

Bioinformatics-Based Screening Approach for the Identification and Characterization of Lipolytic Enzymes from the Marine Diatom Phaeodactylum tricornutum




Abstract
:1. Introduction
2. Results and Discussion
2.1. Retrieval of Sequences and Annotation Screening Confirm the Existence of a High Number of Putative Lipases in P. tricornutum
2.2. Multiple Sequence Alignment Allows the Identification of Conserved Domains and Residues

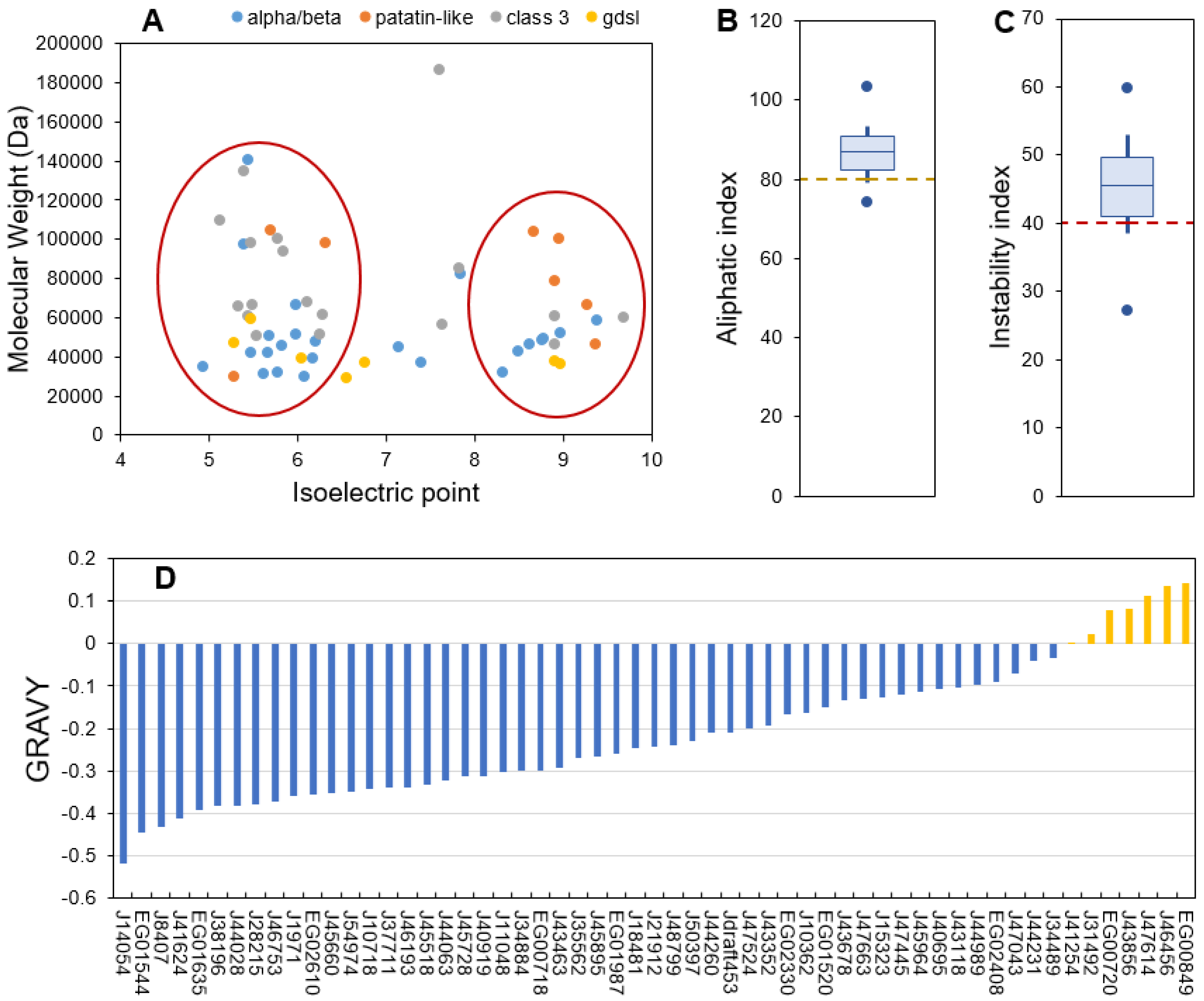
2.3. Physico-Chemical Characteristics Provide General Information about Candidate Lipolytic Enzymes and Insigths of In Vivo Function

2.4. Predicted Subcellular Localization Varies across Families and Is Mainly Cytosolic
2.5. Transcription Data Reveal Contrasting Regulation across All Candidate Lipases
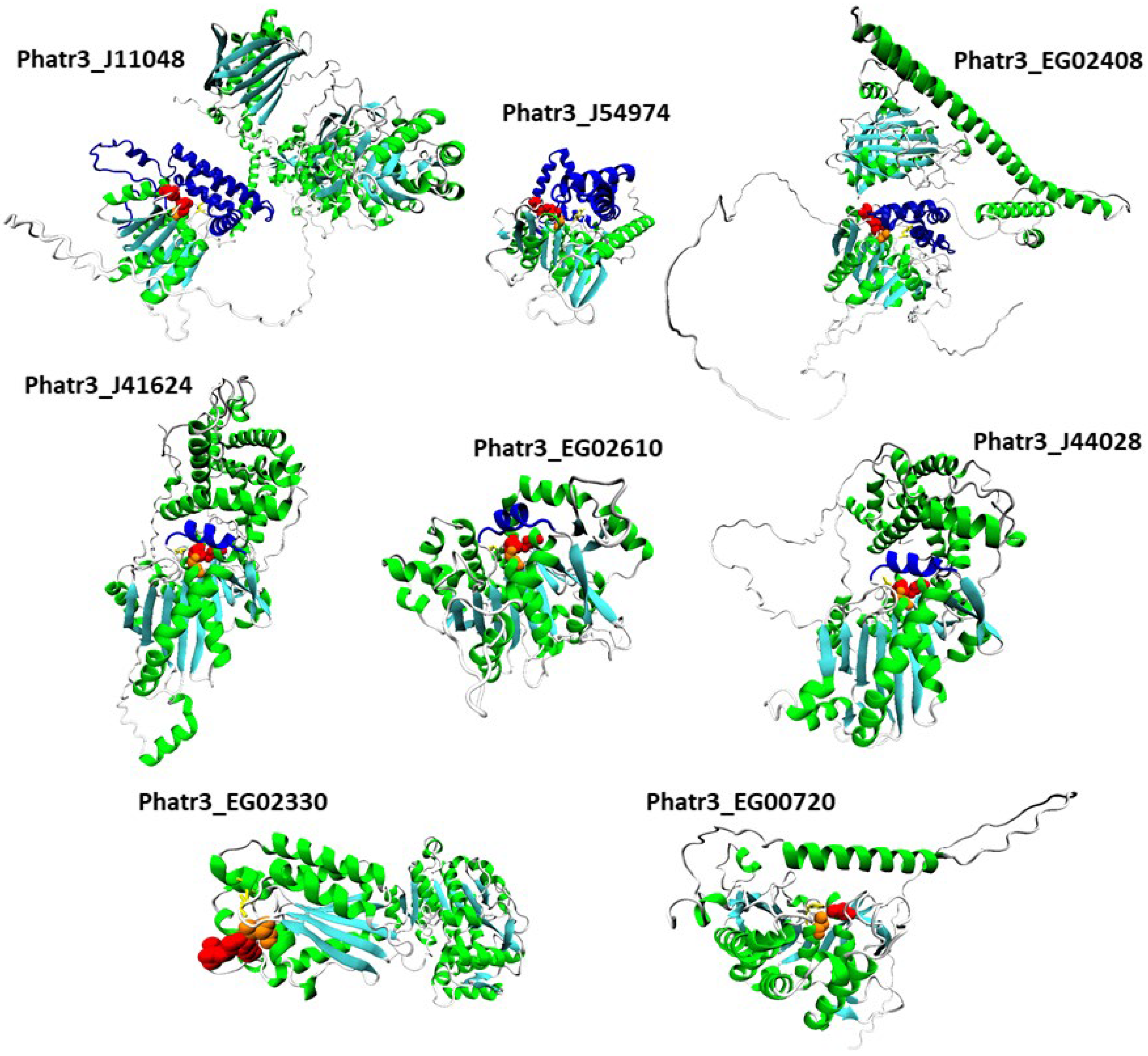
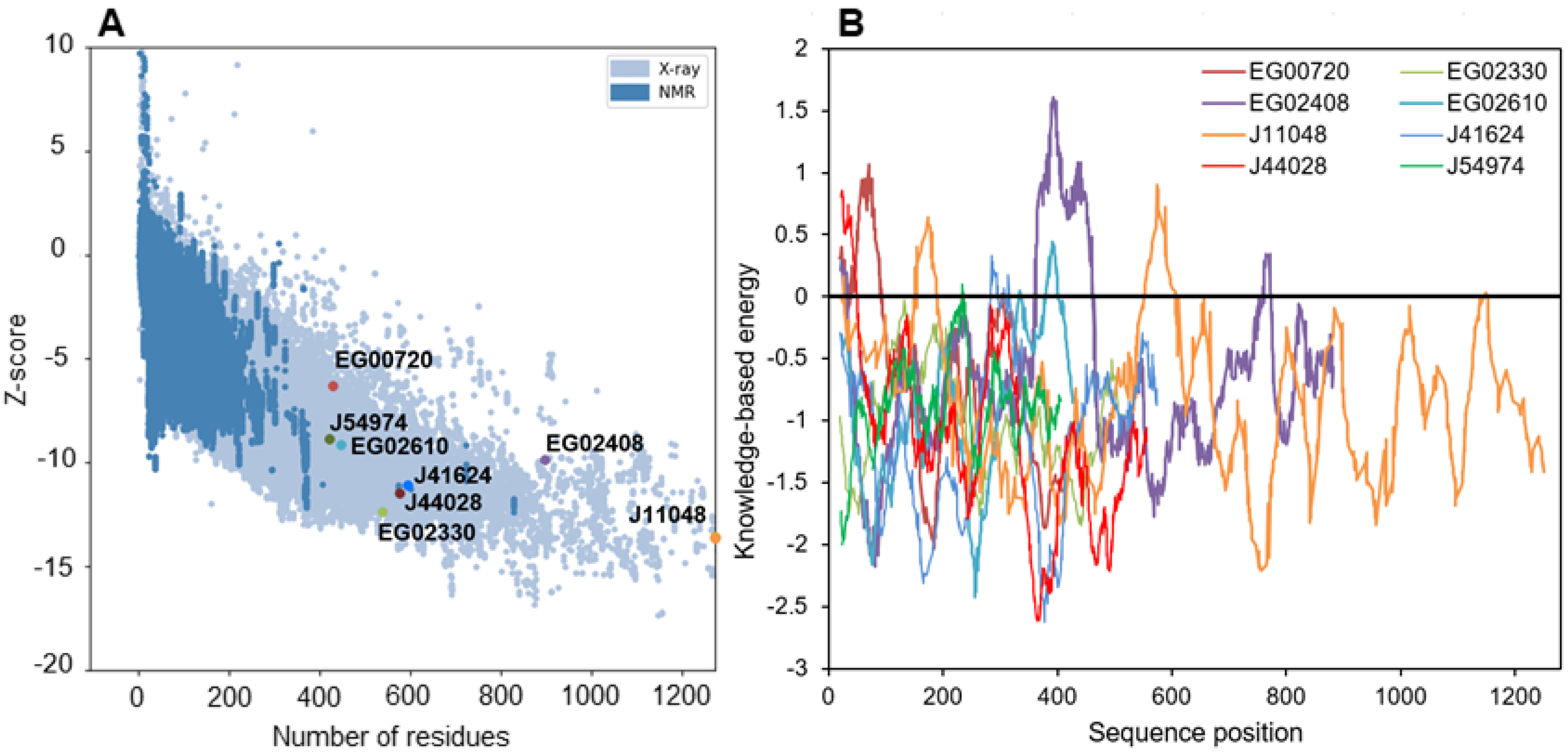
2.6. Tertiary Structure Prediction of the Most Regulated Candidate Lipases Reveals a Functional Lipase Fold and Additional Domains That May Regulate Catalytic Activity

3. Materials and Methods
3.1. Steps for the Screening Procedure and Retrieval of Sequences
3.2. Multiple Alignment of Sequences
3.3. Determination of Physico-Chemical Characteristics
3.4. Subcellular Localization Prediction and Post-Translational Modification
3.5. Tertiary Structure Prediction and Quality Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schoefs, B.; Van de Vijver, B.; Wetzel, C.E.; Ector, L. Introduction: From Diatom Species Identification to Ecological and Biotechnological Applications. Bot. Lett. 2020, 167, 2–6. [Google Scholar] [CrossRef]
- Bertrand, J.; Coste, C.; Le Cohu, R.; Renon, J.-P.; Ector, L. Étude préliminaire sur la présence de diatomées sur les lichens. Bot. Lett. 2016, 163, 93–115. [Google Scholar] [CrossRef]
- Nelson, D.M.; Tréguer, P.; Brzezinski, M.A.; Leynaert, A.; Quéguiner, B. Production and Dissolution of Biogenic Silica in the Ocean: Revised Global Estimates, Comparison with Regional Data and Relationship to Biogenic Sedimentation. Glob. Biogeochem. Cycles 1995, 9, 359–372. [Google Scholar] [CrossRef]
- Field, C.B.; Behrenfeld, M.J.; Randerson, J.T.; Falkowski, P.G. Primary Production of the Biosphere: Integrating Terrestrial and Oceanic Components. Science 1998, 281, 237–240. [Google Scholar] [CrossRef]
- Wagner, H.; Jakob, T.; Fanesi, A.; Wilhelm, C. Towards an Understanding of the Molecular Regulation of Carbon Allocation in Diatoms: The Interaction of Energy and Carbon Allocation. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160410. [Google Scholar] [CrossRef]
- Sayanova, O.; Mimouni, V.; Ulmann, L.; Morant, A.; Pasquet, V.; Schoefs, B.; Napier, J.A. Modulation of Lipid Biosynthesis by Stress in Diatoms. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160407. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Mayama, S.; Nemoto, M.; Fukuda, Y.; Muto, M.; Yoshino, T.; Matsunaga, T.; Tanaka, T. Morphological and Molecular Phylogenetic Analysis of the High Triglyceride-Producing Marine Diatom, Fistulifera Solaris Sp. Nov. (Bacillariophyceae): Fistulifera Solaris Sp. Nov. Phycol. Res. 2014, 62, 257–268. [Google Scholar] [CrossRef]
