
New Azaphilones from the Marine-Derived Fungus Penicillium sclerotiorum E23Y-1A with Their Anti-Inflammatory and Antitumor Activities

 ,
,  ,
, 
Abstract
:1. Introduction
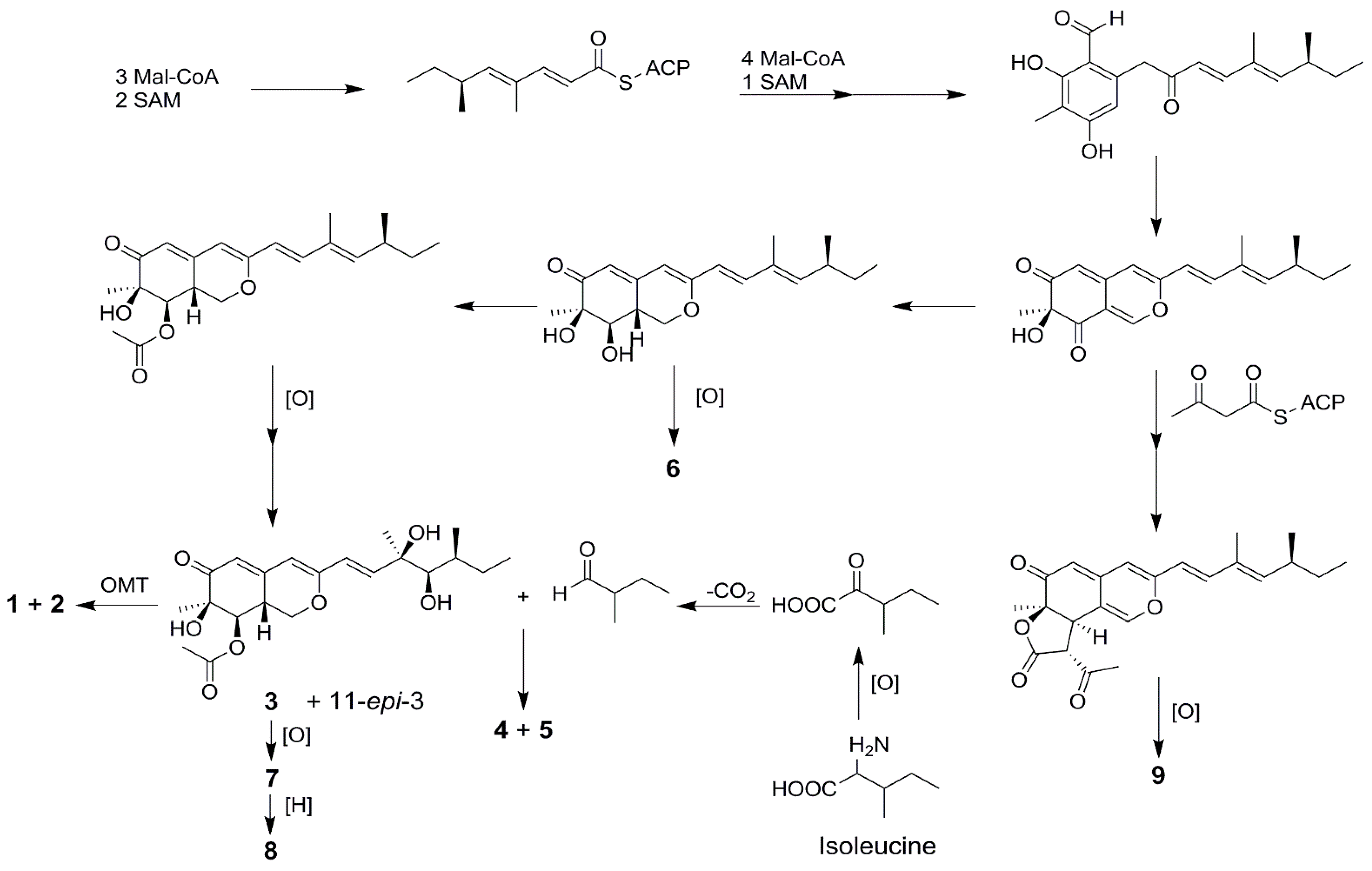
2. Results and Discussion
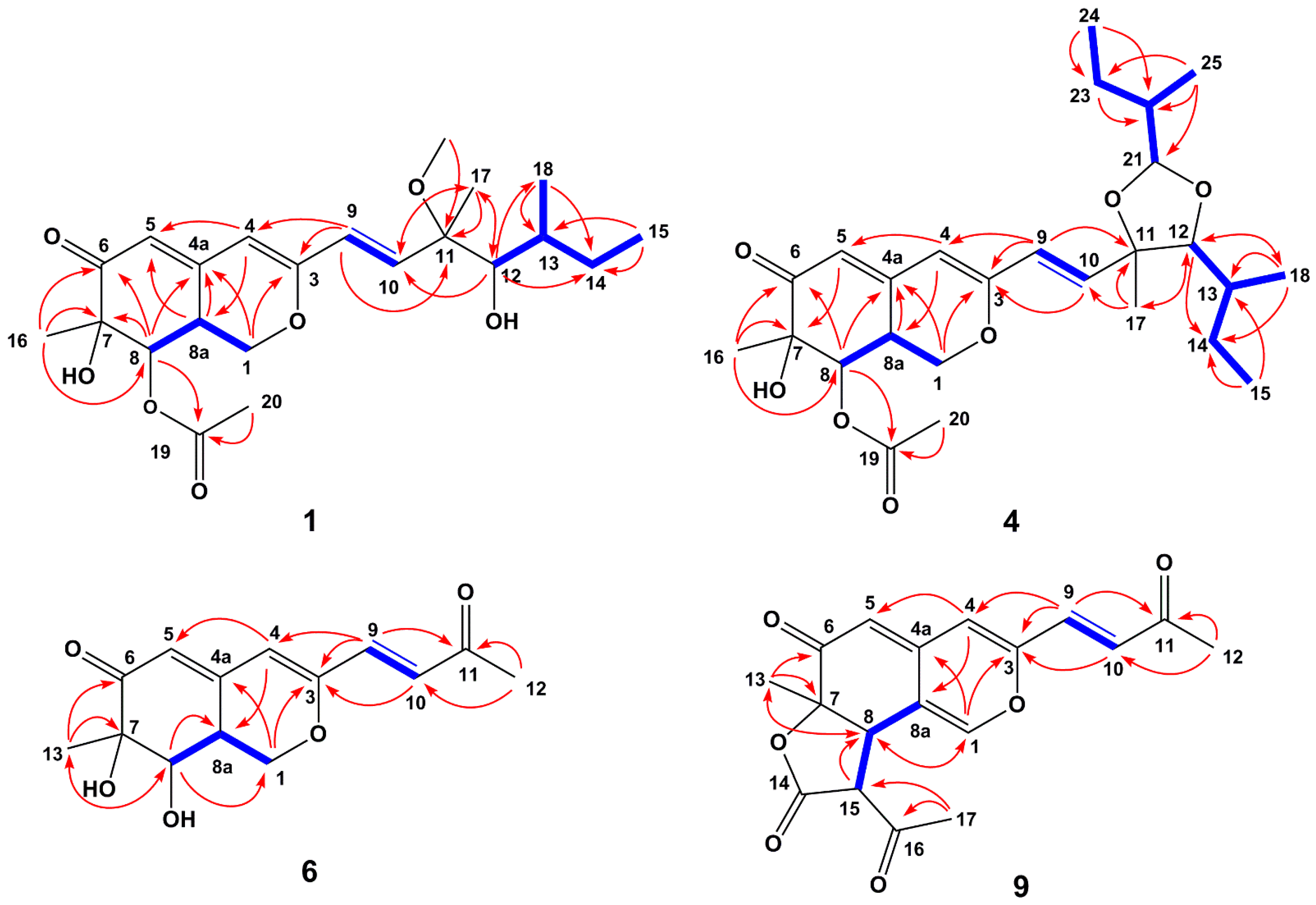
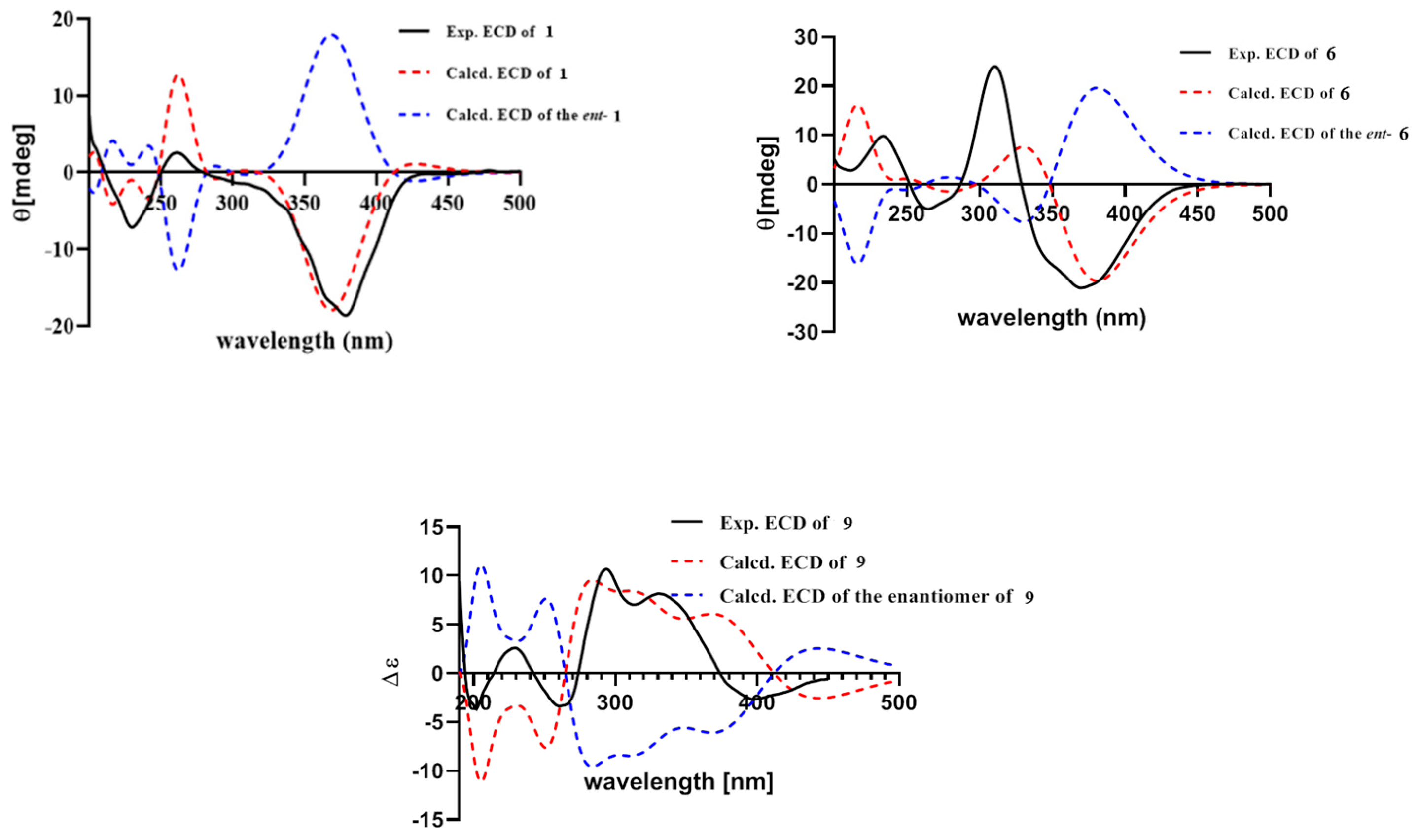
2.1. Structure Elucidation of New Compounds

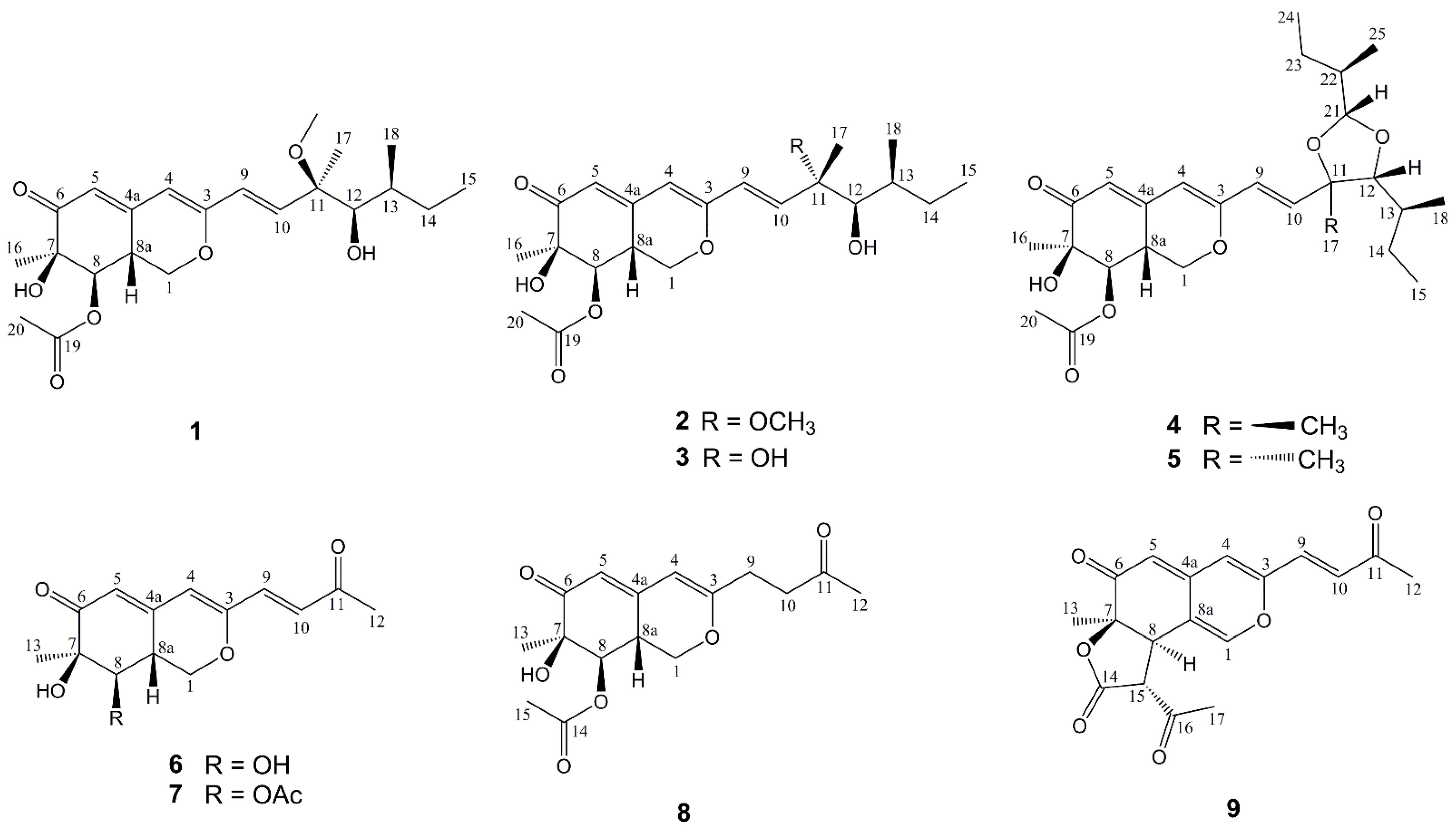{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 b | 4 a | 5 a | 6 a | 7 a | 8 a | 9 a |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4.35, dd (10.8, 5.2) | 4.35, dd (10.8, 5.2) | 4.35, dd (10.7, 5.2) | 4.36, dd (10.7, 5.3) | 4.35, dd (10.8, 5.2) | 4.86, dd, (10.9, 5.5) | 4.39, dd, (10.8, 5.3) | 4.28, dd, (10.7, 5.3) | 7.41, s |
| 3.80, dd (13.7,10.8) | 3.80, dd (13.6, 10.8) | 3.81, dd (13.6,10.7) | 3.79, dd (13.6, 10.7) | 3.80, dd (13.6, 10.7) | 3.81, dd, (13.4, 10.9) | 3.82, dd, (13.6, 10.9) | 3.73, dd, (13.8, 10.8) | ||
| 4 | 5.67, s | 5.67, s | 5.67, s | 5.67, s | 5.68, s | 5.94, s | 5.96, s | 5.52, s | 6.37, s |
| 5 | 5.80, brs | 5.79, s | 5.79, d (2.1) | 5.80, d (2.1) | 5.79, d (2.1) | 5.88, brs | 5.91, brs | 5.70, s | 5.53, s |
| 8 | 4.98, d (10.0) | 4.97, d (10.1) | 4.98, d (10.0) | 4.98, d (10.1) | 4.98, d (10.0) | 3.47, d (9.3) | 4.99, d (10.0) | 4.95, d (9.9) | 3.85, d (12.2) |
| 8a | 3.35, m | 3.35, m | 3.35, m | 3.34, m | 3.34, m | 3.04, m | 3.37, m | 3.27, m | |
| 9 | 6.00, d (15.9) | 5.98, d (16.1) | 6.19, d (15.6) | 6.12, d (15.5) | 6.18, d (15.4) | 6.85, d (15.6) | 6.84, d (15.7) | 2.51, t (7.3) | 6.93, d (15.8) |
| 10 | 6.31, d (15.9) | 6.42, d (16.1) | 6.43, d (15.5) | 6.37, d (15.5) | 6.39, d (15.4) | 6.70, d (15.7) | 6.65, d (15.7) | 2.68, m | 6.76, d (15.7) |
