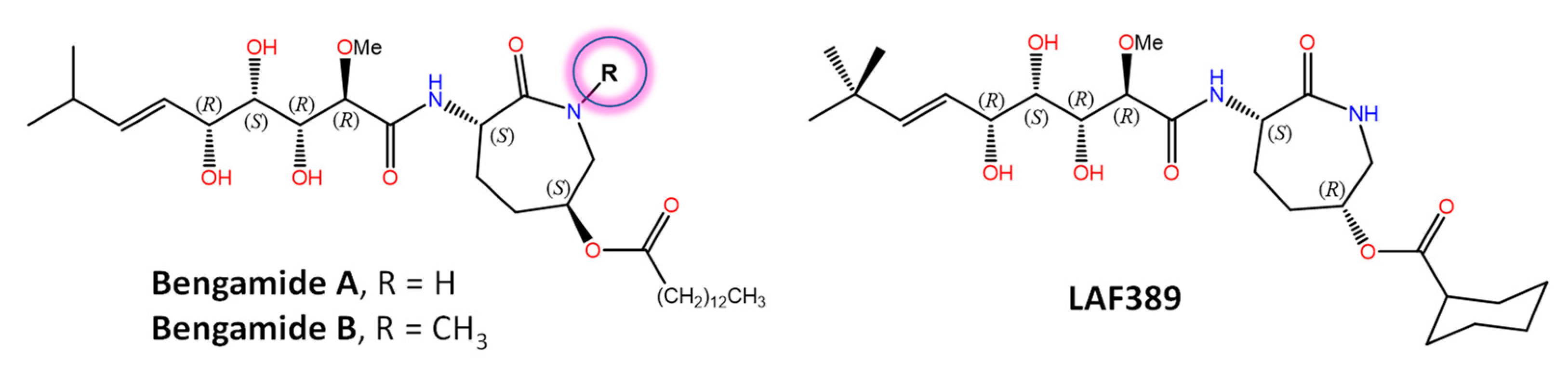
- Quiñoà, E.; Adamczeski, M.; Crews, P.; Bakus, G.J. Bengamides, heterocyclic anthelmintics from a Jaspidae marine sponge. J. Org. Chem. 1986, 51, 4494–4497. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Miao, Z.H. Marine-derived angiogenesis inhibitors for cancer therapy. Mar. Drugs 2013, 11, 903–933. [Google Scholar] [CrossRef] [Green Version]
- White, K.N.; Tenney, K.; Crews, P. The bengamides: A mini-review of natural sources, analogues, biological properties, biosynthetic origins, and future prospects. J. Nat. Prod. 2017, 80, 740–755. [Google Scholar] [CrossRef]
- Towbin, H.; Bair, K.W.; DeCaprio, J.A.; Eck, M.J.; Kim, S.; Kinder, F.R.; Morollo, A.; Mueller, D.R.; Schindler, P.; Song, H.K.; et al. Proteomics-based target identification: Bengamides as a new class of methionine aminopeptidase inhibitors. J. Biol. Chem. 2003, 278, 52964–52971. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Lu, J.P.; Ye, Q.Z. Structural analysis of bengamide derivatives as inhibitors of methionine aminopeptidases. J. Med. Chem. 2012, 55, 8021–8027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.P.; Yuan, X.H.; Yuan, H.; Wang, W.L.; Wan, B.; Franzblau, S.G.; Ye, Q.Z. Inhibition of Mycobacterium tuberculosis methionine aminopeptidases by bengamide derivatives. ChemMedChem 2011, 6, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porras-Alcalá, C.; Moya-Utrera, F.; García-Castro, M.; Sánchez-Ruiz, A.; López-Romero, J.M.; Pino-González, M.S.; Díaz-Morilla, A.; Kitamura, S.; Wolan, D.W.; Prados, J.; et al. The development of the bengamides as new antibiotics against drug-resistant bacteria. Mar. Drugs 2022, 20, 373. [Google Scholar] [CrossRef]
- Liu, S.; Widom, J.; Kemp, C.W.; Crews, C.M.; Clardy, J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science 1998, 282, 1324–1327. [Google Scholar] [CrossRef]