- Chisti, Y. Biodiesel from Microalgae. Biotechnol. Adv. 2007, 25, 294–306. [Google Scholar] [CrossRef]
- Ulmann, L.; Blanckaert, V.; Mimouni, V.; Andersson, M.X.; Schoefs, B.; Chenais, B. Microalgal Fatty Acids and Their Implication in Health and Disease. Mini Rev. Med. Chem. 2017, 17, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Leyland, B.; Zarka, A.; Didi-Cohen, S.; Boussiba, S.; Khozin-Goldberg, I. High Resolution Proteome of Lipid Droplets Isolated from the Pennate Diatom Phaeodactylum Tricornutum (Bacillariophyceae) Strain Pt4 Provides Mechanistic Insights into Complex Intracellular Coordination during Nitrogen Deprivation. J. Phycol. 2020, 56, 1642–1663. [Google Scholar] [CrossRef]
- Lupette, J.; Jaussaud, A.; Seddiki, K.; Morabito, C.; Brugière, S.; Schaller, H.; Kuntz, M.; Putaux, J.-L.; Jouneau, P.-H.; Rébeillé, F.; et al. The Architecture of Lipid Droplets in the Diatom Phaeodactylum Tricornutum. Algal Res. 2019, 38, 101415. [Google Scholar] [CrossRef]
- Huang, B.; Marchand, J.; Blanckaert, V.; Lukomska, E.; Ulmann, L.; Wielgosz-Collin, G.; Rabesaotra, V.; Moreau, B.; Bougaran, G.; Mimouni, V.; et al. Nitrogen and Phosphorus Limitations Induce Carbon Partitioning and Membrane Lipid Remodelling in the Marine Diatom Phaeodactylum Tricornutum. Eur. J. Phycol. 2019, 54, 342–358. [Google Scholar] [CrossRef]
- Alipanah, L.; Winge, P.; Rohloff, J.; Najafi, J.; Brembu, T.; Bones, A.M. Molecular Adaptations to Phosphorus Deprivation and Comparison with Nitrogen Deprivation Responses in the Diatom Phaeodactylum Tricornutum. PLoS ONE 2018, 13, e0193335. [Google Scholar] [CrossRef] [PubMed]
- Mus, F.; Toussaint, J.-P.; Cooksey, K.E.; Fields, M.W.; Gerlach, R.; Peyton, B.M.; Carlson, R.P. Physiological and Molecular Analysis of Carbon Source Supplementation and PH Stress-Induced Lipid Accumulation in the Marine Diatom Phaeodactylum tricornutum. Appl. Microbiol. Biotechnol. 2013, 97, 3625–3642. [Google Scholar] [CrossRef]
- Nunez, M.; Quigg, A. Changes in Growth and Composition of the Marine Microalgae Phaeodactylum Tricornutum and Nannochloropsis Salina in Response to Changing Sodium Bicarbonate Concentrations. J. Appl. Phycol. 2016, 28, 2123–2138. [Google Scholar] [CrossRef]
- Sharma, N.; Fleurent, G.; Awwad, F.; Cheng, M.; Meddeb-Mouelhi, F.; Budge, S.M.; Germain, H.; Desgagné-Penix, I. Red Light Variation an Effective Alternative to Regulate Biomass and Lipid Profiles in Phaeodactylum Tricornutum. Appl. Sci. 2020, 10, 2531. [Google Scholar] [CrossRef]
- Duarte, B.; Feijão, E.; Goessling, J.W.; Caçador, I.; Matos, A.R. Pigment and Fatty Acid Production under Different Light Qualities in the Diatom Phaeodactylum Tricornutum. Appl. Sci. 2021, 11, 2550. [Google Scholar] [CrossRef]
- Dinamarca, J.; Levitan, O.; Kumaraswamy, G.K.; Lun, D.S.; Falkowski, P.G. Overexpression of a Diacylglycerol Acyltransferase Gene in Phaeodactylum Tricornutum Directs Carbon towards Lipid Biosynthesis. J. Phycol. 2017, 53, 405–414. [Google Scholar] [CrossRef]