| 12 | 3.39, d (2.7) | 3.45, s | 3.49, d (2.1) | 3.38, d (8.8) | 3.30, d (9.6) | 2.33, s | 1.36, s | 2.17, s | 2.36, s |
| 13 | 1.53, m | 1.47, m | 1.53, m | 1.47, m | 1.64, m | 1.49, s | 1.49, s | 1.34, s | 1.58, s |
| 14 | 1.38, m | 1.40, m | 1.41, m | 1.62, m | 1.39, m | ||||
| 1.24, m | 1.28, m | 1.30, m | 1.07, m | 1.01, m | |||||
| 15 | 0.85, t (7.4) | 0.87, t (7.4) | 0.90, t (7.4) | 0.88, t (7.4) | 0.90, t (7.3) | 2.23, s | 2.20, s | 3.75, d (12.3) | |
| 16 | 1.35, s | 1.34, s | 1.35, s | 1.36, s | 1.36, s | ||||
| 17 | 1.29, s | 1.31, s | 1.35, s | 1.41, s | 1.24, s | 2.48, s | |||
| 18 | 0.90, d (6.8) | 0.83, d (6.5) | 0.86, d (6.8) | 0.97, d (6.6) | 1.04, d (6.5) | ||||
| 20 | 2.21, s | 2.20, s | 2.21, s | 2.21, s | 2.21, s | ||||
| OCH3 | 3.16, s | 3.16, s | |||||||
| 21 | 4.71, d (5.0) | 4.74, d (4.0) | |||||||
| 22 | 1.62, m | 1.60, m | |||||||
| 23 | 1.60, m | 1.60, m | |||||||
| 1.20, m | 1.20, m | ||||||||
| 24 | 0.93, t (7.3) | 0.93, t (7.3) | |||||||
| 25 | 0.98, d (6.6) | 0.94, d (6.6) |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 68.0 | 68.0 | 68.1 | 68.1 | 68.0 | 69.1 | 68.2 | 68.3 | 147.1 |
| 3 | 159.6 | 159.6 | 160.0 | 160.0 | 160.0 | 158.1 | 157.7 | 166.4 | 153.5 |
| 4 | 105.0 | 104.8 | 104.7 | 104.8 | 105.0 | 110.9 | 110.7 | 101.3 | 114.3 |
| 4a | 151.1 | 151.1 | 151.1 | 151.2 | 151.0 | 149.8 | 149.2 | 150.9 | 142.8 |
| 5 | 116.9 | 116.9 | 116.7 | 116.7 | 116.7 | 119.1 | 119.5 | 115.5 | 109.1 |
| 6 | 194.6 | 194.6 | 194.4 | 194.4 | 194.4 | 196.7 | 194.5 | 194.5 | 191.8 |
| 7 | 74.4 | 74.4 | 74.4 | 74.5 | 74.5 | 74.5 | 74.6 | 74.4 | 82.8 |