- Bailey, L. Effect of fumagillin upon Nosema apis (Zander). Nature 1953, 171, 212–213. [Google Scholar] [CrossRef]
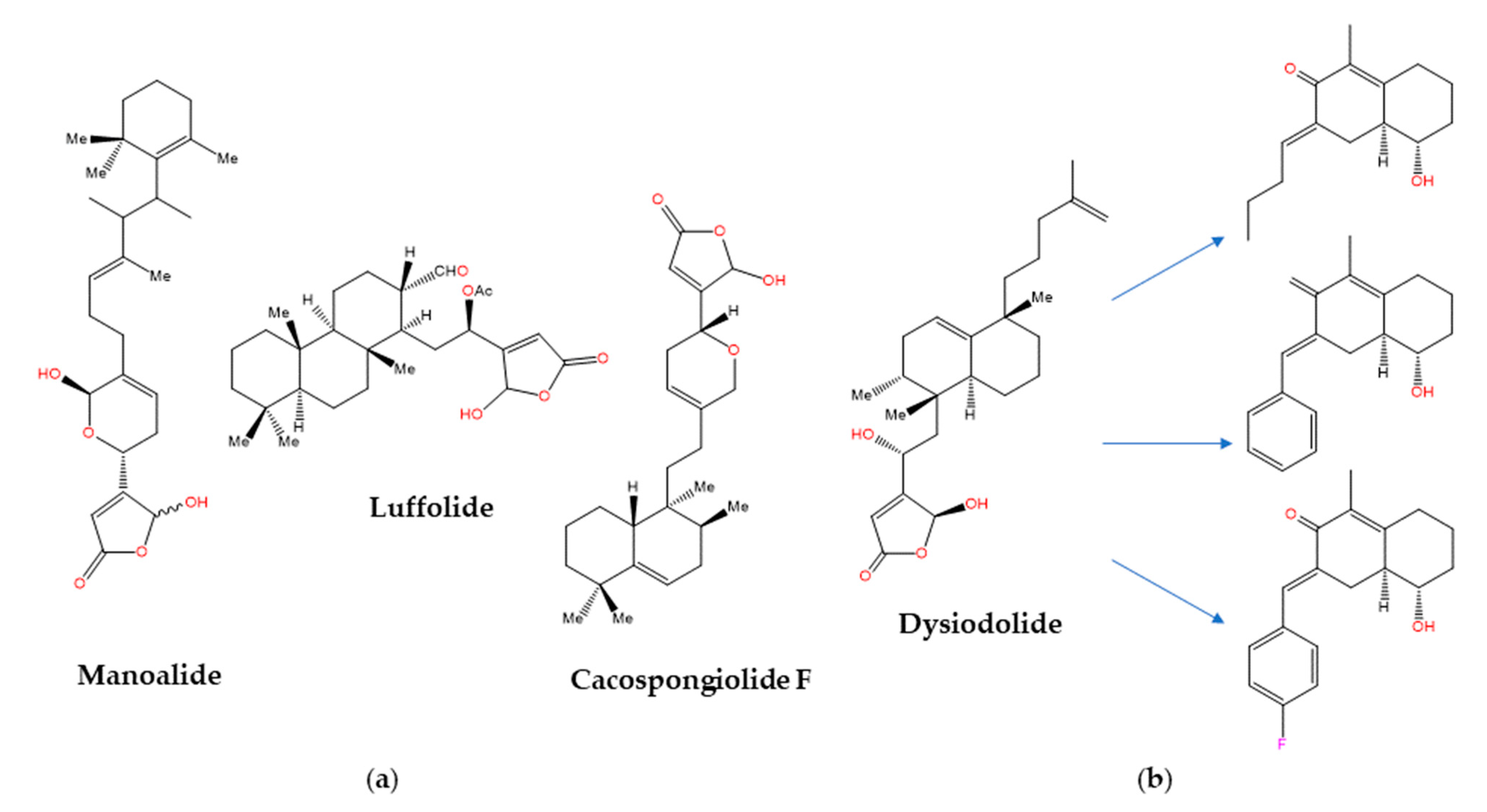
- Rateb, M.E.; Houssen, W.E.; Schumacher, M.; Harrison, W.T.; Diederich, M.; Ebel, R.; Jaspars, M. Bioactive diterpene derivatives from the marine sponge Spongionella sp. J. Nat. Prod. 2009, 72, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Potts, B.C.; Faulkner, D.J.; Jacobs, R.S. Phospholipase A2 inhibitors from marine organisms. J. Nat. Prod. 1992, 55, 1701–1717. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, J.A.; Alfonso, A.; Leirós, M.; Alonso, E.; Rateb, M.E.; Jaspars, M.; Houssen, W.E.; Ebel, R.; Tabudravu, J.; Botana, L.M. Identification of Spongionella compounds as cyclosporine A mimics. Pharmacol. Res. 2016, 107, 407–414. [Google Scholar] [CrossRef]
- Li, K.; Gustafson, K.R. Sesterterpenoids: Chemistry, biology, and biosynthesis. Nat. Prod. Rep. 2021, 38, 1251–1281. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.S.; Lin, W.; Proksch, P. Bioactive sesterterpenes and triterpenes from marine sponges: Occurrence and pharmacological significance. Mar. Drugs 2010, 8, 313–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posadas, I.; De Rosa, S.; Terencio, M.C.; Paya, M.; Alcaraz, M.J. Cacospongionolide B suppresses the expression of inflammatory enzymes and tumour necrosis factor-alpha by inhibiting nuclear factor-kappa B activation. Br. J. Pharmacol. 2003, 138, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Gunasekera, S.P.; McCarthy, P.J.; Kelly-Borges, M.; Lobkovsky, E.; Clardy, J. Dysidiolide: A novel protein phosphatase inhibitor from the Caribbean sponge Dysidea etheria de Laubenfels. J. Am. Chem. Soc. 1996, 118, 8759–8760. [Google Scholar] [CrossRef]
