
Isolation and Characterization of NpCI, a New Metallocarboxypeptidase Inhibitor from the Marine Snail Nerita peloronta with Anti-Plasmodium falciparum Activity

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
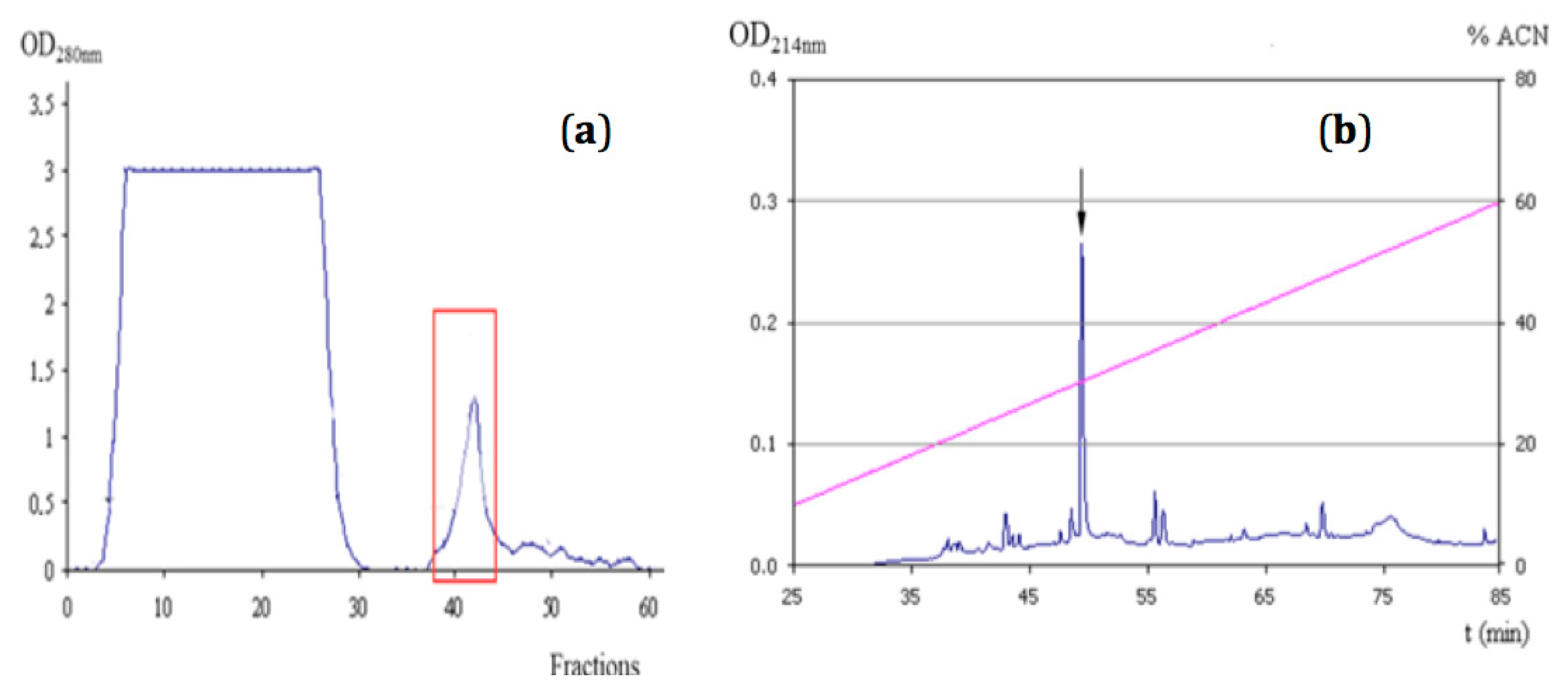
2.1. Purification of NpCI
2.2. Molecular Characterization of NpCI
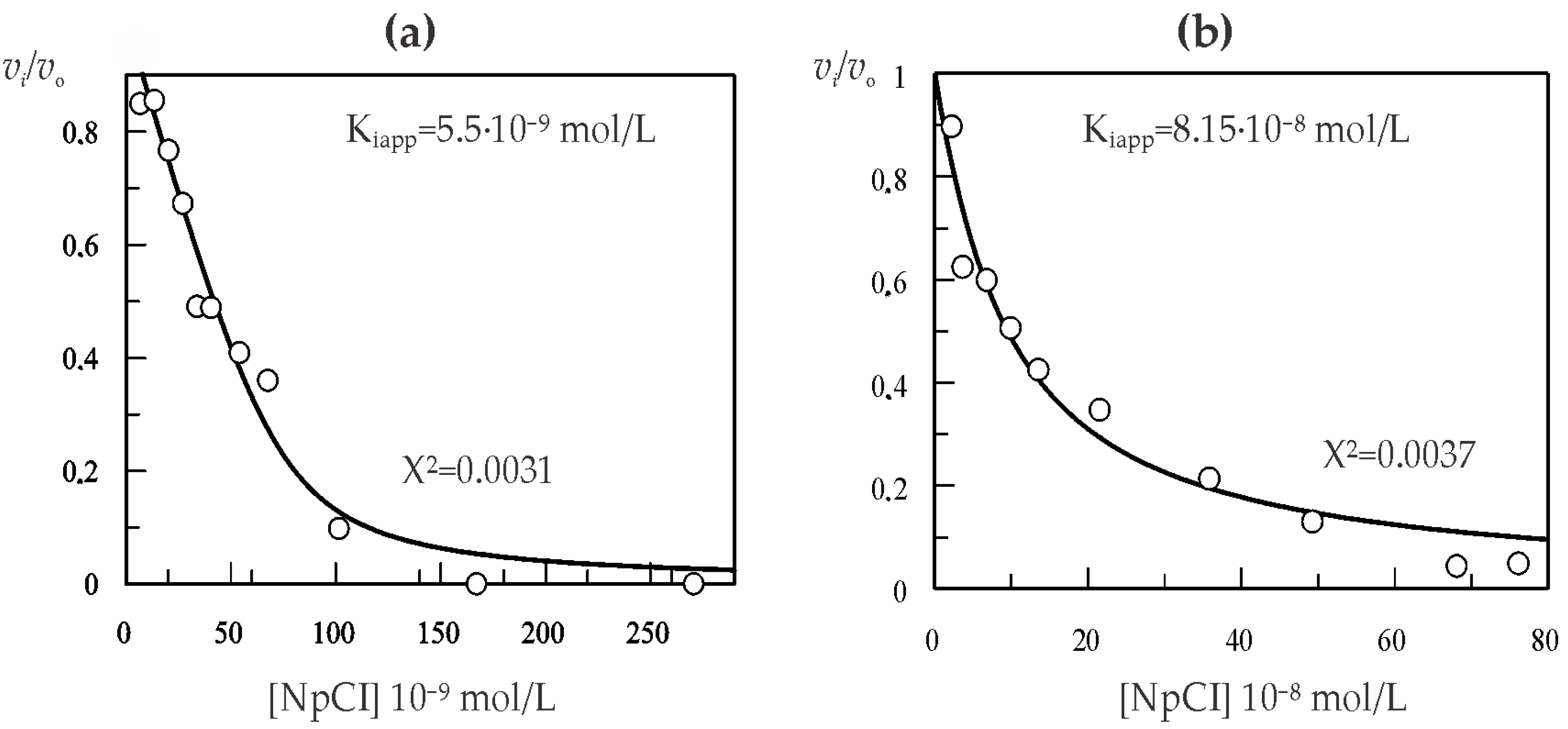
2.3. Kinetic Characterization of NpCI
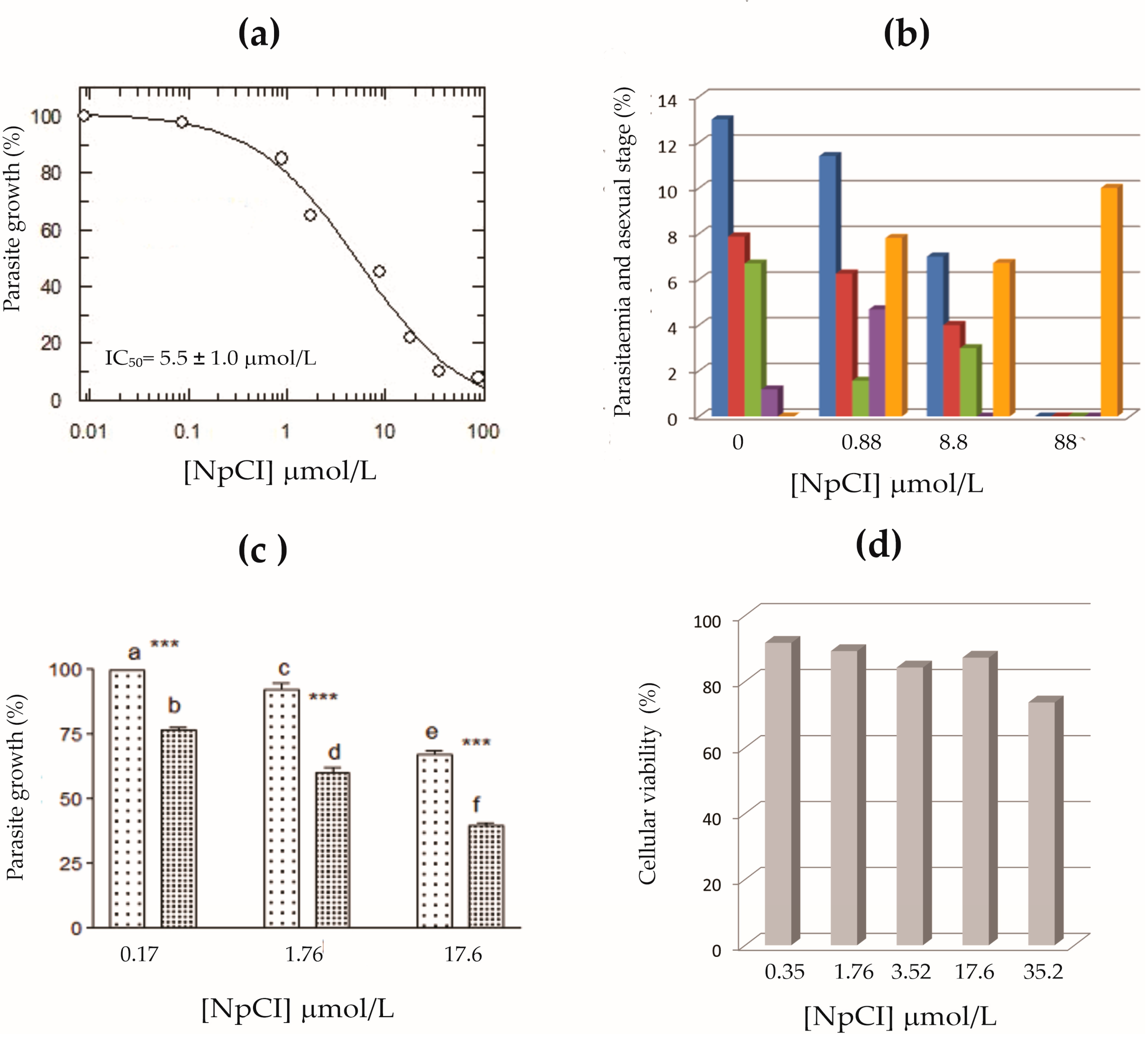
2.4. Inhibitory Effect of Purified NpCI on P. falciparum Growth
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Purification of Nerita peloronta Carboxypeptidase Inhibitor (NpCI)
4.2.2. Molecular Characterization of NpCI
4.2.3. Kinetic Characterization of NpCI
4.2.4. Analysis of NpCI Fractions on P. falciparum Growth
4.2.5. Cytotoxicity Effect of NpCI Fractions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Arolas, J.L.; Vendrell, J.; Aviles, F.X.; Fricker, L.D. Metallocarboxypeptidases: Emerging Drug Targets in Biomedicine. Curr. Pharm. Des. 2007, 13, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Bulinski, J.C. Post-Translational Regulation of the Microtubule Cytoskeleton: Mechanisms and Functions. Nat. Rev. Mol. Cell Biol. 2011, 12, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Pallares, I.; Covaleda, G.X.; Aviles, F.; Vendrell, J. Metallocarboxypeptidases and Their Inhibitors: Recent Developments in Biomedically Relevant Protein and Organic Ligands. Curr. Med. Chem. 2013, 20, 1595–1608. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46, 624–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, G.M.; Nau, H.; Biemann, K.; Grahn, D.T.; Ericsson, L.H.; Neurath, H. The Amino Acid Sequence of a Carboxypeptidase Inhibitor from Potatoes. Biochemistry 1975, 14, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Hass, G.M.; Hermodson, M.A. Amino Acid Sequence of a Carboxypeptidase Inhibitor from Tomato Fruit. Biochemistry 1981, 20, 2256–2260. [Google Scholar] [CrossRef] [PubMed]