| 8 | 74.5 | 74.5 | 74.6 | 74.5 | 74.5 | 74.4 | 74.3 | 74.5 | 42.9 |
| 8a | 34.7 | 34.7 | 36.4 | 34.8 | 34.8 | 36.7 | 34.7 | 34.4 | 114.4 |
| 9 | 125.0 | 125.0 | 122.3 | 123.2 | 122.5 | 135.0 | 134.8 | 28.5 | 131.9 |
| 10 | 139.5 | 138.4 | 140.0 | 138.9 | 140.8 | 130.3 | 130.4 | 40.1 | 131.0 |
| 11 | 80.4 | 80.6 | 76.1 | 80.8 | 80.7 | 197.8 | 197.7 | 206.6 | 196.8 |
| 12 | 79.5 | 80.2 | 79.4 | 90.4 | 88.8 | 28.7 | 28.5 | 30.1 | 29.0 |
| 13 | 35.4 | 35.7 | 34.8 | 35.2 | 34.5 | 20.5 | 19.9 | 20.1 | 23.3 |
| 14 | 29.0 | 29.2 | 28.9 | 25.6 | 25.4 | 170.5 | 170.6 | 168.3 | |
| 15 | 11.9 | 12.0 | 12.0 | 11.2 | 11.1 | 20.8 | 20.8 | 57.2 | |
| 16 | 20.0 | 20.0 | 20.1 | 20.1 | 20.1 | 200.0 | |||
| 17 | 16.4 | 18.5 | 26.7 | 25.2 | 21.3 | 30.3 | |||
| 18 | 14.0 | 13.8 | 13.4 | 16.0 | 16.4 | ||||
| 19 | 170.6 | 170.6 | 170.6 | 170.6 | 170.5 | ||||
| 20 | 20.8 | 20.8 | 20.8 | 20.8 | 20.8 | ||||
| OCH3 | 50.4 | 50.6 | |||||||
| 21 | 105.3 | 105.8 | |||||||
| 22 | 38.8 | 38.7 | |||||||
| 23 | 24.8 | 24.5 | |||||||
| 24 | 11.6 | 11.8 | |||||||
| 25 | 14.1 | 13.6 |
2.2. Bioassay
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Identification, Fermentation, and Extract
3.3. Isolation and Purification
3.4. NMR Calculation
3.5. ECD Calculation
3.6. Anti-Inflammatory Activity Test
3.7. Cytotoxic Detection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2022, 39, 1122–1171. [Google Scholar] [CrossRef] [PubMed]
- Julianti, E.; Abrian, I.A.; Wibowo, M.S.; Azhari, M.; Tsurayya, N.; Izzati, F.; Juanssilfero, A.B.; Bayu, A.; Rahmawati, S.I.; Putra, M.Y. Secondary metabolites from marine-derived fungi and actinobacteria as potential sources of novel colorectal cancer drugs. Mar. Drugs 2022, 20, 67. [Google Scholar] [CrossRef] [PubMed]