- Marcos, I.S.; Escola, M.A.; Moro, R.F.; Basabe, P.; Diez, D.; Sanz, F.; Mollinedo, F.; de la Iglesia-Vicente, J.; Sierra, B.G.; Urones, J.G. Synthesis of novel antitumoural analogues of dysidiolide from ent-halimic acid. Bioorg. Med. Chem. 2007, 15, 5719–5737. [Google Scholar] [CrossRef]
- Dekker, F.J.; Koch, M.A.; Waldmann, H. Protein structure similarity clustering (PSSC) and natural product structure as inspiration sources for drug development and chemical genomics. Curr. Opin. Chem. Biol. 2005, 9, 232–239. [Google Scholar] [CrossRef]
- Koch, M.A.; Wittenberg, L.O.; Basu, S.; Jeyaraj, D.A.; Gourzoulidou, E.; Reinecke, K.; Odermatt, A.; Waldmann, H. Compound library development guided by protein structure similarity clustering and natural product structure. Proc. Natl. Acad. Sci. USA 2004, 101, 16721–16726. [Google Scholar] [CrossRef] [Green Version]
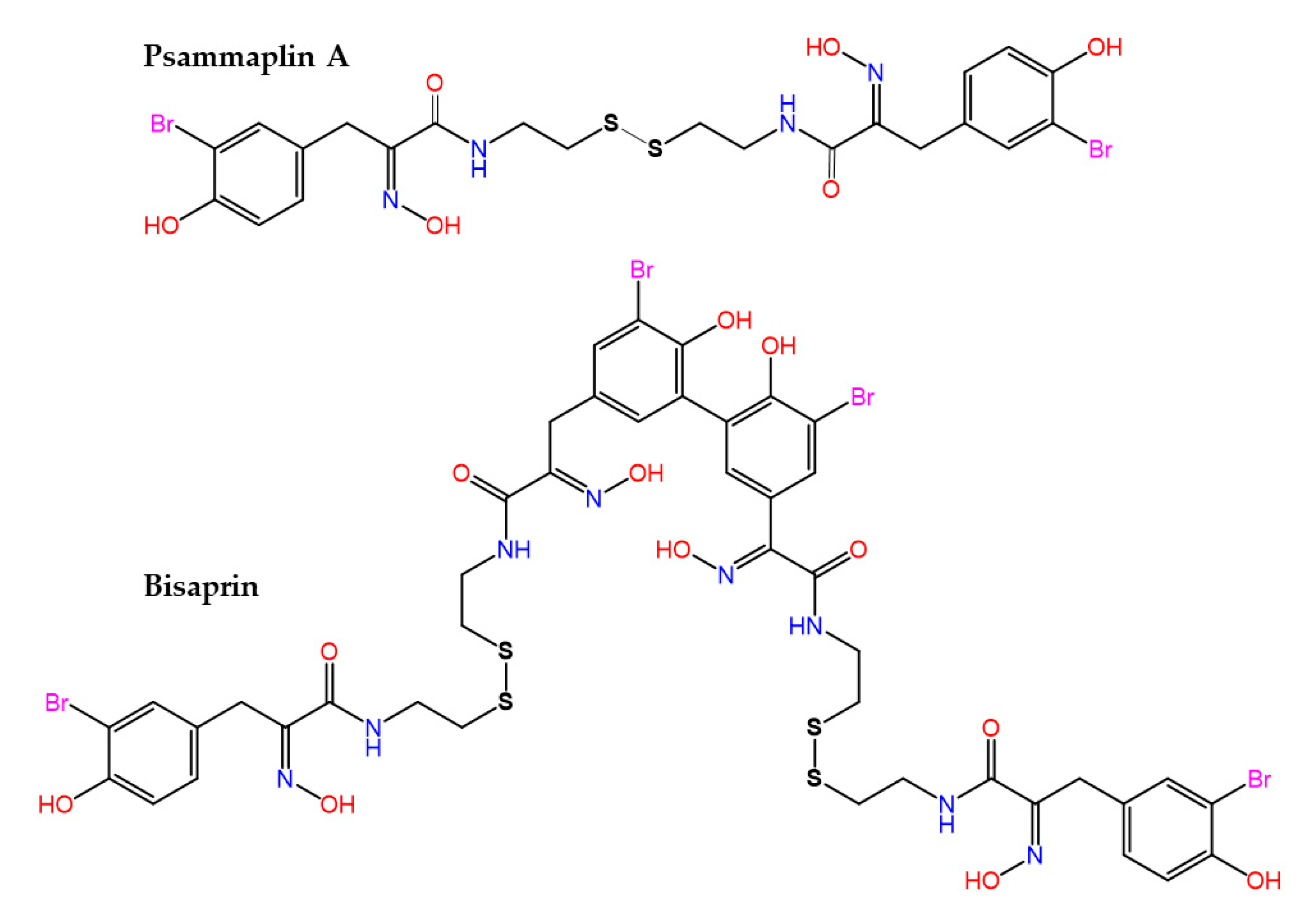
- Pina, I.C.; Gautschi, J.T.; Wang, G.Y.; Sanders, M.L.; Schmitz, F.J.; France, D.; Cornell-Kennon, S.; Sambucetti, L.C.; Remiszewski, S.W.; Perez, L.B.; et al. Psammaplins from the sponge Pseudoceratina purpurea: Inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 2003, 68, 3866–3873. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Shin, J.; Kwon, H.J. Psammaplin A is a natural prodrug that inhibits class I histone deacetylase. Exp. Mol. Med. 2007, 39, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sayed, A.M. The Pherobase: Database of Pheromones and Semiochemicals; 2022. Available online: https://pherolist.org/ (accessed on 20 December 2022).
- Oluwabusola, E.T.; Katermeran, N.P.; Poh, W.H.; Goh, T.M.B.; Tan, L.T.; Diyaolu, O.; Tabudravu, J.; Ebel, R.; Rice, S.A.; Jaspars, M. Inhibition of the quorum sensing system, elastase production and biofilm formation in Pseudomonas aeruginosa by psammaplin A and bisaprasin. Molecules 2022, 27, 1721. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
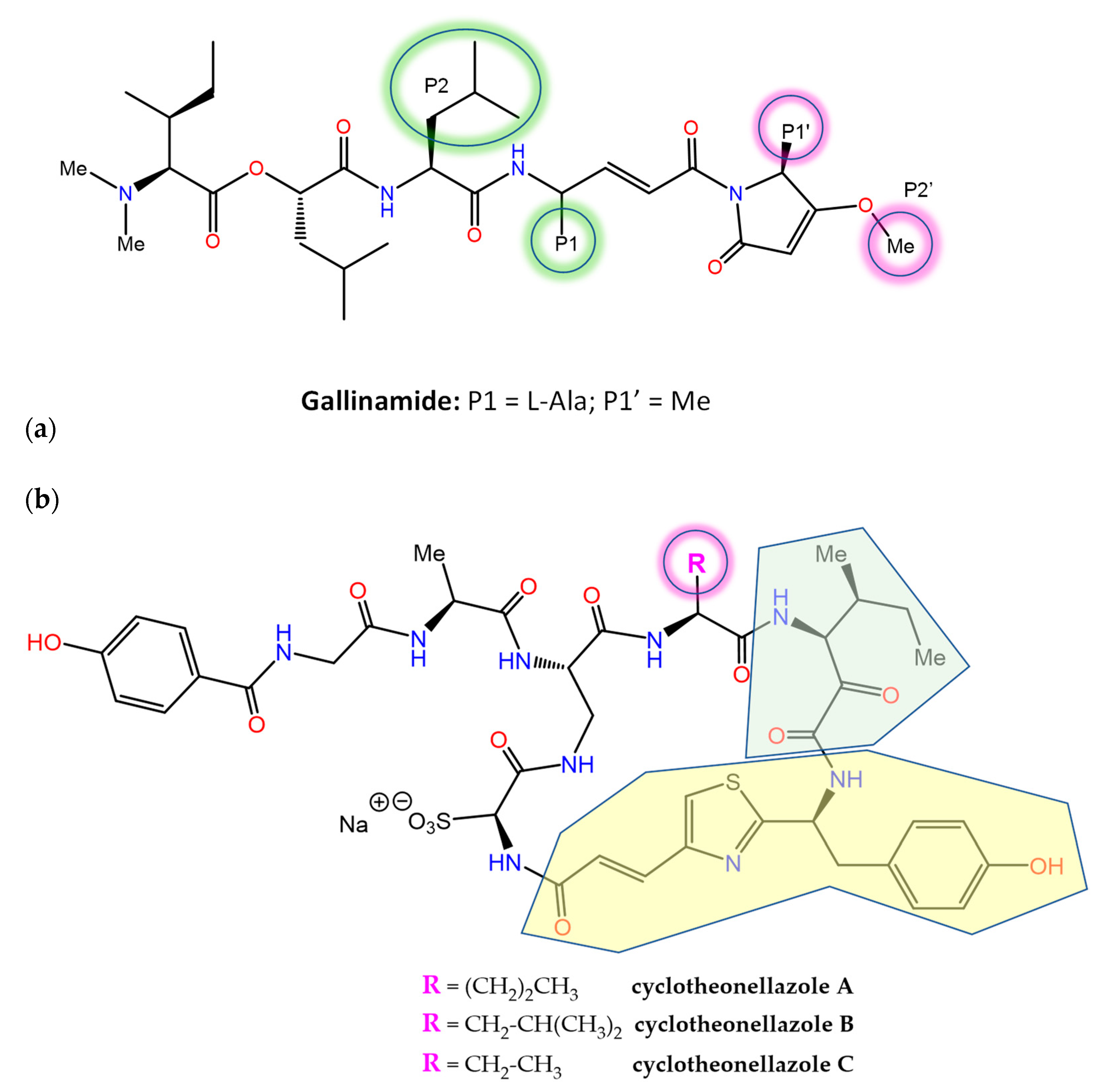