- Arolas, J.L.; Lorenzo, J.; Rovira, A.; Castellà, J.; Aviles, F.X.; Sommerhoff, C.P. A Carboxypeptidase Inhibitor from the Tick Rhipicephalus bursa. J. Biol. Chem. 2005, 280, 3441–3448. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Zhou, J.; Liao, M.; Hatta, T.; Harnnoi, T.; Umemiya, R.; Inoue, N.; Xuan, X.; Fujisaki, K. Characterization of a Carboxypeptidase Inhibitor from the Tick Haemaphysalis longicornis. J. Insect Physiol. 2007, 53, 1079–1087. [Google Scholar] [CrossRef]
- Reverter, D.; Vendrell, J.; Canals, F.; Horstmann, J.; Avilés, F.X.; Fritz, H.; Sommerhoff, C.P. A Carboxypeptidase Inhibitor from the Medical Leech Hirudo medicinalis. J. Biol. Chem. 1998, 273, 32927–32933. [Google Scholar] [CrossRef] [Green Version]
- Homandberg, G.A.; Litwiller, R.D.; Peanasky, R.J. Carboxypeptidase Inhibitors from Ascaris suum: The Primary Structure. Arch. Biochem. Biophys. 1989, 270, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Covaleda, G.; Alonso Del Rivero, M.; Chávez, M.A.; Avilés, F.X.; Reverter, D. Crystal Structure of Novel Metallocarboxypeptidase Inhibitor from Marine Mollusk Nerita versicolor in Complex with Human Carboxypeptidase A4. J. Biol. Chem. 2012, 287, 9250–9258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covaleda-Cortés, G.; Hernández, M.; Trejo, S.A.; Mansur, M.; Rodríguez-Calado, S.; García-Pardo, J.; Lorenzo, J.; Vendrell, J.; Chávez, M.Á.; Alonso-Del-Rivero, M.; et al. Characterization, Recombinant Production and Structure-Function Analysis of NvCI, A Picomolar Metallocarboxypeptidase Inhibitor from the Marine Snail Nerita versicolor. Mar. Drugs 2019, 17, 511. [Google Scholar] [CrossRef] [Green Version]