- Haslam, R.P.; Hamilton, M.L.; Economou, C.K.; Smith, R.; Hassall, K.L.; Napier, J.A.; Sayanova, O. Overexpression of an Endogenous Type 2 Diacylglycerol Acyltransferase in the Marine Diatom Phaeodactylum Tricornutum Enhances Lipid Production and Omega-3 Long-Chain Polyunsaturated Fatty Acid Content. Biotechnol. Biofuels 2020, 13, 87. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, Y.; Ding, W.; Hu, H.; Liu, J. Lipid Production Is More than Doubled by Manipulating a Diacylglycerol Acyltransferase in Algae. GCB Bioenergy 2021, 13, 185–200. [Google Scholar] [CrossRef]
- Zhu, B.-H.; Tu, C.-C.; Shi, H.-P.; Yang, G.-P.; Pan, K.-H. Overexpression of Endogenous Delta-6 Fatty Acid Desaturase Gene Enhances Eicosapentaenoic Acid Accumulation in Phaeodactylum Tricornutum. Process Biochem. 2017, 57, 43–49. [Google Scholar] [CrossRef]
- Wang, X.; Dong, H.-P.; Wei, W.; Balamurugan, S.; Yang, W.-D.; Liu, J.-S.; Li, H.-Y. Dual Expression of Plastidial GPAT1 and LPAT1 Regulates Triacylglycerol Production and the Fatty Acid Profile in Phaeodactylum Tricornutum. Biotechnol. Biofuels 2018, 11, 318. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, S.-F.; Li, R.-Y.; Yang, W.-D.; Liu, J.-S.; Lin, C.S.K.; Balamurugan, S.; Li, H.-Y. TAG Pathway Engineering via GPAT2 Concurrently Potentiates Abiotic Stress Tolerance and Oleaginicity in Phaeodactylum Tricornutum. Biotechnol. Biofuels 2020, 13, 160. [Google Scholar] [CrossRef]
- Xue, J.; Niu, Y.-F.; Huang, T.; Yang, W.-D.; Liu, J.-S.; Li, H.-Y. Genetic Improvement of the Microalga Phaeodactylum Tricornutum for Boosting Neutral Lipid Accumulation. Metab. Eng. 2015, 27, 1–9. [Google Scholar] [CrossRef]
- Zhu, B.-H.; Zhang, R.-H.; Lv, N.-N.; Yang, G.-P.; Wang, Y.-S.; Pan, K.-H. The Role of Malic Enzyme on Promoting Total Lipid and Fatty Acid Production in Phaeodactylum Tricornutum. Front. Plant Sci. 2018, 9, 826. [Google Scholar] [CrossRef]
- Zhang, R.; Zhu, B.; Tu, C.; Li, Y.; Zhao, Y.; Pan, K. The Combined Effect of Nitrogen Deprivation and Overexpression of Malic Enzyme Gene on Growth and Lipid Accumulation in Phaeodactylum Tricornutum. J. Appl. Phycol. 2021, 33, 3637–3645. [Google Scholar] [CrossRef]
- Conte, M.; Lupette, J.; Seddiki, K.; Meï, C.; Dolch, L.-J.; Gros, V.; Barette, C.; Rébeillé, F.; Jouhet, J.; Maréchal, E. Screening for Biologically Annotated Drugs That Trigger Triacylglycerol Accumulation in the Diatom Phaeodactylum. Plant Physiol. 2018, 177, 532–552. [Google Scholar] [CrossRef]
- Zhu, B.-H.; Shi, H.-P.; Yang, G.-P.; Lv, N.-N.; Yang, M.; Pan, K.-H. Silencing UDP-Glucose Pyrophosphorylase Gene in Phaeodactylum Tricornutum Affects Carbon Allocation. New Biotechnol. 2016, 33, 237–244. [Google Scholar] [CrossRef]
- Daboussi, F.; Leduc, S.; Maréchal, A.; Dubois, G.; Guyot, V.; Perez-Michaut, C.; Amato, A.; Falciatore, A.; Juillerat, A.; Beurdeley, M.; et al. Genome Engineering Empowers the Diatom Phaeodactylum Tricornutum for Biotechnology. Nat. Commun. 2014, 5, 3831. [Google Scholar] [CrossRef]
- Barka, F.; Angstenberger, M.; Ahrendt, T.; Lorenzen, W.; Bode, H.B.; Büchel, C. Identification of a Triacylglycerol Lipase in the Diatom Phaeodactylum Tricornutum. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2016, 1861, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Zienkiewicz, K.; Zienkiewicz, A. Degradation of Lipid Droplets in Plants and Algae—Right Time, Many Paths, One Goal. Front. Plant Sci. 2020, 11, 579019. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Romero, I.T.; Warakanont, J.; Li-Beisson, Y. Lipid Catabolism in Microalgae. New Phytol. 2018, 218, 1340–1348. [Google Scholar] [CrossRef]