- Miller, B.; Friedman, A.J.; Choi, H.; Hogan, J.; McCammon, J.A.; Hook, V.; Gerwick, W.H. The marine cyanobacterial metabolite gallinamide A is a potent and selective inhibitor of human cathepsin L. J. Nat. Prod. 2014, 77, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, P.D.; Miller, B.W.; McCall, L.I.; Almaliti, J.; Reher, R.; Hirata, K.; Le, T.; Siqueira-Neto, J.L.; Hook, V.; Gerwick, W.H. Design of gallinamide a analogs as potent inhibitors of the cysteine proteases human cathepsin l and Trypanosoma cruzi cruzain. J. Med. Chem. 2019, 62, 9026–9044. [Google Scholar] [CrossRef]
- Barbosa Da Silva, E.; Sharma, V.; Hernandez-Alvarez, L.; Tang, A.H.; Stoye, A.; O’Donoghue, A.J.; Gerwick, W.H.; Payne, R.J.; McKerrow, J.H.; Podust, L.M. Intramolecular interactions enhance the potency of gallinamide A analogues against Trypanosoma cruzi. J. Med. Chem. 2022, 65, 4255–4269. [Google Scholar] [CrossRef]
- Klenchin, V.A.; King, R.; Tanaka, J.; Marriott, G.; Rayment, I. Structural basis of swinholide A binding to actin. Chem. Biol. 2005, 12, 287–291. [Google Scholar] [CrossRef] [Green Version]
- Issac, M.; Aknin, M.; Gauvin-Bialecki, A.; De Voogd, N.; Ledoux, A.; Frederich, M.; Kashman, Y.; Carmeli, S. Cyclotheonellazoles A-C, potent protease inhibitors from the marine sponge Theonella aff. swinhoei. J. Nat. Prod. 2017, 80, 1110–1116. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, M.; Xu, H.; Zhang, T.; Zhang, S.; Zhao, X.; Jiang, P.; Li, J.; Ye, B.; Sun, Y.; et al. Elastase inhibitor cyclotheonellazole A: Total synthesis and in vivo biological evaluation for acute lung injury. J. Med. Chem. 2022, 65, 2971–2987. [Google Scholar] [CrossRef]
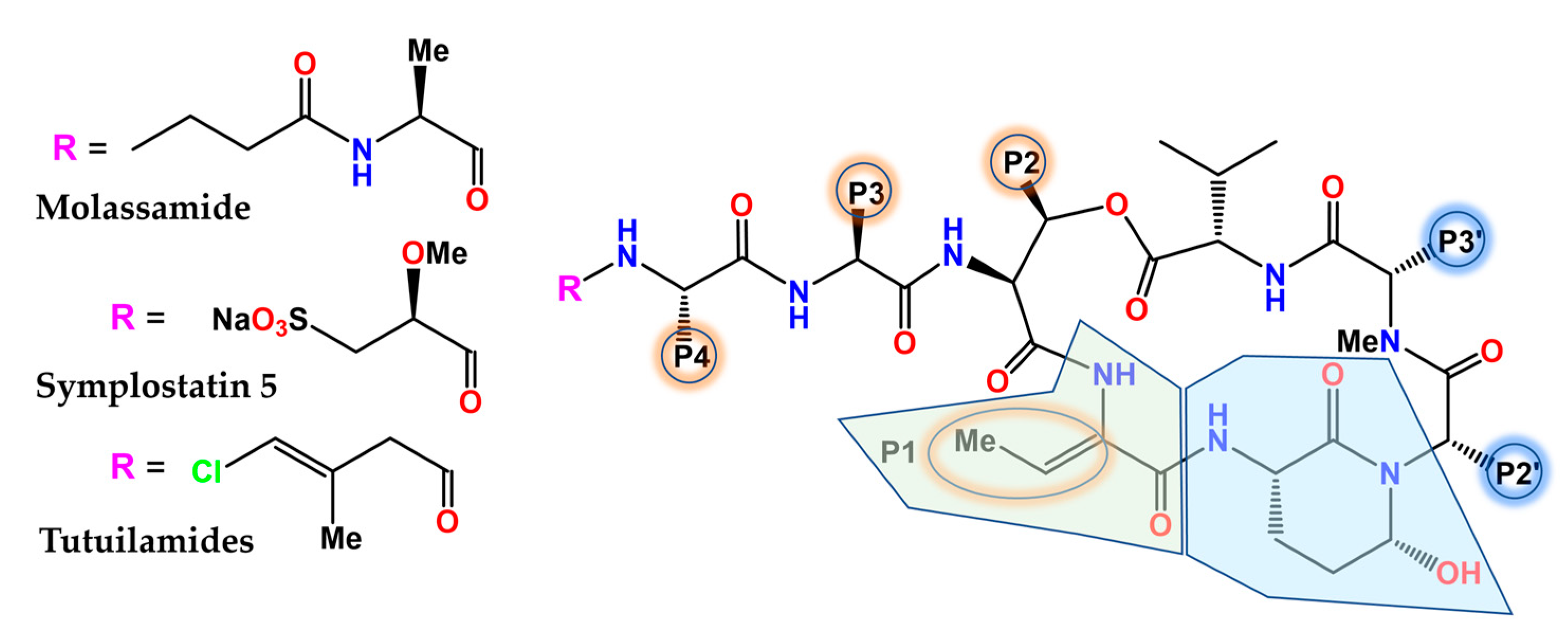