- Alonso-del-Rivero, M.; Trejo, S.A.; Reytor, M.L.; Rodriguez-De-La-Vega, M.; Delfin, J.; Diaz, J.; González-González, Y.; Canals, F.; Chavez, M.A.; Aviles, F.X. Tri-Domain Bifunctional Inhibitor of Metallocarboxypeptidases A and Serine Proteases Isolated from Marine Annelid Sabellastarte magnifica. J. Biol. Chem. 2012, 287, 15427–15438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uratini, Y.; Takiguchi-Hayashi, K.; Miyasaka, N.; Sato, M.; Jin, M.; Arimatsu, Y. Latexin, a Carboxypeptidase A Inhibitor, Is Expressed in Rat Peritoneal Mast Cells and Is Associated with Granular Structures Distinct from Secretory Granules and Lysosomes. Biochem. J. 2000, 346, 817–826. [Google Scholar] [CrossRef]
- Pallarès, I.; Bonet, R.; García-Castellanos, R.; Ventura, S.; Avilés, F.X.; Vendrell, J.; Gomis-Rüth, F.X. Structure of Human Carboxypeptidase A4 with Its Endogenous Protein Inhibitor, Latexin. Proc. Natl. Acad. Sci. USA 2005, 102, 3978–3983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konecny, F. Evaluation of Two Recombinant Plasminogen Activators in Massive Pulmonary Embolism Model and Potato Carboxypeptidase Inhibitor (PCI) Role in Inhibition of Thrombin Activatable Fibrinolysis Inhibitor TAFIa in Lungs. Recent Pat. Endocr. Metab. Immune Drug Discov. 2008, 2, 45–56. [Google Scholar] [CrossRef]
- Mao, S.S.; Holahan, M.A.; Bailey, C.; Wu, G.; Colussi, D.; Carroll, S.S.; Cook, J.J. Demonstration of Enhanced Endogenous Fibrinolysis in Thrombin Activatable Fibrinolysis Inhibitor-Deficient Mice. Blood Coagul. Fibrinolysis 2005, 16, 407–415. [Google Scholar] [CrossRef]