- Casas-Godoy, L.; Gasteazoro, F.; Duquesne, S.; Bordes, F.; Marty, A.; Sandoval, G. Lipases: An Overview. In Lipases and Phospholipases; Sandoval, G., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1835, pp. 3–38. ISBN 978-1-4939-8671-2. [Google Scholar]
- Bauer, T.L.; Buchholz, P.C.F.; Pleiss, J. The Modular Structure of α/Β-hydrolases. FEBS J. 2020, 287, 1035–1053. [Google Scholar] [CrossRef]
- Eastmond, P.J. SUGAR-DEPENDENT1 Encodes a Patatin Domain Triacylglycerol Lipase That Initiates Storage Oil Breakdown in Germinating Arabidopsis Seeds. Plant Cell 2006, 18, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat Mobilization in Adipose Tissue Is Promoted by Adipose Triglyceride Lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [PubMed]
- Bielen, A.; Ćetković, H.; Long, P.F.; Schwab, H.; Abramić, M.; Vujaklija, D. The SGNH-Hydrolase of Streptomyces Coelicolor Has (Aryl)Esterase and a True Lipase Activity. Biochimie 2009, 91, 390–400. [Google Scholar] [CrossRef]
- Akoh, C.C.; Lee, G.-C.; Liaw, Y.-C.; Huang, T.-H.; Shaw, J.-F. GDSL Family of Serine Esterases/Lipases. Prog. Lipid Res. 2004, 43, 534–552. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, Y.; Hu, H. Identification of the Triacylglycerol Lipase in the Chloroplast Envelope of the Diatom Phaeodactylum Tricornutum. Algal Res. 2018, 33, 440–447. [Google Scholar] [CrossRef]
- Calleja, P.; Leterrier, M.; Maréchal, E. Modified Algae Strain and Method of Triacylglycerol Accumulation Using Said Strain. U.S. Patent 9,938,545, 10 April 2018. [Google Scholar]
- Shu, Q.; Pan, Y.; Hu, H. CGI-58 Protein Acts as a Positive Regulator of Triacylglycerol Accumulation in Phaeodactylum Tricornutum. J. Microbiol. Biotechnol. 2023, 33, 1–9. [Google Scholar] [CrossRef]
- Trentacoste, E.M.; Shrestha, R.P.; Smith, S.R.; Gle, C.; Hartmann, A.C.; Hildebrand, M.; Gerwick, W.H. Metabolic Engineering of Lipid Catabolism Increases Microalgal Lipid Accumulation without Compromising Growth. Proc. Natl. Acad. Sci. USA 2013, 110, 19748–19753. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Watanabe, K.; Kaha, M.; Yabu, Y.; Yoshino, T.; Matsumoto, M.; Tanaka, T. Assessment on the Oil Accumulation by Knockdown of Triacylglycerol Lipase in the Oleaginous Diatom Fistulifera Solaris. Sci. Rep. 2021, 11, 20905. [Google Scholar] [CrossRef]
- Chandra, P.; Enespa; Singh, R.; Arora, P.K. Microbial Lipases and Their Industrial Applications: A Comprehensive Review. Microb. Cell Factories 2020, 19, 42. [Google Scholar] [CrossRef]