- Köcher, S.; Resch, S.; Kessenbrock, T.; Schrapp, L.; Ehrmann, M.; Kaiser, M. From dolastatin 13 to cyanopeptolins, micropeptins, and lyngbyastatins: The chemical biology of Ahp-cyclodepsipeptides. Nat. Prod. Rep. 2020, 37, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Luo, D.; Seabra, G.M.; Luesch, H. Ahp-Cyclodepsipeptides as tunable inhibitors of human neutrophil elastase and kallikrein 7: Total synthesis of tutuilamide A, serine protease selectivity profile and comparison with lyngbyastatin 7. Bioorg. Med. Chem. 2020, 28, 115756. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Dufresne, C.; Cerny, R.L.; Herald, D.L.; Schmidt, J.M.; Kizu, H. Antineoplastic agent. 174. Isolation and structure of the cytostatic depsipeptide dolastatin 13 from the sea hare Dolabella auricularia. J. Am. Chem. Soc. 1989, 111, 5015–5017. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Miller, M.W.; Kwan, J.C.; Luesch, H.; Paul, V.J. Molassamide, a depsipeptide serine protease inhibitor from the marine cyanobacterium Dichothrix utahensis. J. Nat. Prod. 2010, 73, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Taori, K.; Biggs, J.S.; Jakoncic, J.; Ostrov, D.A.; Paul, V.J.; Luesch, H. Potent elastase inhibitors from cyanobacteria: Structural basis and mechanisms mediating cytoprotective and anti-inflammatory effects in bronchial epithelial cells. J. Med. Chem. 2013, 56, 1276–1290. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Dolios, G.; Petrick, L. Reproducible untargeted metabolomics workflow for exhaustive MS2 data acquisition of MS1 features. J. Cheminform. 2022, 14, 6. [Google Scholar] [CrossRef]
- Keller, L.; Canuto, K.M.; Liu, C.; Suzuki, B.M.; Almaliti, J.; Sikandar, A.; Naman, C.B.; Glukhov, E.; Luo, D.; Duggan, B.M.; et al. Tutuilamides A-C: Vinyl-chloride-containing cyclodepsipeptides from marine cyanobacteria with potent elastase inhibitory properties. ACS Chem. Biol. 2020, 15, 751–757. [Google Scholar] [CrossRef]