- Al-Saleem, M.S.M.; Hassan, W.H.B.; El Sayed, Z.I.; Abdel-Aal, M.M.; Abdel-Mageed, W.M.; Abdelsalam, E.; Abdelaziz, S. Metabolic profiling and in vitro assessment of the biological activities of the ethyl acetate extract of Penicillium chrysogenum “Endozoic of Cliona sp. Marine Sponge” from the Red Sea (Egypt). Mar. Drugs 2022, 20, 326. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.P.; Chen, D.; Xie, C.L.; Liu, Q.; Zhong, T.H.; Shao, Z.; Liu, G.; Luo, L.Z.; Yang, X.W. Anti-food allergic compounds from Penicillium griseofulvum MCCC 3A00225, a deep-sea-derived fungus. Mar. Drugs 2021, 19, 224. [Google Scholar] [CrossRef] [PubMed]
- McCauley, E.P.; Piña, I.C.; Thompson, A.D.; Bashir, K.; Weinberg, M.; Kurz, S.L.; Crews, P. Highlights of marine natural products having parallel scaffolds found from marine-derived bacteria, sponges, and tunicates. J. Antibiot. 2020, 73, 504–525. [Google Scholar] [CrossRef]
- Liu, L.; Zheng, Y.Y.; Shao, C.L.; Wang, C.Y. Metabolites from marine invertebrates and their symbiotic microorganisms: Molecular diversity discovery, mining, and application. Mar. Life Sci. Tech. 2019, 1, 60–94. [Google Scholar] [CrossRef] [Green Version]
- Pang, K.L.; Overy, D.P.; Jones, E.B.G.; Calado, M.D.L.; Burgaud, G.; Walker, A.K.; Johnson, J.A.; Kerr, R.G.; Cha, H.J.; Bills, G.F. ‘Marine fungi’ and ‘marine-derived fungi’ in natural product chemistry research Toward a new consensual definition. Fungal Biol. Rev. 2016, 30, 163–175. [Google Scholar] [CrossRef]
- Ding, B.; Yin, Y.; Zhang, F.L.; Li, Z.Y. Recovery and phylogenetic diversity of culturable fungi associated with marine sponges Clathrina luteoculcitella and Holoxea sp. in the South China Sea. Mar. Biotechnol. 2011, 13, 713–721. [Google Scholar] [CrossRef]