- Siegler, H.; Valerius, O.; Ischebeck, T.; Popko, J.; Tourasse, N.J.; Vallon, O.; Khozin-Goldberg, I.; Braus, G.H.; Feussner, I. Analysis of the Lipid Body Proteome of the Oleaginous Alga Lobosphaera Incisa. BMC Plant Biol. 2017, 17, 98. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Benning, C.; Kuo, M.-H. Rapid Triacylglycerol Turnover in Chlamydomonas Reinhardtii Requires a Lipase with Broad Substrate Specificity. Eukaryot. Cell 2012, 11, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-H.; Uygun, S.; Roston, R.; Shiu, S.-H.; Benning, C. Recovery from N Deprivation Is a Transcriptionally and Functionally Distinct State in Chlamydomonas. Plant Physiol. 2018, 176, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, A.; Maheswari, U.; Dorrell, R.G.; Vieira, F.R.J.; Maumus, F.; Kustka, A.; McCarthy, J.; Allen, A.E.; Kersey, P.; Bowler, C.; et al. Integrative Analysis of Large Scale Transcriptome Data Draws a Comprehensive Landscape of Phaeodactylum Tricornutum Genome and Evolutionary Origin of Diatoms. Sci. Rep. 2018, 8, 4834. [Google Scholar] [CrossRef]
- Rydel, T.J.; Williams, J.M.; Krieger, E.; Moshiri, F.; Stallings, W.C.; Brown, S.M.; Pershing, J.C.; Purcell, J.P.; Alibhai, M.F. The Crystal Structure, Mutagenesis, and Activity Studies Reveal That Patatin Is a Lipid Acyl Hydrolase with a Ser-Asp Catalytic Dyad. Biochemistry 2003, 42, 6696–6708. [Google Scholar] [CrossRef]
- Kumari, A.; Gupta, R. Phenylalanine to Leucine Point Mutation in Oxyanion Hole Improved Catalytic Efficiency of Lip12 from Yarrowia Lipolytica. Enzyme Microb. Technol. 2013, 53, 386–390. [Google Scholar] [CrossRef]
- Ben Hlima, H.; Dammak, M.; Karray, A.; Drira, M.; Michaud, P.; Fendri, I.; Abdelkafi, S. Molecular and Structural Characterizations of Lipases from Chlorella by Functional Genomics. Mar. Drugs 2021, 19, 70. [Google Scholar] [CrossRef]
- Nomaguchi, T.; Maeda, Y.; Liang, Y.; Yoshino, T.; Asahi, T.; Tanaka, T. Comprehensive Analysis of Triacylglycerol Lipases in the Oleaginous Diatom Fistulifera Solaris JPCC DA0580 with Transcriptomics under Lipid Degradation. J. Biosci. Bioeng. 2018, 126, 258–265. [Google Scholar] [CrossRef]
- Holmes, R. Comparative Studies of Adipose Triglyceride Lipase Genes and Proteins: An Ancient Gene in Vertebrate Evolution. Open Access Bioinform. 2012, 15, 1–15. [Google Scholar] [CrossRef]
- Pleiss, J.; Fischer, M.; Peiker, M.; Thiele, C.; Schmid, R.D. Lipase Engineering Database Understanding and Exploiting Sequence–Structure–Function Relationships. J. Mol. Catal. B Enzym. 2000, 10, 491–508. [Google Scholar] [CrossRef]
- Abida, H.; Dolch, L.-J.; Meï, C.; Villanova, V.; Conte, M.; Block, M.A.; Finazzi, G.; Bastien, O.; Tirichine, L.; Bowler, C.; et al. Membrane Glycerolipid Remodeling Triggered by Nitrogen and Phosphorus Starvation in Phaeodactylum Tricornutum. Plant Physiol. 2015, 167, 118–136. [Google Scholar] [CrossRef]
- Nobusawa, T.; Yamakawa-Ayukawa, K.; Saito, F.; Nomura, S.; Takami, A.; Ohta, H. A Homolog of Arabidopsis SDP1 Lipase in Nannochloropsis Is Involved in Degradation of de Novo-Synthesized Triacylglycerols in the Endoplasmic Reticulum. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 1185–1193. [Google Scholar] [CrossRef]
- Yang, Z.-K.; Ma, Y.-H.; Zheng, J.-W.; Yang, W.-D.; Liu, J.-S.; Li, H.-Y. Proteomics to Reveal Metabolic Network Shifts towards Lipid Accumulation Following Nitrogen Deprivation in the Diatom. J. Appl. Phycol. 2014, 26, 10. [Google Scholar] [CrossRef] [PubMed]
- Kiraga, J.; Mackiewicz, P.; Mackiewicz, D.; Kowalczuk, M. The Relationships between the Isoelectric Point and: Length of Proteins, Taxonomy and Ecology of Organisms. BMC Genom. 2007, 8, 16. [Google Scholar] [CrossRef]