- Willemse, J.L.; Heylen, E.; Nesheim, M.E.; Hendriks, D.F. Carboxypeptidase U (TAFIa): A New Drug Target for Fibrinolytic Therapy. J. Thromb. Haemost. 2009, 7, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, A.; Listwan, P.; Cowieson, N.; Huber, T.; Ravasi, T.; Wells, C.A.; Flanagan, J.U.; Kellie, S.; Hume, D.A.; Kobe, B.; et al. An Inflammatory Role for the Mammalian Carboxypeptidase Inhibitor Latexin: Relationship to Cystatins and the Tumor Suppressor TIG1. Structure 2005, 13, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Howard, D.; Rector, K.; Swiderski, C.; Brandon, J.; Schook, L.; Mehta, J.; Bryson, J.S.; Bondada, S.; Liang, Y. Latexin Is Down-Regulated in Hematopoietic Malignancies and Restoration of Expression Inhibits Lymphoma Growth. PLoS ONE 2012, 7, e44979. [Google Scholar] [CrossRef]
- Aviles Puigvert, F.X.; Lorenzo Rivera, J.; Rodriguez-Vera, M.; QuerolMurillo, E.; Bautista Marugán, M.; Díez Martín, A.; Bautista Santa Cruz, J.M. Therapeutic Agents for Treatment of Malaria. WO/2008077977 A1 patent, 3 July 2008. [Google Scholar]
- Rodriguez De La Vega, M.; Sevilla, R.G.; Hermoso, A.; Lorenzo, J.; Tanco, S.; Diez, A.; Fricker, L.D.; Bautista, J.M.; Avilés, F.X. Nna1-like Proteins Are Active Metallocarboxypeptidases of a New and Diverse M14 Subfamily. FASEB J. 2007, 20, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez de La Vega Otazo, M.; Lorenzo, J.; Tort, O.; Avilés, F.X.; Bautista, J.M. Functional Segregation and Emerging Role of Cilia-Related Cytosolic Carboxypeptidases (CCPs). FASEB J. 2013, 27, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Tort, O.; Tanco, S.; Rocha, C.; Bièche, I.; Seixas, C.; Bosc, C.; Andrieux, A.; Moutin, M.J.; Avilés, F.X.; Lorenzo, J.; et al. The Cytosolic Carboxypeptidases CCP2 and CCP3 Catalyze Posttranslational Removal of Acidic Amino Acids. Mol. Biol. Cell 2014, 25, 3017–3027. [Google Scholar] [CrossRef] [PubMed]