- Reher, R.; Aron, A.T.; Fajtova, P.; Stincone, P.; Wagner, B.; Perez-Lorente, A.I.; Liu, C.; Shalom, I.Y.B.; Bittremieux, W.; Wang, M.; et al. Native metabolomics identifies the rivulariapeptolide family of protease inhibitors. Nat. Commun. 2022, 13, 4619. [Google Scholar] [CrossRef]
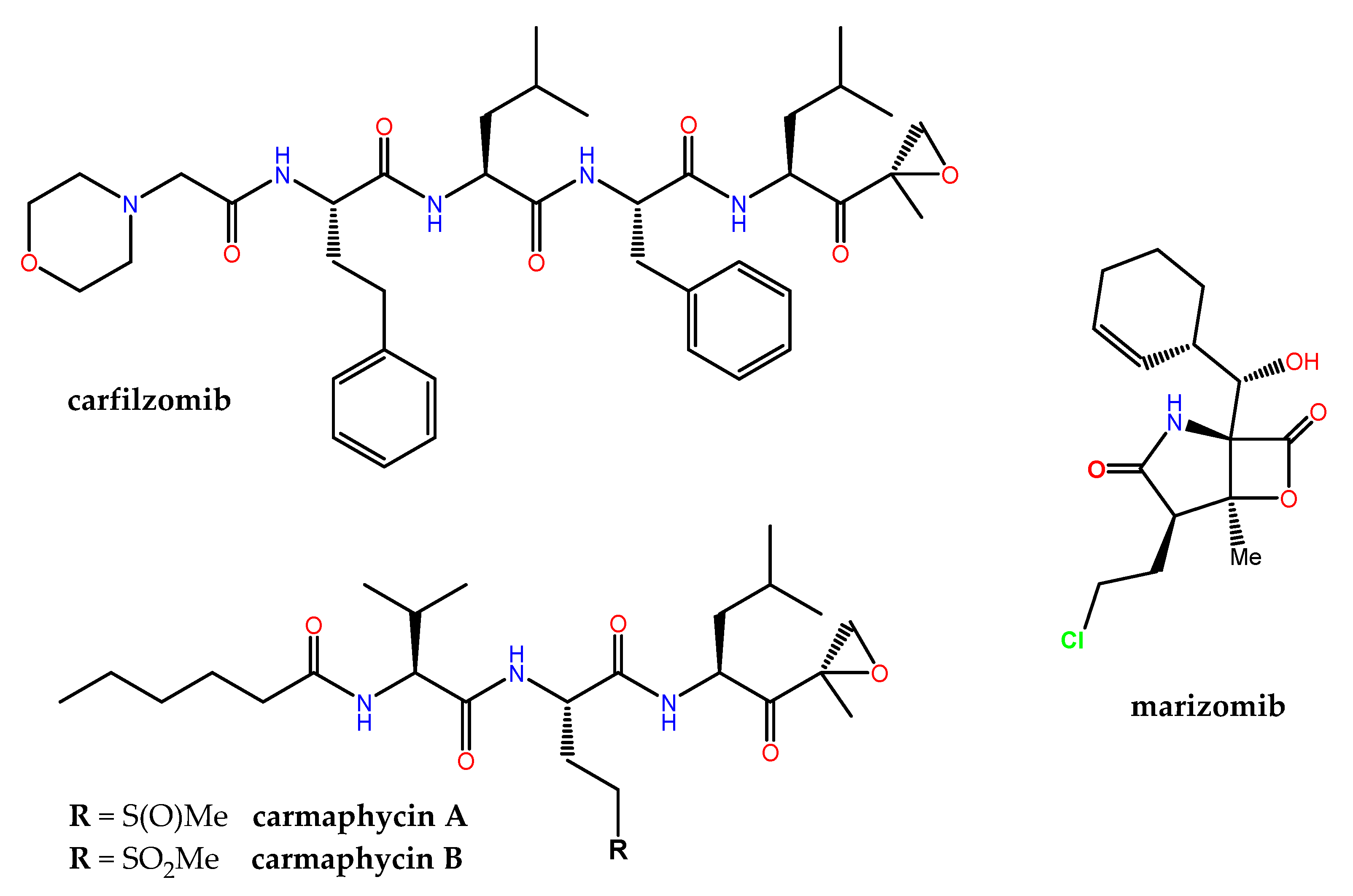
- Hanada, M.; Sugawara, K.; Kaneta, K.; Toda, S.; Nishiyama, Y.; Tomita, K.; Yamamoto, H.; Konishi, M.; Oki, T. Epoxomicin, a new antitumor agent of microbial origin. J. Antibiot. 1992, 45, 1746–1752. [Google Scholar] [CrossRef] [Green Version]
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720. [Google Scholar] [CrossRef]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C., Jr.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbell, G.E.; Tepe, J.J. Natural product scaffolds as inspiration for the design and synthesis of 20S human proteasome inhibitors. RSC Chem. Biol. 2020, 1, 305–332. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Unno, Y.; Tanaka, M.; Sasaki, T.; Yamano, A.; Hirokawa, T.; Kameda, T.; Asai, A.; Arisawa, M.; Shuto, S. Investigation of the noncovalent binding mode of covalent proteasome inhibitors around the transition state by combined use of cyclopropylic strain-based conformational restriction and computational modeling. J. Med. Chem. 2013, 56, 5829–5842. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, N.; Zhang, T.; Zhang, B.; Sajeevan, T.P.; Joseph, V.; Armstrong, L.; He, S.; Yan, X.; Naman, C.B. A systematic review of recently reported marine derived natural product kinase inhibitors. Mar. Drugs 2019, 17, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnel, R.B.; Scheuer, P.J. 11-Hydroxystaurosporine: A highly cytotoxic, powerful protein kinase C inhibitor from a tunicate. J. Org. Chem. 1992, 57, 6327–6329. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Janne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Lo, Y.C.; Liu, T.; Morrissey, K.M.; Kakiuchi-Kiyota, S.; Johnson, A.R.; Broccatelli, F.; Zhong, Y.; Joshi, A.; Altman, R.B. Computational analysis of kinase inhibitor selectivity using structural knowledge. Bioinformatics 2019, 35, 235–242. [Google Scholar] [CrossRef]