- Ma, H.G.; Liu, Q.; Zhu, G.L.; Liu, H.S.; Zhu, W.M. Marine natural products sourced from marine-derived Penicillium fungi. J. Asian Nat. Prod. Res. 2016, 18, 92–115. [Google Scholar] [CrossRef]
- Yang, X.L.; Liu, J.P.; Mei, J.H.; Jiang, R.; Tu, S.Z.; Deng, H.F.; Liu, J.; Yang, S.M.; Li, J. Origins, structures, and bioactivities of secondary metabolites from marine-derived Penicillium fungi. Mini Rev. Med. Chem. 2021, 21, 2000–2019. [Google Scholar] [CrossRef]
- Yang, J.Y.; Tanga, M.M.; Chen, L.; Lai, X.Y.; Zhuo, X.; Zhou, X.M.; Chen, G.Y. Study on the secondary metabolites of endophytic penicillium sclerotiorum HLL113. Chin. J. Org. Chem. 2022, 42, 896–900. [Google Scholar] [CrossRef]
- Wang, H.C.; Ke, T.Y.; Ko, Y.C.; Lin, J.J.; Chang, J.S.; Cheng, Y.B. Anti-inflammatory azaphilones from the edible alga-derived fungus Penicillium sclerotiorum. Mar. Drugs 2021, 19, 529. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.M.; Yang, S.X.; Qin, J.C. Azaphilonoids: Chemistry and biology. Chem. Rev. 2013, 113, 4755–4811. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zeng, Y.B.; Yin, J.J.; Chang, W.J.; Zhao, X.L.; Mao, Y. Two new azaphilones from the marine-derived fungus Penicillium sclerotiorum E23Y-1A. Phytochem. Lett. 2022, 47, 76–80. [Google Scholar] [CrossRef]
- Wang, Z.; Zeng, Y.B.; Zhao, W.B.; Dai, F.H.; Chang, W.J.; Lv, F. Structures and biological activities of brominated azaphilones produced by Penicillium sclerotiorum E23Y-1A. Phytochem. Lett. 2022, 52, 138–142. [Google Scholar] [CrossRef]