- Ikai, A. Thermostability and Aliphatic Index of Globular Proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [CrossRef]
- Guruprasad, K.; Reddy, B.V.B.; Pandit, M.W. Correlation between Stability of a Protein and Its Dipeptide Composition: A Novel Approach for Predicting in Vivo Stability of a Protein from Its Primary Sequence. Protein Eng. 1990, 4, 155–161. [Google Scholar] [CrossRef]
- Gamage, D.G.; Gunaratne, A.; Periyannan, G.R.; Russell, T.G. Applicability of Instability Index for In Vitro Protein Stability Prediction. Protein Pept. Lett. 2019, 26, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, Z.; Vaites, L.P.; Shen, N.; Mitchell, D.C.; Huttlin, E.L.; Paulo, J.A.; Harry, B.L.; Gygi, S.P. Proteome-Wide Mapping of Short-Lived Proteins in Human Cells. Mol. Cell 2021, 81, 4722–4735.e5. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, H.; Mao, X.; Liu, J. Proteomics Analysis of Lipid Droplets from the Oleaginous Alga Chromochloris Zofingiensis Reveals Novel Proteins for Lipid Metabolism. Genom. Proteom. Bioinform. 2019, 17, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, K.; Yoshida, M.; Suzuki, I.; Watanabe, M.M. Identification of a Major Lipid Droplet Protein in a Marine Diatom Phaeodactylum Tricornutum. Plant Cell Physiol. 2016, 57, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hao, T.-B.; Balamurugan, S.; Yang, W.-D.; Liu, J.-S.; Dong, H.-P.; Li, H.-Y. A Lipid Droplet-Associated Protein Involved in Lipid Droplet Biogenesis and Triacylglycerol Accumulation in the Oleaginous Microalga Phaeodactylum Tricornutum. Algal Res. 2017, 26, 215–224. [Google Scholar] [CrossRef]
- Thazar-Poulot, N.; Miquel, M.; Fobis-Loisy, I.; Gaude, T. Peroxisome Extensions Deliver the Arabidopsis SDP1 Lipase to Oil Bodies. Proc. Natl. Acad. Sci. USA 2015, 112, 4158–4163. [Google Scholar] [CrossRef] [PubMed]
- Leyland, B.; Boussiba, S.; Khozin-Goldberg, I. A Review of Diatom Lipid Droplets. Biology 2020, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Hemsley, P.A.; Grierson, C.S. Multiple Roles for Protein Palmitoylation in Plants. Trends Plant Sci. 2008, 13, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, V.; Andosch, A.; Geretschläger, A.; Affenzeller, M.; Lütz-Meindl, U. Carbon Starvation Induces Lipid Degradation via Autophagy in the Model Alga Micrasterias. J. Plant Physiol. 2017, 208, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Sato, N.; Shinozaki, A.; Watanabe, M. Increase in Peroxisome Number and the Gene Expression of Putative Glyoxysomal Enzymes in Chlamydomonas Cells Supplemented with Acetate. J. Plant Res. 2015, 128, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Jallet, D.; Xing, D.; Hughes, A.; Moosburner, M.; Simmons, M.P.; Allen, A.E.; Peers, G. Mitochondrial Fatty Acid Β-oxidation Is Required for Storage-lipid Catabolism in a Marine Diatom. New Phytol. 2020, 228, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dersch, J.; Puzik, K.; Bäcker, O.; Liu, X.; Stork, S.; Schulz, J.; Heimerl, T.; Klingl, A.; Zauner, S.; et al. The Central Vacuole of the Diatom Phaeodactylum Tricornutum: Identification of New Vacuolar Membrane Proteins and of a Functional Di-Leucine-Based Targeting Motif. Protist 2017, 168, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Scarsini, M.; Thiriet-Rupert, S.; Veidl, B.; Mondeguer, F.; Hu, H.; Marchand, J.; Schoefs, B. The Transition Toward Nitrogen Deprivation in Diatoms Requires Chloroplast Stand-By and Deep Metabolic Reshuffling. Front. Plant Sci. 2022, 12, 760516. [Google Scholar] [CrossRef]