- Dai, X.; Xu, Y.; Qiu, H.; Qian, X.; Lin, M.; Luo, L.; Zhao, Y.; Huang, D.; Zhang, Y.; Chen, Y.; et al. KID: A kinase-focused interaction database and its application in the construction of kinase-focused molecule databases. J. Chem. Inf. Model. 2022, 62, 6022–6034. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.P.A.; Grimm, S.H.; Wijdeven, R.H.M.; Lenselink, E.B.; Neefjes, J.; van Boeckel, C.A.A.; van Westen, G.J.P.; van der Stelt, M. Drug discovery maps, a machine learning model that visualizes and predicts kinome-inhibitor interaction landscapes. J. Chem. Inf. Model. 2019, 59, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Cortistatins A, B, C, and D, anti-angiogenic steroidal alkaloids, from the marine sponge Corticium simplex. J. Am. Chem. Soc. 2006, 128, 3148–3149. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Liau, B.B.; Nitulescu, II; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, S.; Watanabe, Y.; Tanabe, D.; Arai, M.; Suna, H.; Miyamoto, K.; Tsujibo, H.; Tsujikawa, K.; Yamamoto, H.; Kobayashi, M. Structure-activity relationship and biological property of cortistatins, anti-angiogenic spongean steroidal alkaloids. Bioorg. Med. Chem. 2007, 15, 6758–6762. [Google Scholar] [CrossRef]
- Funabashi, M.; Baba, S.; Takatsu, T.; Kizuka, M.; Ohata, Y.; Tanaka, M.; Nonaka, K.; Spork, A.P.; Ducho, C.; Chen, W.C.; et al. Structure-based gene targeting discovery of sphaerimicin, a bacterial translocase I inhibitor. Angew. Chem. Int. Ed. Engl. 2013, 52, 11607–11611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Gui, Y.H.; Xu, Q.H.; Lin, H.W.; Lu, Y.H. Spongiactinospora rosea gen. nov., sp. nov., a new member of the family Streptosporangiaceae. Int. J. Syst. Evol. Microbiol. 