- Gu, B.B.; Wu, J.; Tang, J.; Jiao, W.H.; Li, L.; Sun, F.; Wang, S.P.; Yang, F.; Lin, H.W. Azaphilone and isocoumarin derivatives from the sponge-derived fungus Eupenicillium sp. 6A-9. Tetrahedron Lett. 2018, 59, 3345–3348. [Google Scholar] [CrossRef]
- Son, S.; Ko, S.K.; Kim, J.W.; Lee, J.K.; Jang, M.; Ryoo, I.J.; Hwang, G.J.; Kwon, M.C.; Shin, K.S.; Futamura, Y.; et al. Structures and biological activities of azaphilones produced by Penicillium sp. KCB11A109 from a ginseng field. Phytochemistry 2016, 122, 154–164. [Google Scholar] [CrossRef]
- Pavesi, C.; Flon, V.; Mann, S.; Leleu, S.; Prado, S.; Franck, X. Biosynthesis of azaphilones: A review. Nat. Prod. Rep. 2021, 38, 1058–1071. [Google Scholar] [CrossRef]
- Luo, X.; Lin, X.; Tao, H.; Wang, J.; Li, J.; Yang, B.; Zhou, X.; Liu, Y. Isochromophilones A–F, cytotoxic chloroazaphilones from the marine mangrove endophytic fungus Diaporthe sp. SCSIO 41011. J. Nat. Prod. 2018, 81, 934–941. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Hao, J.-D.; Ning, X.-Y.; Wu, J.-S.; Zhao, D.-L.; Kong, C.-J.; Shao, C.-L.; Wang, C.-Y. Penicilazaphilones D and E: Two new azaphilones from a sponge-derived strain of the fungus Penicillium sclerotiorum. RSC Adv. 2018, 8, 4348–4353. [Google Scholar] [CrossRef]
- Yoshida, E.; Fujimoto, H.; Baba, M.; Yamazaki, M. Four new chlorinated azaphilones, helicusins A–D, closely related to 7-epi-sclerotiorin, from an ascomycetous fungus, Talaromyces helices. Chem. Pharm. Bull. 1995, 43, 1307–1310. [Google Scholar] [CrossRef] [Green Version]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Jinno, M.; Kikuchi, T.; Kajimoto, T.; Numata, A.; Tanaka, R. Three new azaphilones produced by a marine fish-derived Chaetomium globosum. J. Antibiot. 