- Tanco, S.; Tort, O.; Demol, H.; Aviles, F.X.; Gevaert, K.; Van Damme, P.; Lorenzo, J.C. Terminomics Screen for Natural Substrates of Cytosolic Carboxypeptidase 1 Reveals Processing of Acidic Protein C Termini. Mol. Cell. Proteomics 2015, 14, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Covaleda, G.; Trejo, S.A.; Salas-Sarduy, E.; Alonso, M.; Chavez, M.A.; Francesc, X. Intensity Fading MALDI-TOF Mass Spectrometry and Functional Proteomics Assignments to Identify Protease Inhibitors in Marine Invertebrates. J. Proteomics 2017, 2017 165, 75–92. [Google Scholar] [CrossRef]
- Mendiola, J.; Hernández, H.; Sariego, I.; Rojas, L.; Otero, A.; Ramírez, A.; de los Angeles Chávez, M.; Payrol, J.A.; Hernández, A. Antimalarial Activity from Three Ascidians: An Exploration of Different Marine Invertebrate Phyla. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 909–916. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–609. [Google Scholar]
- Garcia-Guerrero, M.C.; Garcia-Pardo, J.; Berenguer, E.; Fernandez-Alvarez, R.; Barfi, G.B.; Lyons, P.J.; Aviles, F.X.; Huber, R.; Lorenzo, J.; Reverter, D. Crystal Structure and Mechanism of Human Carboxypeptidase O: Insights into Its Specific Activity for Acidic Residues. Proc. Natl. Acad. Sci. USA 2018, 115, E3932–E3939. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.F. The Slow-Binding and Slow, Tight-Binding Inhibition of Enzyme-Catalysed Reactions. Trends Biochem. Sci. 1982, 7, 102–105. [Google Scholar] [CrossRef]
- Bieth, J. Theoretical and Practical Aspects of Proteinase Inhibition Kinetics. Methods Enzymol. 1995, 248, 59–84. [Google Scholar]