2019, 69, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Yabe, M.; Mashalidis, E.H.; Sato, T.; Yamamoto, K.; Hikiji, Y.; Katsuyama, A.; Shinohara, M.; Minato, Y.; Takahashi, S.; et al. Synthesis of macrocyclic nucleoside antibacterials and their interactions with MraY. Nat. Commun. 2022, 13, 7575. [Google Scholar] [CrossRef]
- Pereira, R.B.; Evdokimov, N.M.; Lefranc, F.; Valentao, P.; Kornienko, A.; Pereira, D.M.; Andrade, P.B.; Gomes, N.G.M. Marine-derived anticancer agents: Clinical benefits, innovative mechanisms, and new targets. Mar. Drugs 2019, 17, 329. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.; Hopkins, A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef]
- Hight, S.K.; Clark, T.N.; Kurita, K.L.; McMillan, E.A.; Bray, W.; Shaikh, A.F.; Khadilkar, A.; Haeckl, F.P.J.; Carnevale-Neto, F.; La, S.; et al. High-throughput functional annotation of natural products by integrated activity profiling. Proc. Natl. Acad. Sci. USA 2022, 119, e2208458119. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gago, F. Computational Approaches to Enzyme Inhibition by Marine Natural Products in the Search for New Drugs. Mar. Drugs 2023, 21, 100. https://doi.org/10.3390/md21020100
Gago F. Computational Approaches to Enzyme Inhibition by Marine Natural Products in the Search for New Drugs. Marine Drugs. 2023; 21(2):100. https://doi.org/10.3390/md21020100
Chicago/Turabian StyleGago, Federico. 2023. "Computational Approaches to Enzyme Inhibition by Marine Natural Products in the Search for New Drugs" Marine Drugs 21, no. 2: 100. https://doi.org/10.3390/md21020100
APA StyleGago, F. (2023). Computational Approaches to Enzyme Inhibition by Marine Natural Products in the Search for New Drugs. Marine Drugs, 21(2), 100. https://doi.org/10.3390/md21020100