2012, 65, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wu, C.; Long, H.; Chen, R.; Liu, D.; Proksch, P.; Guo, P.; Lin, W. Varioxiranols A−G and 19-O-methyl-22-methoxypre-shamixanthone, PKS and hybrid PKS-derived metabolites from a sponge-associated Emericella variecolor fungus. J. Nat. Prod. 2015, 78, 2461–2470. [Google Scholar] [CrossRef]
- Iwai, T.; Kubota, T.; Kobayashi, J. Absolute configuration of amphidinin A. J. Nat. Prod. 2014, 77, 1541–1544. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef]
- Kim, S.M.; Son, S.; Kim, J.W.; Jeon, E.S.; Ko, S.K.; Ryoo, I.J.; Shin, K.S.; Hirota, H.; Takahashi, S.; Osada, H.; et al. Penidioxolanes A and B, 1, 3-dioxolane containing azaphilone derivatives from marine-derived Penicillium sp. KCB12C078. Nat. Prod. Sci. 2015, 21, 231–236. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Vandermeersch, T.; Flynn, C.J.; Maguire, A.R.; Hutchison, G.R. Confab-systematic generation of diverse low-energy conformers. J. Cheminformatics 2011, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Bannwarth, C.; Ehlert, S.; Grimme, S.J. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019.
- Bruhn, T.; Schaumloffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.Z.; Wang, H.; Li, W.; Yang, L.; Yang, J.L.; Yuan, J.Z.; Wei, Y.M.; Jiang, B.; Mei, W.L.; Dai, H.F. Filarones A and B, new anti-inflammatory dimeric 2-(2-phenethyl) chromones from agarwood of Aquilaria filaria. Phytochem. Lett. 2021, 46, 11–14. [Google Scholar] [CrossRef]
- Mosmann, T.J. Rapid colorimetic assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 53–63. [Google Scholar] [CrossRef] [PubMed]