- Waern, I.; Taha, S.; Lorenzo, J.; Montpeyó, D.; Covaleda-Cortés, G.; Avilés, F.X.; Wernersson, S. Carboxypeptidase Inhibition by NvCI Suppresses Airway Hyperreactivity in a Mouse Asthma Model. Allergy Eur. J. Allergy Clin. Immunol. 2021, 76, 2234–2237. [Google Scholar] [CrossRef]
- Soria-Castro, R.; Meneses-Preza, Y.G.; Rodríguez-López, G.M.; Romero-Ramírez, S.; Sosa-Hernández, V.A.; Cervantes-Díaz, R.; Pérez-Fragoso, A.; Torres-Ruíz, J.; Gómez-Martín, D.; Campillo-Navarro, M.; et al. Severe COVID-19 Is Marked by Dysregulated Serum Levels of Carboxypeptidase A3 and Serotonin. J. Leukoc. Biol. 2021, 110, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Zhang, Z.; Cao, R.; Zang, H.; Pei, W.; Sun, T. CPA4 Promotes EMT in Pancreatic Cancer via Stimulating PI3K-AKT-MTOR Signaling. Onco. Targets Ther. 2020, 13, 8567–8580. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Cai, Z.M.; Li, P.J.; Wei, M.Y.; Wang, C.Y.; Gu, Y.C.; Shao, C.L. Trends of Antimalarial Marine Natural Products: Progresses, Challenges and Opportunities. Nat. Prod. Rep. 2022, 39, 969–990. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.K.; Seo, C.H.; Park, Y. Marine Peptides and Their Anti-Infective Activities. Mar. Drugs 2015, 13, 618–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanglas, L.; Aviles, F.; Huber, R.; Gomis-Rüth, F.X.; Arolas, J.L. Mammalian Metallopeptidase Inhibition at the Defense Barrier of Ascaris Parasite. Proc. Natl. Acad. Sci. USA 2009, 106, 1743–1747. [Google Scholar] [CrossRef] [Green Version]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists: Second Edition; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 1–538. [Google Scholar] [CrossRef]
- PlasmoDB. Available online: https://plasmodb.org/plasmo/app/#genome-browser (accessed on 10 October 2022).