- Alipanah, L.; Rohloff, J.; Winge, P.; Bones, A.M.; Brembu, T. Whole-Cell Response to Nitrogen Deprivation in the Diatom Phaeodactylum Tricornutum. J. Exp. Bot. 2015, 66, 6281–6296. [Google Scholar] [CrossRef]
- Matthijs, M.; Fabris, M.; Broos, S.; Vyverman, W.; Goossens, A. Profiling of the Early Nitrogen Stress Response in the Diatom Phaeodactylum Tricornutum Reveals a Novel Family of RING-Domain Transcription Factors. Plant Physiol. 2016, 170, 489–498. [Google Scholar] [CrossRef]
- Adelfi, M.G.; Vitale, R.M.; d’Ippolito, G.; Nuzzo, G.; Gallo, C.; Amodeo, P.; Manzo, E.; Pagano, D.; Landi, S.; Picariello, G.; et al. Patatin-like Lipolytic Acyl Hydrolases and Galactolipid Metabolism in Marine Diatoms of the Genus Pseudo-Nitzschia. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 181–190. [Google Scholar] [CrossRef]
- Heydarizadeh, P.; Veidl, B.; Huang, B.; Lukomska, E.; Wielgosz-Collin, G.; Couzinet-Mossion, A.; Bougaran, G.; Marchand, J.; Schoefs, B. Carbon Orientation in the Diatom Phaeodactylum Tricornutum: The Effects of Carbon Limitation and Photon Flux Density. Front. Plant Sci. 2019, 10, 471. [Google Scholar] [CrossRef]
- Chepyshko, H.; Lai, C.-P.; Huang, L.-M.; Liu, J.-H.; Shaw, J.-F. Multifunctionality and Diversity of GDSL Esterase/Lipase Gene Family in Rice (Oryza Sativa L. Japonica) Genome: New Insights from Bioinformatics Analysis. BMC Genom. 2012, 13, 309. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Iyer, L.M.; Koonin, E.V.; Aravind, L. Adaptations of the Helix-Grip Fold for Ligand Binding and Catalysis in the START Domain Superfamily. Proteins Struct. Funct. Genet. 2001, 43, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Oberer, M.; Boeszoermenyi, A.; Nagy, H.M.; Zechner, R. Recent Insights into the Structure and Function of Comparative Gene Identification-58. Curr. Opin. Lipidol. 2011, 22, 149–158. [Google Scholar] [CrossRef]
- Ding, L.-N.; Li, M.; Wang, W.-J.; Cao, J.; Wang, Z.; Zhu, K.-M.; Yang, Y.-H.; Li, Y.-L.; Tan, X.-L. Advances in Plant GDSL Lipases: From Sequences to Functional Mechanisms. Acta Physiol. Plant. 2019, 41, 151. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.-H.; Hu, D.-X.; Balamurugan, S.; Lu, Y.; Yang, W.-D.; Liu, J.-S.; Li, H.-Y. Identification of a Putative Patatin-like Phospholipase Domain-Containing Protein 3 (PNPLA3) Ortholog Involved in Lipid Metabolism in Microalga Phaeodactylum Tricornutum. Algal Res. 2015, 12, 274–279. [Google Scholar] [CrossRef]