| Compound | IC50 ± SD (μM) a | ||||
|---|---|---|---|---|---|
| K562 | BEL-7402 | SGC-7901 | A549 | Hela | |
| 1 | – | – | – | – | – |
| 2 | – | – | – | – | – |
| 3 | – | – | – | – | – |
| 4 | 23.94 ± 0.11 | 60.66 ± 0.13 | 46.17 ± 0.17 | 60.16 ± 0.26 | 59.30 ± 0.60 |
| 5 | – | – | – | – | – |
| 6 | – | – | – | – | – |
| 7 | – | – | – | – | – |
| 8 | – | – | – | – | – |
| 9 | – | – | – | – | – |
| cisplatin b | 3.08 ± 0.05 | 4.02 ± 0.06 | 4.11 ± 0.02 | 1.93 ± 0.02 | 11.29 ± 0.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Y.; Wang, Z.; Chang, W.; Zhao, W.; Wang, H.; Chen, H.; Dai, H.; Lv, F. New Azaphilones from the Marine-Derived Fungus Penicillium sclerotiorum E23Y-1A with Their Anti-Inflammatory and Antitumor Activities. Mar. Drugs 2023, 21, 75. https://doi.org/10.3390/md21020075
Zeng Y, Wang Z, Chang W, Zhao W, Wang H, Chen H, Dai H, Lv F. New Azaphilones from the Marine-Derived Fungus Penicillium sclerotiorum E23Y-1A with Their Anti-Inflammatory and Antitumor Activities. Marine Drugs. 2023; 21(2):75. https://doi.org/10.3390/md21020075
Chicago/Turabian StyleZeng, Yanbo, Zhi Wang, Wenjun Chang, Weibo Zhao, Hao Wang, Huiqin Chen, Haofu Dai, and Fang Lv. 2023. "New Azaphilones from the Marine-Derived Fungus Penicillium sclerotiorum E23Y-1A with Their Anti-Inflammatory and Antitumor Activities" Marine Drugs 21, no. 2: 75. https://doi.org/10.3390/md21020075
APA StyleZeng, Y., Wang, Z., Chang, W., Zhao, W., Wang, H., Chen, H., Dai, H., & Lv, F. (2023). New Azaphilones from the Marine-Derived Fungus Penicillium sclerotiorum E23Y-1A with Their Anti-Inflammatory and Antitumor Activities. Marine Drugs, 21(2), 75. https://doi.org/10.3390/md21020075