- Pinheiro, L.C.S.; Feitosa, L.M.; Gandi, M.O.; Silveira, F.F.; Boechat, N. The Development of Novel Compounds Against Malaria: Quinolines, Triazolpyridines, Pyrazolopyridines and Pyrazolopyrimidines. Molecules. 2019, 24, 4095. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Krohn, R.L.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 1985, 7, 6–85. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Schnaible, V.; Wefing, S.; Resemann, A.; Suckau, D.; Bücker, A.; Wolf-Kümmeth, S.; Hoffmann, D. Screening for Disulfide Bonds in Proteins by MALDI In-Source Decay and LIFT-TOF/TOF-MS. Anal. Chem. 2002, 74, 4980–4988. [Google Scholar] [CrossRef]
- Gella, A.; Prada-Dacasa, P.; Carrascal, M.; Urpi, A.; González-Torres, M.; Abian, J.; Sanz, E.; Quintana, A. Mitochondrial Proteome of Affected Glutamatergic Neurons in a Mouse Model of Leigh Syndrome. Front. Cell Dev. Biol. 2020, 8, 660. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mock, W.; Liu, Y.; Stanford, D. Arazoformyl Peptide Surrogates as Spectrophotometric Kinetic Assay Substrates for Carboxypeptidase. A. Anal. Biochem. 1996, 239, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Mock, W.L.; Xhu, C. Catalytic Activity of Carboxypeptidase B and of Carboxypeptidase Y with Anisylazoformyl Substrates. Bioorg. Med. Chem. Lett. 1999, 9, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Leatherbarrow, R.J. Grafit 3.0; Erithacus Software Ltd.: Staines, UK, 1992. [Google Scholar]
- Erlanger, B.; Kokowsky, N.; Cohen, W. The Preparation and Properties of Two New Chromogenic Substrates of Trypsin. Arch. Biochem. Biophys. 1961, 95, 271–278. [Google Scholar] [CrossRef]
- Wells, J.A.; Cunningham, B.C.; Graycart, T.P.; Estell, D.A. Recruitment of Substrate-Specificity Properties from One Enzyme into a Related One by Protein Engineering. Biochemistry 1987, 84, 5167–5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schechter, I.; Berger, A. On the Size of the Active Site in Proteases I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–161. [Google Scholar] [CrossRef]
- Moneriz, C.; Marín-García, P.; García-Granados, A.; Bautista, J.M.; Diez, A.; Puyet, A. Parasitostatic Effect of Maslinic Acid. I. Growth Arrest of Plasmodium falciparum Intraerythrocytic Stages. Malar. J. 2011, 10, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radfar, A.; Méndez, D.; Moneriz, C.; Linares, M.; Marín-García, P.; Puyet, A.; Diez, A.; Bautista, J.M. Synchronous Culture of Plasmodium falciparum at High Parasitemia Levels. Nat. Protoc. 2009, 4, 1899–1915. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | [Prots] (mg/mL) | Inhibitory Activity (Ut) | Specific Activity (U/mg) | Yield (%) | Purification (Times) |
|---|---|---|---|---|---|
| TCA-clarified extract | 3.010 | 0.903 | 0.300 | 100 | 1 |
| Affinity chromatography | 0.183 | 0.724 | 3.956 | 80.2 | 13 |
| RP-HPLC | 0.023 | 0.710 | 30.87 | 78. 6 | 103 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrera-Muñoz, A.; Sierra-Gómez, Y.; Covaleda-Cortés, G.; Reytor, M.L.; González-González, Y.; Bautista, J.M.; Avilés, F.X.; Alonso-del-Rivero, M. Isolation and Characterization of NpCI, a New Metallocarboxypeptidase Inhibitor from the Marine Snail Nerita peloronta with Anti-Plasmodium falciparum Activity. Mar. Drugs 2023, 21, 94. https://doi.org/10.3390/md21020094
Cabrera-Muñoz A, Sierra-Gómez Y, Covaleda-Cortés G, Reytor ML, González-González Y, Bautista JM, Avilés FX, Alonso-del-Rivero M. Isolation and Characterization of NpCI, a New Metallocarboxypeptidase Inhibitor from the Marine Snail Nerita peloronta with Anti-Plasmodium falciparum Activity. Marine Drugs. 2023; 21(2):94. https://doi.org/10.3390/md21020094
Chicago/Turabian StyleCabrera-Muñoz, Aymara, Yusvel Sierra-Gómez, Giovanni Covaleda-Cortés, Mey L. Reytor, Yamile González-González, José M. Bautista, Francesc Xavier Avilés, and Maday Alonso-del-Rivero. 2023. "Isolation and Characterization of NpCI, a New Metallocarboxypeptidase Inhibitor from the Marine Snail Nerita peloronta with Anti-Plasmodium falciparum Activity" Marine Drugs 21, no. 2: 94. https://doi.org/10.3390/md21020094
APA StyleCabrera-Muñoz, A., Sierra-Gómez, Y., Covaleda-Cortés, G., Reytor, M. L., González-González, Y., Bautista, J. M., Avilés, F. X., & Alonso-del-Rivero, M. (2023). Isolation and Characterization of NpCI, a New Metallocarboxypeptidase Inhibitor from the Marine Snail Nerita peloronta with Anti-Plasmodium falciparum Activity. Marine Drugs, 21(2), 94. https://doi.org/10.3390/md21020094