- Van Tol, H.M.; Armbrust, E.V. Genome-Scale Metabolic Model of the Diatom Thalassiosira Pseudonana Highlights the Importance of Nitrogen and Sulfur Metabolism in Redox Balance. PLoS ONE 2021, 16, e0241960. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting Sequence Signals in Targeting Peptides Using Deep Learning. Life Sci. Alliance 2019, 2, e201900429. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Dyrløv Bendtsen, J.; Nielsen, H.; von Heijne, G.; Brunak, S. Improved Prediction of Signal Peptides: SignalP 3.0. J. Mol. Biol. 2004, 340, 783–795. [Google Scholar] [CrossRef]
- Gschloessl, B.; Guermeur, Y.; Cock, J.M. HECTAR: A Method to Predict Subcellular Targeting in Heterokonts. BMC Bioinform. 2008, 9, 393. [Google Scholar] [CrossRef]
- Gruber, A.; Rocap, G.; Kroth, P.G.; Armbrust, E.V.; Mock, T. Plastid Proteome Prediction for Diatoms and Other Algae with Secondary Plastids of the Red Lineage. Plant J. 2015, 81, 519–528. [Google Scholar] [CrossRef]
- Claros, M.G.; Vincens, P. Computational Method to Predict Mitochondrially Imported Proteins and Their Targeting Sequences. Eur. J. Biochem. 1996, 241, 779–786. [Google Scholar] [CrossRef]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Almagro Armenteros, J.J.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM Predicts Alpha and Beta Transmembrane Proteins Using Deep Neural Networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Xie, Y.; Zheng, Y.; Li, H.; Luo, X.; He, Z.; Cao, S.; Shi, Y.; Zhao, Q.; Xue, Y.; Zuo, Z.; et al. GPS-Lipid: A Robust Tool for the Prediction of Multiple Lipid Modification Sites. Sci. Rep. 2016, 6, 28249. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ID (Phatr3) | Family | Regulation (log2) | GRAVY | Predicted Localization |
|---|---|---|---|---|
| J54974 | α/β | −3.35 | −0.35 | Cytosol |
| EG02408 | α/β | −3.32 | −0.091 | Chloroplast |
| EG00720 | Patatin | −2.80 | 0.078 | ER/Cytosol |
| J40695 | α/β | 2.03 | −0.108 | Cytosol |
| J41624 | Class 3 | 2.09 | −0.411 | ER and/or vacuole |
| EG02610 | Class 3 | 2.96 | −0.357 | Cytosol |
| J11048 | α/β | 2.98 | −0.304 | Cytosol |
| J44028 | Class 3 | 3.02 | −0.381 | Chloroplast |
| EG02330 | GDSL | 3.41 | −0.168 | Cytosol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murison, V.; Hérault, J.; Schoefs, B.; Marchand, J.; Ulmann, L. Bioinformatics-Based Screening Approach for the Identification and Characterization of Lipolytic Enzymes from the Marine Diatom Phaeodactylum tricornutum. Mar. Drugs 2023, 21, 125. https://doi.org/10.3390/md21020125
Murison V, Hérault J, Schoefs B, Marchand J, Ulmann L. Bioinformatics-Based Screening Approach for the Identification and Characterization of Lipolytic Enzymes from the Marine Diatom Phaeodactylum tricornutum. Marine Drugs. 2023; 21(2):125. https://doi.org/10.3390/md21020125
Chicago/Turabian StyleMurison, Victor, Josiane Hérault, Benoît Schoefs, Justine Marchand, and Lionel Ulmann. 2023. "Bioinformatics-Based Screening Approach for the Identification and Characterization of Lipolytic Enzymes from the Marine Diatom Phaeodactylum tricornutum" Marine Drugs 21, no. 2: 125. https://doi.org/10.3390/md21020125
APA StyleMurison, V., Hérault, J., Schoefs, B., Marchand, J., & Ulmann, L. (2023). Bioinformatics-Based Screening Approach for the Identification and Characterization of Lipolytic Enzymes from the Marine Diatom Phaeodactylum tricornutum. Marine Drugs, 21(2), 125. https://doi.org/10.3390/md21020125